
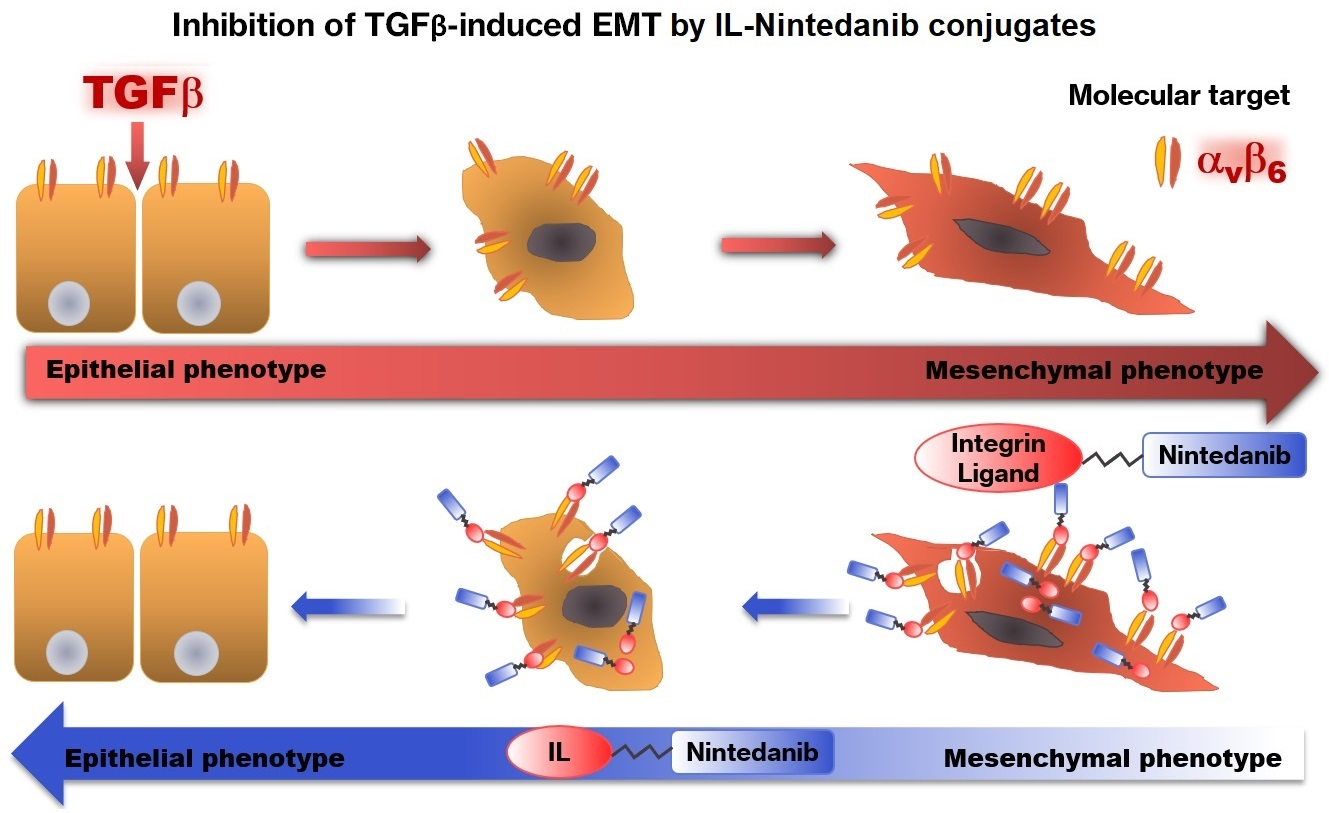
Nintedanib-αVβ6 Integrin Ligand Conjugates Reduce TGFβ-Induced EMT in Human Non-Small Cell Lung Cancer

 ,
, 
 ,
, 
 ,
,  , , , and
, , , and 
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Synthesis and Characterization of Conjugated Compounds
2.2. Integrin Expression
2.3. Inhibition of Adhesion
2.4. Cell Internalization of Conjugated Compounds 1–3 and Nintedanib
2.5. Tyrosine Kinase Inhibition Activity
2.6. Inhibition of α-Smooth Muscle Actin Protein Expression
2.7. Uptake and Inhibition of Tumor Spheroid Growth and Migration
2.8. Inhibition of the Epithelial-Mesenchymal Transition
2.9. Inhibition of Clonogenic Activity
3. Discussion
4. Materials and Methods
4.1. Synthesis and Characterization of Conjugated Compounds
4.2. Cell Cultures
4.3. Determination of αvβ6 Expression
4.4. Inhibition of Cell Adhesion to Fibronectin
4.5. Evaluation of Cell Internalization of Nintedanib and Conjugated Compounds 1–3
4.6. MTT Assay
4.7. Western Blot Assay
4.8. Inhibition of Multicellular Tumor Spheroids (MCTS) Migration and Growth
4.9. Immunofluorescence
4.10. Invasion Assay
4.11. Colonies
4.12. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liu, S.; Kurzrock, R. Toxicity of targeted therapy: Implications for response and impact of genetic polymorphisms. Cancer Treat. Rev. 2014, 40, 883–891. [Google Scholar] [CrossRef] [PubMed]
- Craene, B.; Berx, G. Regulatory networks defining EMT during cancer initiation and progression. Nat. Rev. Cancer 2013, 13, 97–110. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R. EMT: When epithelial cells decide to become mesenchymal-like cells. J. Clin. Investig. 2009, 119, 1417–1419. [Google Scholar] [CrossRef]
- Hass, R.; von der Ohe, J.; Ungefroren, H. The Intimate Relationship Among EMT, MET and TME: A T(ransdifferentiation) E(nhancing) M(ix) to Be Exploited for Therapeutic Purposes. Cancers 2020, 12, 3674. [Google Scholar] [CrossRef] [PubMed]
- Fuxe, J.; Karlsson, M.C. TGF-β-induced epithelial-mesenchymal transition: A link between cancer and inflammation. Semin. Cancer Biol. 2012, 22, 455–461. [Google Scholar] [CrossRef]
- Chattopadhyay, I.; Ambati, R.; Gundamaraju, R. Exploring the Crosstalk between Inflammation and Epithelial-Mesenchymal Transition in Cancer. Mediat. Inflamm. 2021, 2021, 9918379. [Google Scholar] [CrossRef]
- Tossetta, G.; Paolinelli, F.; Avellini, C.; Salvolini, E.; Ciarmela, P.; Lorenzi, T.; Emanuelli, M.; Toti, P.; Giuliante, R.; Gesuita, R.; et al. IL-1β and TGF-β weaken the placental barrier through destruction of tight junctions: An in vivo and in vitro study. Placenta 2014, 35, 509–516. [Google Scholar] [CrossRef]
- Kyuno, D.; Takasawa, A.; Kikuchi, S.; Takemasa, I.; Osanai, M.; Kojima, T. Role of tight junctions in the epithelial-to-mesenchymal transition of cancer cells. Biochim. Biophys. Acta. Biomembr. 2021, 1863, 183503. [Google Scholar] [CrossRef]
- Tiwari, N.; Gheldof, A.; Tatari, M.; Christofori, G. EMT as the ultimate survival mechanism of cancer cells. Semin. Cancer Biol. 2012, 22, 194–207. [Google Scholar] [CrossRef]
- Goteri, G.; Altobelli, E.; Tossetta, G.; Zizzi, A.; Avellini, C.; Licini, C.; Lorenzi, T.; Castellucci, M.; Ciavattini, A.; Marzioni, D. High temperature requirement A1, transforming growth factor beta1, phosphoSmad2 and Ki67 in eutopic and ectopic endometrium of women with endometriosis. Eur. J. Histochem. 2015, 59, 2570. [Google Scholar] [CrossRef]
- Shibue, T.; Weinberg, R.A. EMT, CSCs, and drug resistance: The mechanistic link and clinical implications. Nat. Rev. Clin. Oncol. 2017, 14, 611–629. [Google Scholar] [CrossRef]
- Doolin, M.T.; Smith, I.M.; Stroka, K.M. Fibroblast to myofibroblast transition is enhanced by increased cell density. Mol. Biol. Cell 2021, 32, ar41. [Google Scholar] [CrossRef] [PubMed]
- Kuzet, S.E.; Gaggioli, C. Fibroblast activation in cancer: When seed fertilizes soil. Cell Tissue Res. 2016, 365, 607–619. [Google Scholar] [CrossRef] [PubMed]
- Platel, V.; Faure, S.; Corre, I.; Clere, N. Endothelial-to-Mesenchymal Transition (EndoMT): Roles in Tumorigenesis, Metastatic Extravasation and Therapy Resistance. J. Oncol. 2019, 2019, 8361945. [Google Scholar] [CrossRef]
- Bianchini, F.; Peppicelli, S.; Fabbrizzi, P.; Biagioni, A.; Mazzanti, B.; Menchi, G.; Calorini, L.; Pupi, A.; Trabocchi, A. Triazole RGD antagonist reverts TGFβ1-induced endothelial-to-mesenchymal transition in endothelial precursor cells. Mol. Cell. Biochem. 2017, 424, 99–110. [Google Scholar] [CrossRef]
- Xu, X.; Zhang, Y.; Wang, X.; Li, S.; Tang, L. Substrate Stiffness Drives Epithelial to Mesenchymal Transition and Proliferation through the NEAT1-Wnt/β-Catenin Pathway in Liver Cancer. Int. J. Mol. Sci. 2021, 22, 12066. [Google Scholar] [CrossRef]
- Busilacchi, E.M.; Costantini, A.; Mancini, G.; Tossetta, G.; Olivieri, J.; Poloni, A.; Viola, N.; Butini, L.; Campanati, A.; Goteri, G.; et al. Nilotinib Treatment of Patients Affected by Chronic Graft-versus-Host Disease Reduces Collagen Production and Skin Fibrosis by Downmodulating the TGF-β and p-SMAD Pathway. Biol. Blood Marrow Transplant. 2020, 26, 823–834. [Google Scholar] [CrossRef] [PubMed]
- Davis, F.M.; Stewart, T.A.; Thompson, E.W.; Monteith, G.R. Targeting EMT in cancer: Opportunities for pharmacological intervention. Trends Pharmacol. Sci. 2014, 35, 479–488. [Google Scholar] [CrossRef] [PubMed]
- Schabath, M.B.; Cote, M.L. Cancer Progress and Priorities: Lung Cancer. Cancer Epidemiol. Biomark. Prev. 2019, 28, 1563–1579. [Google Scholar] [CrossRef]
- Planchard, D.; Popat, S.; Kerr, K.; Novello, S.; Smit, E.F.; Faivre-Finn, C.; Mok, T.S.; Reck, M.; Van Schil, P.E.; Hellmann, M.D.; et al. ESMO Guidelines Committee. Metastatic non-small cell lung cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2018, 29, iv192–iv237. [Google Scholar] [CrossRef]
- Duma, N.; Santana-Davila, R.; Molina, J.R. Non-Small Cell Lung Cancer: Epidemiology, Screening, Diagnosis, and Treatment. Mayo. Clin Proc. 2019, 94, 1623–1640. [Google Scholar] [CrossRef] [PubMed]
- Corral, J.; Majem, M.; Rodríguez-Abreu, D.; Carcereny, E.; Cortes, Á.A.; Llorente, M.; López Picazo, J.M.; García, Y.; Domine, M.; López Criado, M.P. Efficacy of nintedanib and docetaxel in patients with advanced lung adenocarcinoma treated with first-line chemotherapy and second-line immunotherapy in the nintedanib NPU program. Clin. Transl. Oncol. 2019, 21, 1270–1279. [Google Scholar] [CrossRef] [PubMed]
- Jakobsen, K.R.; Demuth, C.; Sorensen, B.S.; Nielsen, A.L. The role of epithelial to mesenchymal transition in resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small cell lung cancer. Transl. Lung Cancer Res. 2016, 5, 172–182. [Google Scholar] [CrossRef] [PubMed]
- Kasai, H.; Allen, J.T.; Mason, R.M.; Kamimura, T.; Zhang, Z. TGF-Β1 Induces Human Alveolar Epithelial to Mesen-chymal Cell Transition (EMT). Respir. Res. 2005, 6, 56. [Google Scholar] [CrossRef]
- Tsoukalas, N.; Aravantinou-Fatorou, E.; Tolia, M.; Giaginis, C.; Galanopoulos, M.; Kiakou, M.; Kostakis, I.D.; Dana, E.; Vamvakaris, I.; Korogiannos, A.; et al. Epithelial–Mesenchymal Transition in Non Small-Cell Lung Cancer. Anticancer. Res. 2017, 37, 1773–1778. [Google Scholar] [CrossRef]
- Shi, M.; Zhu, J.; Wang, R.; Chen, X.; Mi, L.; Walz, T.; Springer, T.A. Latent TGF-β structure and activation. Nature 2011, 474, 343–349. [Google Scholar] [CrossRef]
- Sigrist, C.J.; Bridge, A.; Le Mercier, P. A potential role for integrins in host cell entry by SARS-CoV-2. Antivir. Res. 2020, 177, 104759. [Google Scholar] [CrossRef]
- Munger, J.S.; Huang, X.; Kawakatsu, H.; Griffiths, M.J.; Dalton, S.L.; Wu, J.; Pittet, J.F.; Kaminski, N.; Garat, C.; Matthay, M.A.; et al. The integrin alpha v beta 6 binds and activates latent TGF beta 1: A mechanism for regulating pulmonary inflammation and fibrosis. Cell 1999, 96, 319–328. [Google Scholar] [CrossRef] [PubMed]
- Dutta, A.; Li, J.; Lu, H.; Akech, J.; Pratap, J.; Wang, T.; Zerlanko, B.J.; FitzGerald, T.J.; Jiang, Z.; Birbe, R.; et al. Integrin αvβ6 promotes an osteolytic program in cancer cells by upregulating MMP2. Cancer Res. 2014, 74, 1598–1608. [Google Scholar] [CrossRef]
- Thomas, G.J.; Nystrom, M.L.; Marshall, J.F. Alphavbeta6 Integrin in Wound Healing and Cancer of the Oral Cavity. J. Oral Pathol. Med. 2006, 35, 1–10. [Google Scholar] [CrossRef]
- Vogetseder, A.; Thies, S.; Ingold, B.; Roth, P.; Weller, M.; Schraml, P.; Goodman, S.L.; Moch, H. Av-Integrin Isoform Expression in Primary Human Tumors and Brain Metastases: αv-Integrins in Metastatic Cancer to Brain. Int. J. Cancer 2013, 133, 2362–2371. [Google Scholar] [CrossRef] [PubMed]
- Elayadi, A.N.; Samli, K.N.; Prudkin, L.; Liu, Y.-H.; Bian, A.; Xie, X.-J.; Wistuba, I.I.; Roth, J.A.; McGuire, M.J.; Brown, K.C. A Peptide Selected by Biopanning Identifies the Integrin αvβ6 as a Prognostic Biomarker for Nonsmall Cell Lung Cancer. Cancer Res. 2007, 67, 5889–5895. [Google Scholar] [CrossRef] [PubMed]
- Sartori, A.; Portioli, E.; Battistini, L.; Calorini, L.; Pupi, A.; Vacondio, F.; Arosio, D.; Bianchini, F.; Zanardi, F. Synthesis of Novel c(AmpRGD)–Sunitinib Dual Conjugates as Molecular Tools Targeting the αvβ3 Integrin/VEGFR2 Couple and Impairing Tumor-Associated Angiogenesis. J. Med. Chem. 2017, 60, 248–262. [Google Scholar] [CrossRef] [PubMed]
- Bianchini, F.; Portioli, E.; Ferlenghi, F.; Vacondio, F.; Andreucci, E.; Biagioni, A.; Ruzzolini, J.; Peppicelli, S.; Lulli, M.; Calorini, L.; et al. Cell-Targeted c(AmpRGD)-Sunitinib Molecular Conjugates Impair Tumor Growth of Melanoma. Cancer Lett. 2019, 446, 25–37. [Google Scholar] [CrossRef]
- Bugatti, K.; Andreucci, E.; Monaco, N.; Battistini, L.; Peppicelli, S.; Ruzzolini, J.; Curti, C.; Zanardi, F.; Bianchini, F.; Sartori, A. Nintedanib-Containing Dual Conjugates Targeting αVβ6 Integrin and Tyrosine Kinase Receptors as Potential Antifibrotic Agents. ACS Omega 2022, 7, 17658–17669. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Gu, X.; Zhang, C.; Lu, Q.; Chen, H.; Xu, L. Blocking M2 Muscarinic Receptor Signaling Inhibits Tumor Growth and Reverses Epithelial-Mesenchymal Transition (EMT) in Non-Small Cell Lung Cancer (NSCLC). Cancer Biol. Ther. 2015, 16, 634–643. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Chen, L.; Liu, L.; Niu, X. EMT-Mediated Acquired EGFR-TKI Resistance in NSCLC: Mechanisms and Strategies. Front. Oncol. 2019, 9, 1044. [Google Scholar] [CrossRef]
- Bugatti, K.; Bruno, A.; Arosio, D.; Sartori, A.; Curti, C.; Augustijn, L.; Zanardi, F.; Battistini, L. Shifting Towards αVβ6 Integrin Ligands Using Novel Aminoproline-Based Cyclic Peptidomimetics. Chem. A Eur. J. 2020, 26, 13468–13475. [Google Scholar] [CrossRef]
- Bugatti, K.; Sartori, A.; Battistini, L.; Ruzzolini, J.; Nediani, C.; Curti, C.; Bianchini, F.; Zanardi, F. Nintedanib-αVβ3 Integrin Ligand Dual-Targeting Conjugates towards Precision Treatment of Melanoma. Eur. J. Org. Chem. 2022, 2022, e202200765. [Google Scholar] [CrossRef]
- Byers, L.A.; Diao, L.; Wang, J.; Saintigny, P.; Girard, L.; Peyton, M.; Shen, L.; Fan, Y.; Giri, U.; Tumula, P.K.; et al. An Epithelial–Mesenchymal Transition Gene Signature Predicts Resistance to EGFR and PI3K Inhibitors and Identifies Axl as a Therapeutic Target for Overcoming EGFR Inhibitor Resistance. Clin. Cancer Res. 2013, 19, 279–290. [Google Scholar] [CrossRef]
- Rodón, J.; Carducci, M.; Sepulveda-Sánchez, J.M.; Azaro, A.; Calvo, E.; Seoane, J.; Braña, I.; Sicart, E.; Gueorguieva, I.; Cleverly, A.; et al. Pharmacokinetic, Pharmacodynamic and Biomarker Evaluation of Transforming Growth Factor-β Receptor I Kinase Inhibitor, Galunisertib, in Phase 1 Study in Patients with Advanced Cancer. Investig. New Drugs 2015, 33, 357–370. [Google Scholar] [CrossRef]
- Yan, P.; Zhu, H.; Yin, L.; Wang, L.; Xie, P.; Ye, J.; Jiang, X.; He, X. Integrin Avβ6 Promotes Lung Cancer Proliferation and Metastasis through Upregulation of IL-8–Mediated MAPK/ERK Signaling. Transl. Oncol. 2018, 11, 619–627. [Google Scholar] [CrossRef]
- He, S.; Yin, T.; Li, D.; Gao, X.; Wan, Y.; Ma, X.; Ye, T.; Guo, F.; Sun, J.; Lin, Z.; et al. Enhanced interaction between natural killer cells and lung cancer cells: Involvement in gefitinib-mediated immunoregulation. J. Transl. Med. 2013, 11, 186. [Google Scholar] [CrossRef]
- Van der Bijl, I.; Nawaz, K.; Kazlauskaite, U.; van Stalborch, A.M.; Tol, S.; Jimenez Orgaz, A.; van den Bout, I.; Reinhard, N.R.; Sonnenberg, A.; Margadant, C. Reciprocal integrin/integrin antagonism through kindlin-2 and Rho GTPases regulates cell cohesion and collective migration. Matrix Biol. 2020, 93, 60–78. [Google Scholar] [CrossRef]
- Madamanchi, A.; Zijlstra, A.; Zutter, M.M. Flipping the switch: Integrin switching provides metastatic competence. Sci. Signal. 2014, 7, pe9. [Google Scholar] [CrossRef]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef]
- Russo, M.A.; Paolillo, M.; Sanchez-Hernandez, Y.; Curti, D.; Ciusani, E.; Serra, M.; Colombo, L.; Schinelli, S. A small-molecule RGD-integrin antagonist inhibits cell adhesion, cell migration and induces anoikis in glioblastoma cells. Int. J. Oncol. 2013, 42, 83–92. [Google Scholar] [CrossRef]
- Van Hove, I.; Hu, T.T.; Beets, K.; Van Bergen, T.; Etienne, I.; Stitt, A.W.; Vermassen, E.; Feyen, J.H.M. Targeting RGD-binding integrins as an integrative therapy for diabetic retinopathy and neovascular age-related macular degeneration. Prog. Retin. Eye Res. 2021, 85, 100966. [Google Scholar] [CrossRef]
- Kariya, Y.; Oyama, M.; Suzuki, T.; Kariya, Y. αvβ3 Integrin induces partial EMT independent of TGF-β signaling. Commun. Biol. 2021, 4, 490. [Google Scholar] [CrossRef]
- Moncelet, D.; Bouchaud, V.; Mellet, P.; Ribot, E.; Miraux, S.; Franconi, J.M.; Voisin, P. Cellular density effect on RGD ligand internalization in glioblastoma for MRI application. PLoS ONE 2013, 8, e82777. [Google Scholar] [CrossRef]
- Zhou, S.; Meng, F.; Du, S.; Qian, H.; Ding, N.; Sha, H.; Zhu, M.; Yu, X.; Wang, L.; Liu, B.; et al. Bifunctional iRGD-anti-CD3 enhances antitumor potency of T cells by facilitating tumor infiltration and T-cell activation. J. Immunother. Cancer 2021, 9, e001925. [Google Scholar] [CrossRef]
- Friedland, J.C.; Lee, M.H.; Boettiger, D. Mechanically Activated Integrin Switch Controls α5β1 Function. Science 2009, 323, 642–644. [Google Scholar] [CrossRef]
- Le Saux, G.; Magenau, A.; Gunaratnam, K.; Kilian, K.A.; Böcking, T.; Gooding, J.J.; Gaus, K. Spacing of Integrin Ligands Influences Signal Transduction in Endothelial Cells. Biophys. J. 2011, 101, 764–773. [Google Scholar] [CrossRef]
- Halaby, R. Influence of lysosomal sequestration on multidrug resistance in cancer cells. Cancer Drug Resist. 2019, 2, 31–42. [Google Scholar] [CrossRef]
- Branco, H.; Oliveira, J.; Antunes, C.; Santos, L.L.; Vasconcelos, M.H.; Xavier, C.P.R. Pirfenidone Sensitizes NCI-H460 Non-Small Cell Lung Cancer Cells to Paclitaxel and to a Combination of Paclitaxel with Carboplatin. Int. J. Mol. Sci. 2022, 23, 3631. [Google Scholar] [CrossRef]
- Andreucci, E.; Pietrobono, S.; Peppicelli, S.; Ruzzolini, J.; Bianchini, F.; Biagioni, A.; Stecca, B.; Calorini, L. SOX2 as a novel contributor of oxidative metabolism in melanoma cells. Cell Commun. Signal. 2018, 16, 87. [Google Scholar] [CrossRef]
- Chatzisideri, T.; Leonidis, G.; Karampelas, T.; Skavatsou, E.; Velentza-Almpani, A.; Bianchini, F.; Tamvakopoulos, C.; Sarli, V. Integrin-Mediated Targeted Cancer Therapy Using c(RGDyK)-Based Conjugates of Gemcitabine. J. Med. Chem. 2022, 65, 271–284. [Google Scholar] [CrossRef]
- Ruzzolini, J.; Peppicelli, S.; Bianchini, F.; Andreucci, E.; Urciuoli, S.; Romani, A.; Tortora, K.; Caderni, G.; Nediani, C.; Calorini, L. Cancer Glycolytic Dependence as a New Target of Olive Leaf Extract. Cancers 2020, 12, 317. [Google Scholar] [CrossRef]
- Gilazieva, Z.; Ponomarev, A.; Rutland, C.; Rizvanov, A.; Solovyeva, V. Promising Applications of Tumor Spheroids and Organoids for Personalized Medicine. Cancers 2020, 12, 2727. [Google Scholar] [CrossRef]










Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Andreucci, E.; Bugatti, K.; Peppicelli, S.; Ruzzolini, J.; Lulli, M.; Calorini, L.; Battistini, L.; Zanardi, F.; Sartori, A.; Bianchini, F. Nintedanib-αVβ6 Integrin Ligand Conjugates Reduce TGFβ-Induced EMT in Human Non-Small Cell Lung Cancer. Int. J. Mol. Sci. 2023, 24, 1475. https://doi.org/10.3390/ijms24021475
Andreucci E, Bugatti K, Peppicelli S, Ruzzolini J, Lulli M, Calorini L, Battistini L, Zanardi F, Sartori A, Bianchini F. Nintedanib-αVβ6 Integrin Ligand Conjugates Reduce TGFβ-Induced EMT in Human Non-Small Cell Lung Cancer. International Journal of Molecular Sciences. 2023; 24(2):1475. https://doi.org/10.3390/ijms24021475
Chicago/Turabian StyleAndreucci, Elena, Kelly Bugatti, Silvia Peppicelli, Jessica Ruzzolini, Matteo Lulli, Lido Calorini, Lucia Battistini, Franca Zanardi, Andrea Sartori, and Francesca Bianchini. 2023. "Nintedanib-αVβ6 Integrin Ligand Conjugates Reduce TGFβ-Induced EMT in Human Non-Small Cell Lung Cancer" International Journal of Molecular Sciences 24, no. 2: 1475. https://doi.org/10.3390/ijms24021475
APA StyleAndreucci, E., Bugatti, K., Peppicelli, S., Ruzzolini, J., Lulli, M., Calorini, L., Battistini, L., Zanardi, F., Sartori, A., & Bianchini, F. (2023). Nintedanib-αVβ6 Integrin Ligand Conjugates Reduce TGFβ-Induced EMT in Human Non-Small Cell Lung Cancer. International Journal of Molecular Sciences, 24(2), 1475. https://doi.org/10.3390/ijms24021475