
C/EBPβ Regulates TFAM Expression, Mitochondrial Function and Autophagy in Cellular Models of Parkinson’s Disease

 ,
,  , , , , ,
, , , , ,
Abstract
1. Introduction
2. Results
2.1. C/EBPβ Is Involved in the Regulation of Mitochondrial Genes in the Context of Parkinson’s Disease
2.2. C/EBPβ Regulates the Activity of Human TFAM Promoter
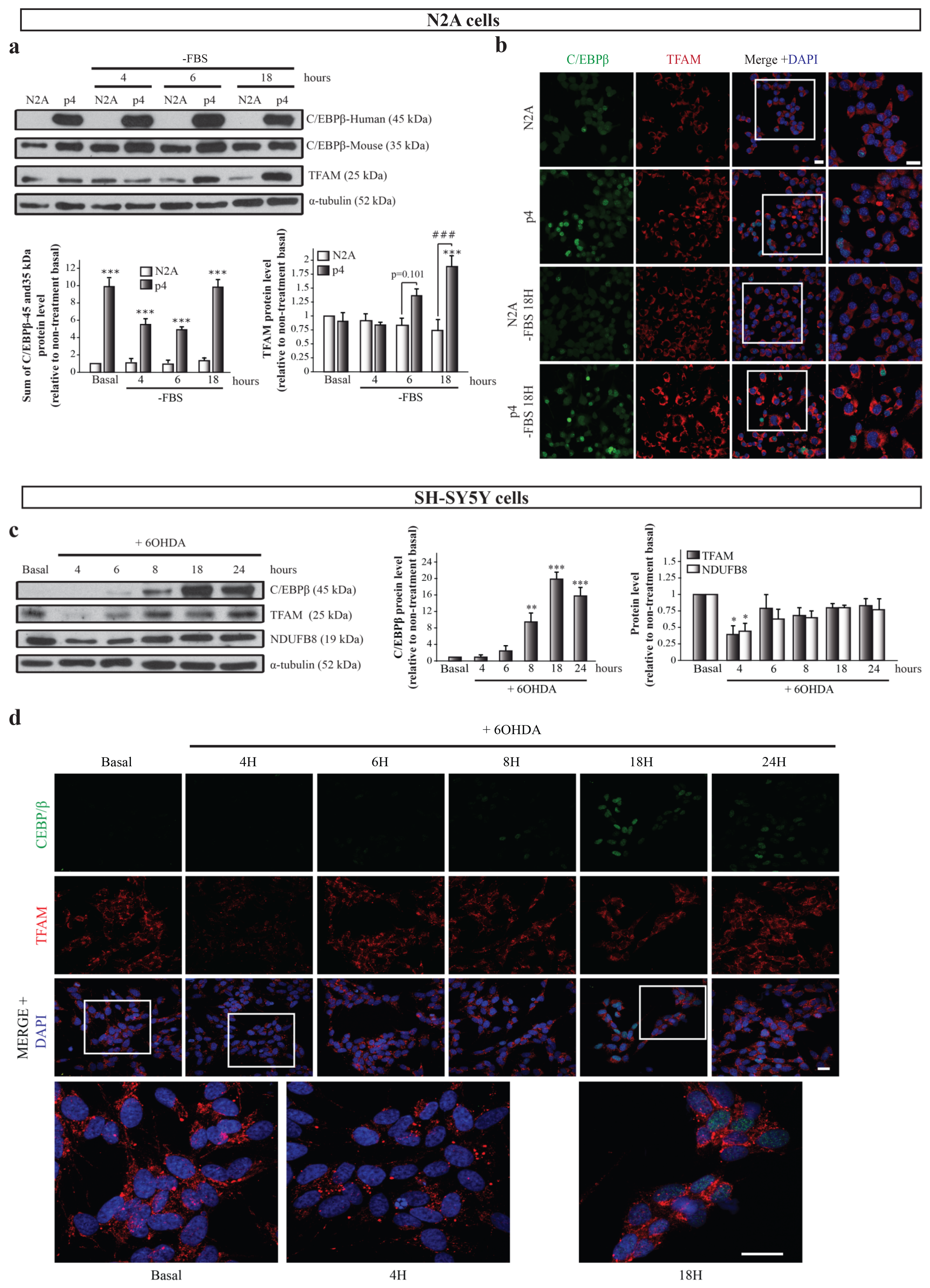
2.3. Possible Link between C/EBPβ and TFAM Protein Levels in Neuronal Differentiation and Oxidative Stress
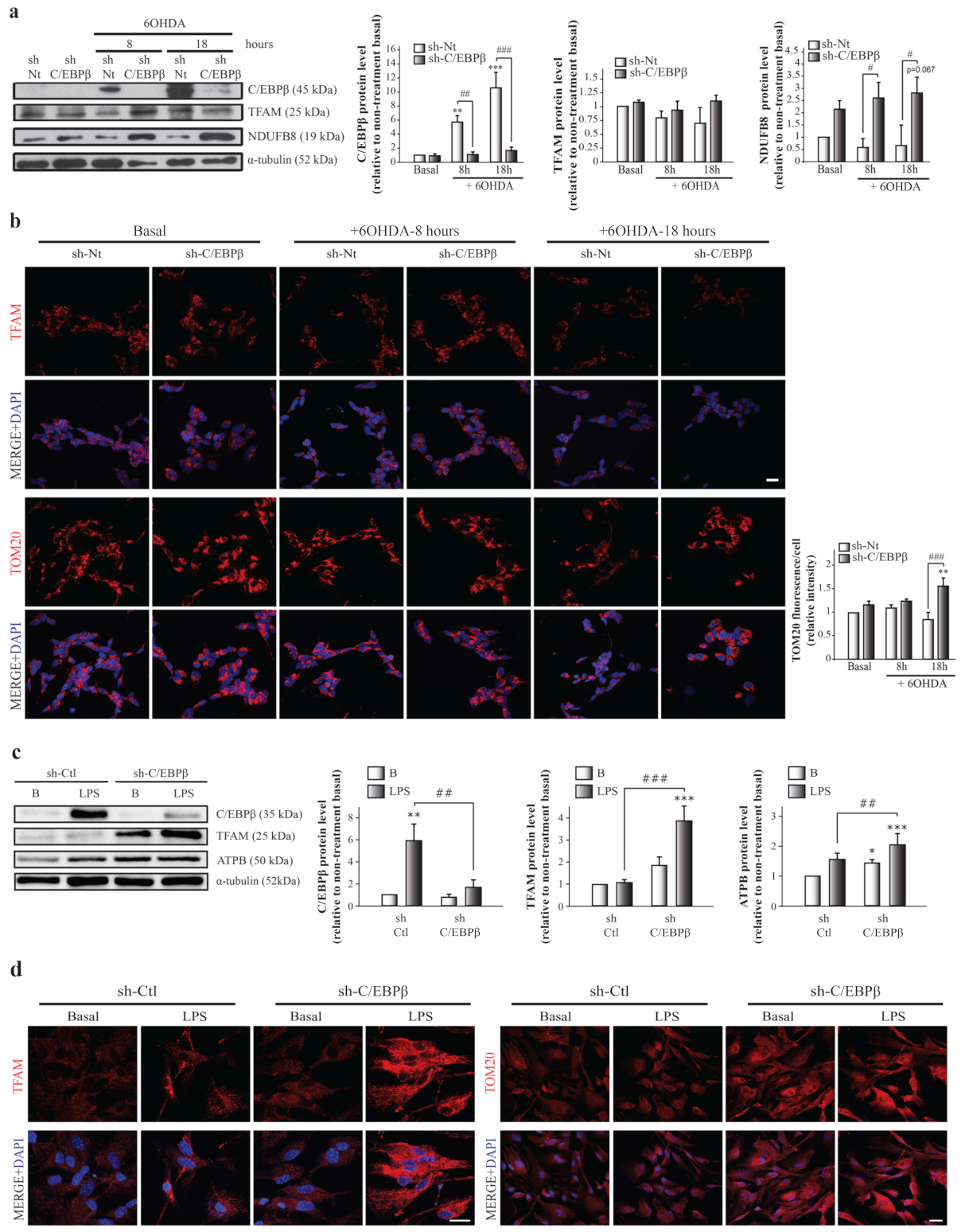
2.4. C/EBPβ Silencing Produces TFAM and Other Mitochondrial Markers Accumulation in Cellular Models of Parkinson’s Disease and Neuroinflammation
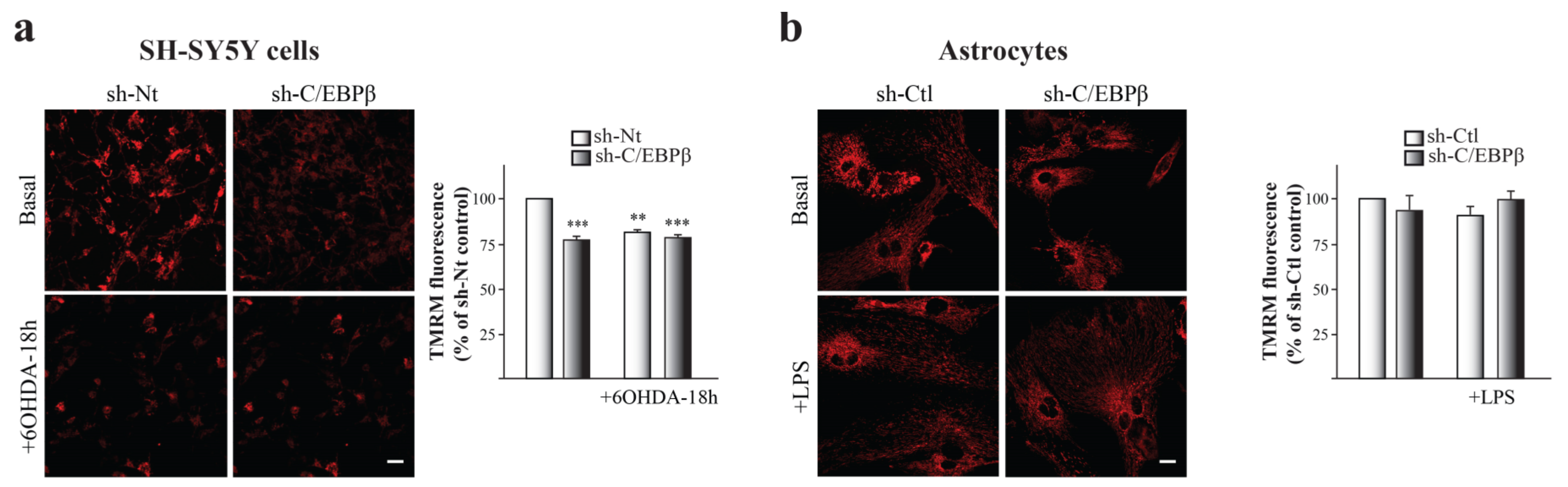
2.5. Mitochondria Accumulation does Not Result in an Increase on Mitochondrial Function
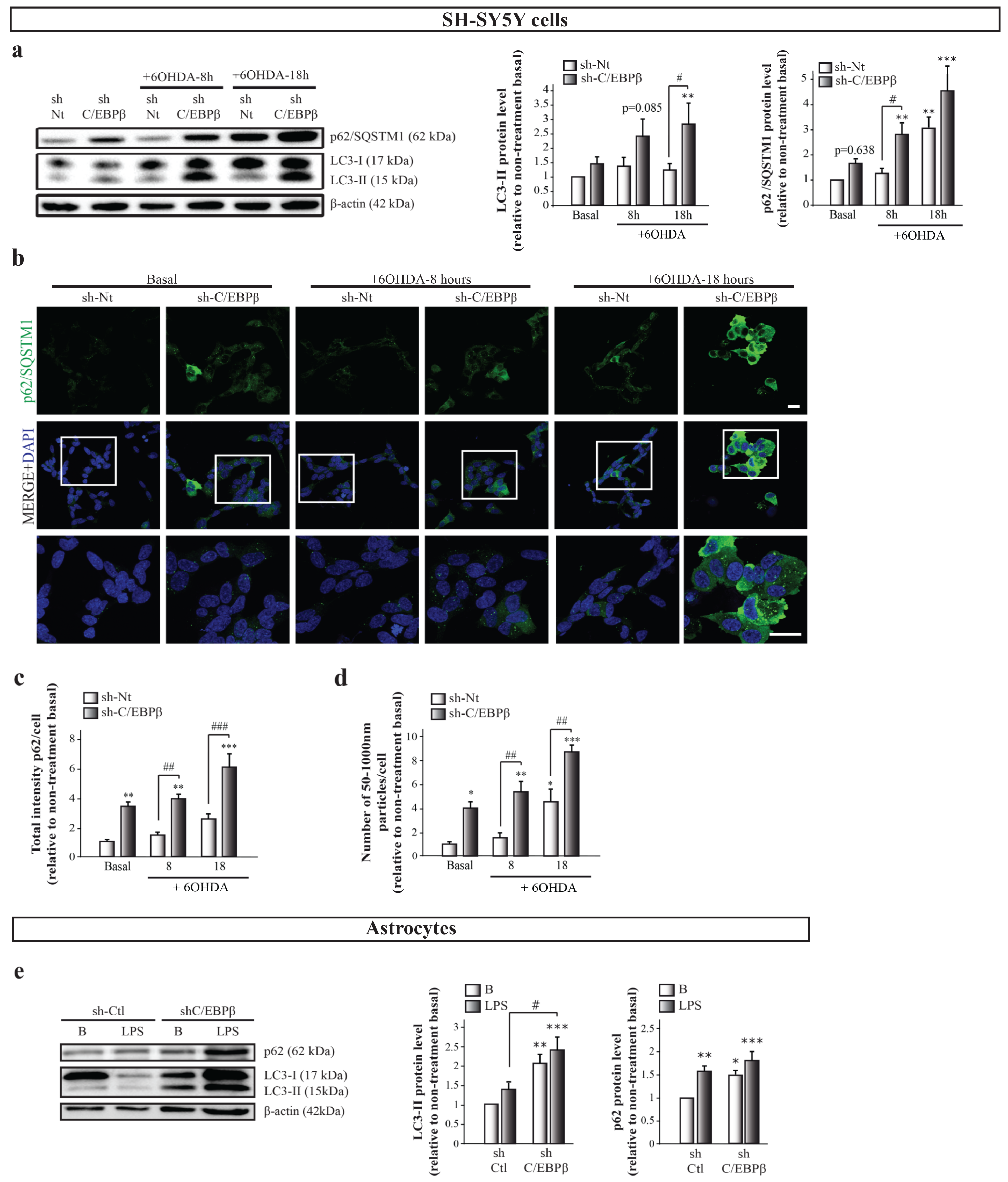
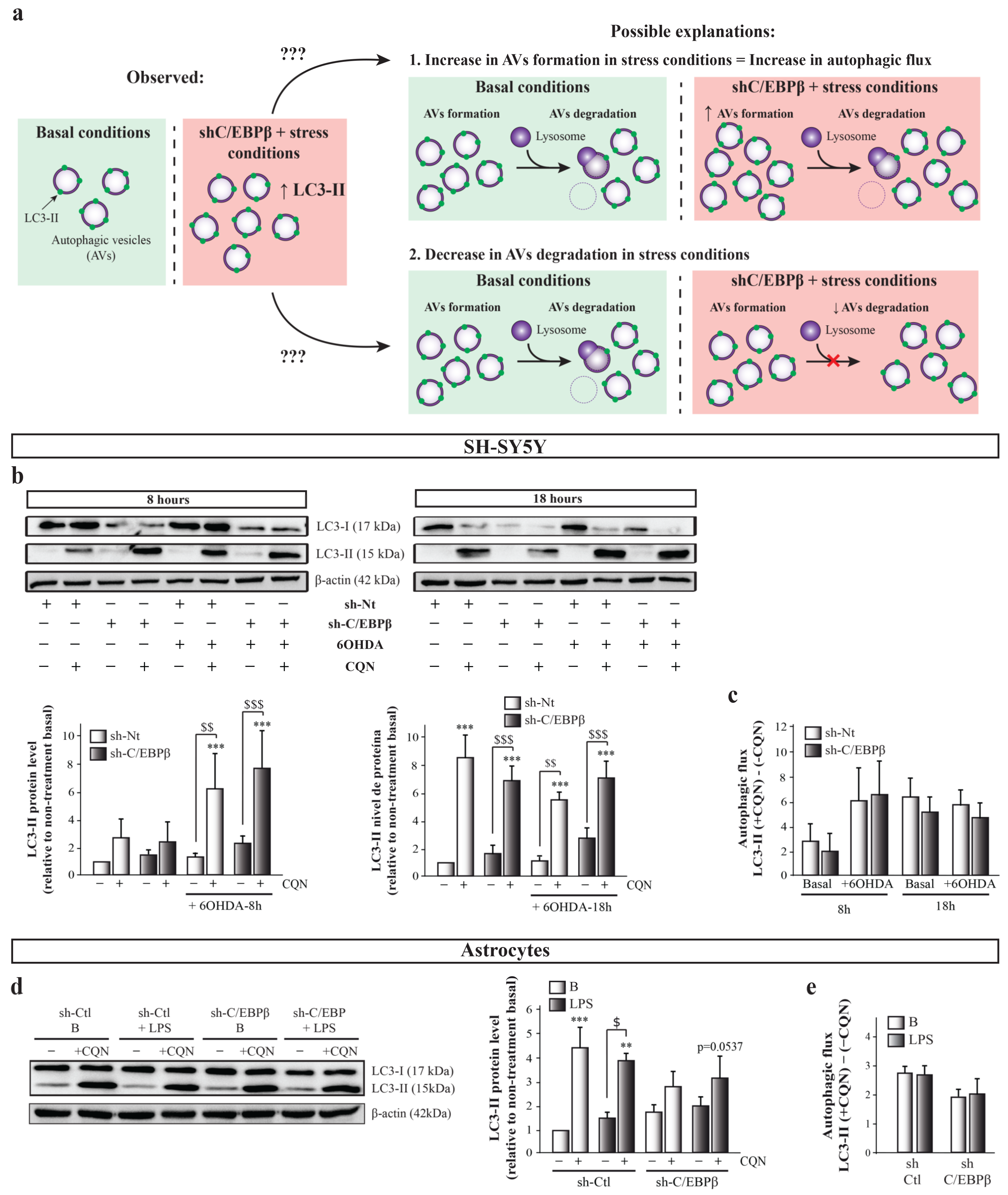
2.6. C/EBPβ Depletion Causes the Alteration in Autophagy in Cellular Models of Parkinson’s Disease and Neuroinflammation
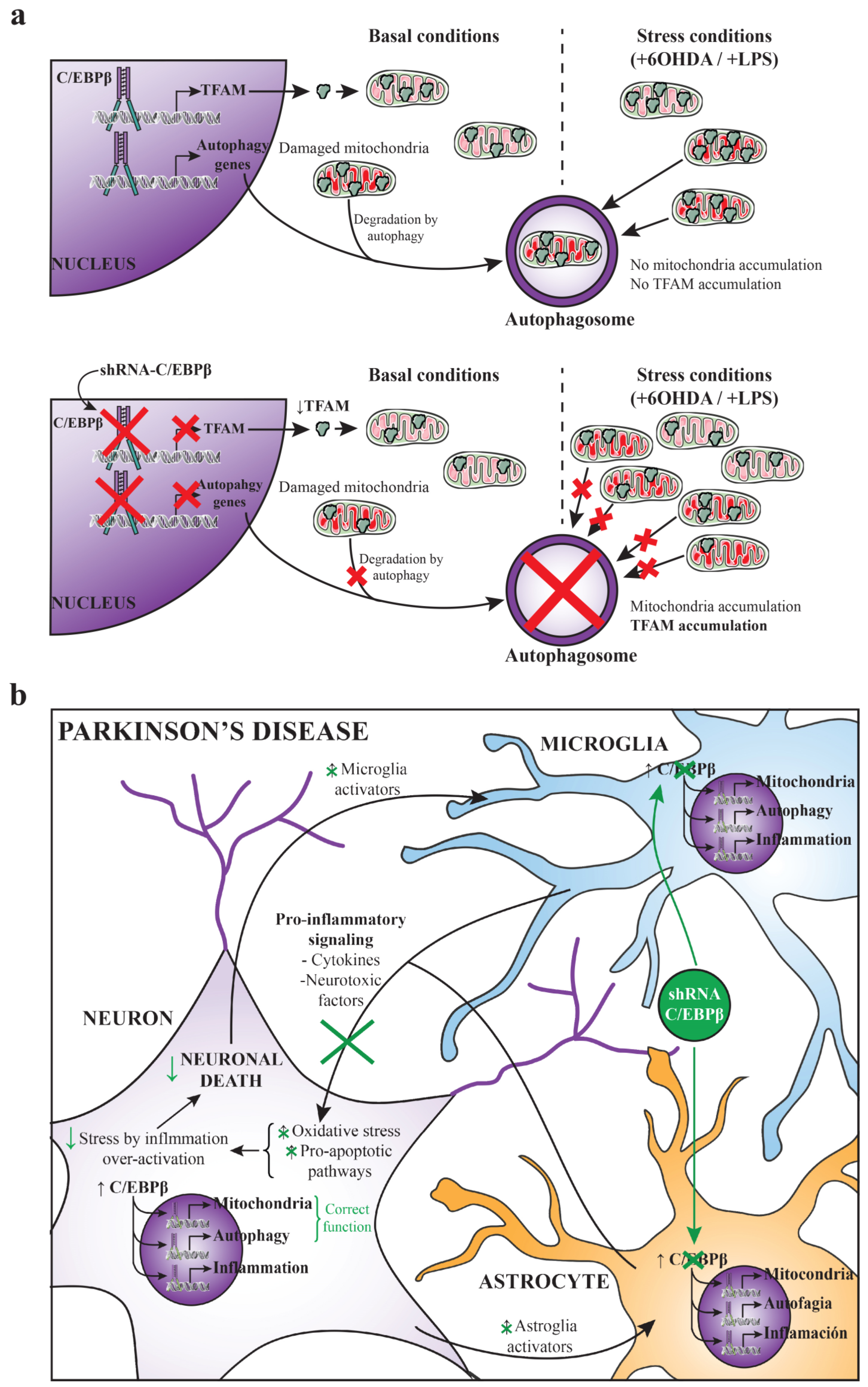
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Real-Time Quantitative PCR
4.3. Cell Cultures
4.3.1. Cell Lines
4.3.2. Primary Astroglial Cultures
4.4. TFAM Promoter Cloning and Luciferase Assay
4.5. Lentiviral-Mediated Gene Silencing
4.6. Immunoblot Analysis
4.7. Immunocytochemistry
4.8. Mitochondrial Membrane Potential (ΔΨm) Measurement
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Berg, D.; Postuma, R.B.; Bloem, B.; Chan, P.; Dubois, B.; Gasser, T.; Goetz, C.G.; Halliday, G.M.; Hardy, J.; Lang, A.E.; et al. Time to Redefine PD? Introductory Statement of the MDS Task Force on the Definition of Parkinson’s Disease. Mov. Disord. 2014, 29, 454–462. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Braak, E.; del Tredici, K.; Rüb, U.; de Vos, R.A.I.; Steur, E.N.H.J. Staging of Brain Pathology Related to Sporadic Parkinson’s Disease. Neurobiol. Aging 2003, 24, 197–211. [Google Scholar] [CrossRef] [PubMed]
- Kowal, S.L.; Dall, T.M.; Chakrabarti, R.; Storm, M.V.; Jain, A. The Current and Projected Economic Burden of Parkinson’s Disease in the United States. Mov. Disord. 2013, 28, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Schulz, R.; Beach, S.R. Caregiving as a Risk Factor for Mortality: The Caregiver Health Effects Study. J. Am. Med. Assoc 1999, 282, 2215–2219. [Google Scholar] [CrossRef] [PubMed]
- Van Bulck, M.; Sierra-Magro, A.; Alarcon-Gil, J.; Perez-Castillo, A.; Morales-Garcia, J.A. Novel Approaches for the Treatment of Alzheimer’s and Parkinson’s Disease. Int. J. Mol. Sci. 2019, 20, 719. [Google Scholar] [CrossRef]
- Michel, P.P.; Hirsch, E.; Hunot, S. Understanding Dopaminergic Cell Death Pathways in Parkinson Disease. Neuron 2016, 90, 675–691. [Google Scholar] [CrossRef]
- Troncoso-Escudero, P.; Parra, A.; Nassif, M.; Vidal, R.L. Outside in: Unraveling the Role of Neuroinflammation in the Progression of Parkinson’s Disease. Front. Neurol. 2018, 9, 860. [Google Scholar] [CrossRef]
- Wong, Y.C.; Krainc, D. α-Synuclein Toxicity in Neurodegeneration: Mechanism and Therapeutic Strategies. Nat. Med. 2017, 23, 1–13. [Google Scholar] [CrossRef]
- Nadeau, S.; Hein, P.; Fernandes, K.J.L.; Peterson, A.C.; Miller, F.D. A Transcriptional Role for C/EBP β in the Neuronal Response to Axonal Injury. Mol. Cell. Neurosci. 2005, 29, 525–535. [Google Scholar] [CrossRef]
- Taubenfeld, S.M.; Milekic, M.H.; Monti, B.; Alberini, C.M. The Consolidation of New but Not Reactivated Memory Requires Hippocampal C/EBPbeta. Nat. Neurosci. 2001, 4, 813–818. [Google Scholar] [CrossRef]
- Alberini, C.M.; Ghirardi, M.; Metz, R.; Kandel, E.R. C/EBP Is an Immediate-Early Gene Required for the Consolidation of Long-Term Facilitation in Aplysia. Cell 1994, 76, 1099–1114. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.Y.; Stern, S.A.; Garcia-Osta, A.; Saunier-Rebori, B.; Pollonini, G.; Bambah-Mukku, D.; Blitzer, R.D.; Alberini, C.M. A Critical Role for IGF-II in Memory Consolidation and Enhancement. Nature 2011, 469, 491–499. [Google Scholar] [CrossRef] [PubMed]
- Milekic, M.H.; Pollonini, G.; Alberini, C.M. Temporal Requirement of C/EBP in the Amygdala Following Reactivation but Not Acquisition of Inhibitory Avoidance. Learn. Mem. 2007, 14, 504–511. [Google Scholar] [CrossRef] [PubMed]
- Ménard, C.; Hein, P.; Paquin, A.; Savelson, A.; Yang, X.M.; Lederfein, D.; Barnabé-Heider, F.; Mir, A.A.; Sterneck, E.; Peterson, A.C.; et al. An Essential Role for a MEK-C/EBP Pathway during Growth Factor-Regulated Cortical Neurogenesis. Neuron 2002, 36, 597–610. [Google Scholar] [CrossRef] [PubMed]
- Cortés-Canteli, M.; Pignatelli, M.; Santos, A.; Perez-Castillo, A. CCAAT/Enhancer-Binding Protein Beta Plays a Regulatory Role in Differentiation and Apoptosis of Neuroblastoma Cells. J. Biol. Chem. 2002, 277, 5460–5467. [Google Scholar] [CrossRef]
- Cortes-Canteli, M.; Aguilar-Morante, D.; Sanz-Sancristobal, M.; Megias, D.; Santos, A.; Perez-Castillo, A. Role of C/EBPβ Transcription Factor in Adult Hippocampal Neurogenesis. PLoS ONE 2011, 6, e24842. [Google Scholar] [CrossRef]
- Cortés-Canteli, M.; Wagner, M.; Ansorge, W.; Pérez-Castillo, A. Microarray Analysis Supports A Role for CCAAT/Enhancer-Binding Protein-β in Brain Injury. J. Biol. Chem. 2004, 279, 14409–14417. [Google Scholar] [CrossRef]
- Fields, J.; Ghorpade, A. C/EBPβ Regulates Multiple IL-1β-Induced Human Astrocyte Inflammatory Genes. J. Neuroinflamm. 2012, 9, 177. [Google Scholar] [CrossRef]
- Hernandez-Encinas, E.; Aguilar-Morante, D.; Cortes-Canteli, M.; Morales-Garcia, J.A.; Gine, E.; Santos, A.; Perez-Castillo, A. CCAAT/Enhancer Binding Protein β Directly Regulates the Expression of the Complement Component 3 Gene in Neural Cells: Implications for the pro-Inflammatory Effects of This Transcription Factor. J. Neuroinflamm. 2015, 12, 14. [Google Scholar] [CrossRef]
- Sandhir, R.; Berman, N.E.J. Age-Dependent Response of CCAAT/Enhancer Binding Proteins Following Traumatic Brain Injury in Mice. Neurochem. Int. 2010, 56, 188–193. [Google Scholar] [CrossRef]
- Dentesano, G.; Straccia, M.; Ejarque-Ortiz, A.; Tusell, J.M.; Serratosa, J.; Saura, J.; Solà, C. Inhibition of CD200R1 Expression by C/EBP Beta in Reactive Microglial Cells. J. Neuroinflamm. 2012, 9, 165. [Google Scholar] [CrossRef]
- Straccia, M.; Gresa-Arribas, N.; Dentesano, G.; Ejarque-Ortiz, A.; Tusell, J.M.; Serratosa, J.; Solà, C.; Saura, J. Pro-Inflammatory Gene Expression and Neurotoxic Effects of Activated Microglia Are Attenuated by Absence of CCAAT/Enhancer Binding Protein β. J. Neuroinflamm. 2011, 8, 156. [Google Scholar] [CrossRef]
- Jana, M.; Anderson, J.A.; Saha, R.N.; Liu, X.; Pahan, K. Regulation of Inducible Nitric Oxide Synthase in Proinflammatory Cytokine-Stimulated Human Primary Astrocytes. Free Radic Biol. Med. 2005, 38, 655–664. [Google Scholar] [CrossRef]
- Cortes-Canteli, M.; Luna-Medina, R.; Sanz-Sancristobal, M.; Alvarez-Barrientos, A.; Santos, A.; Perez-Castillo, A. CCAAT/Enhancer Binding Protein Beta Deficiency Provides Cerebral Protection Following Excitotoxic Injury. J. Cell Sci. 2008, 121, 1224–1234. [Google Scholar] [CrossRef]
- Hernandez-Encinas, E.; Aguilar-Morante, D.; Morales-Garcia, J.A.; Gine, E.; Sanz-SanCristobal, M.; Santos, A.; Perez-Castillo, A. Complement Component 3 (C3) Expression in the Hippocampus after Excitotoxic Injury: Role of C/EBPβ. J. Neuroinflamm. 2016, 13, 276. [Google Scholar] [CrossRef]
- Colangelo, V.; Schurr, J.; Ball, M.J.; Pelaez, R.P.; Bazan, N.G.; Lukiw, W.J. Gene Expression Profiling of 12633 Genes in Alzheimer Hippocampal CA1: Transcription and Neurotrophic Factor down-Regulation and up-Regulation of Apoptotic and pro-Inflammatory Signaling. J. Neurosci. Res. 2002, 70, 462–473. [Google Scholar] [CrossRef]
- Strohmeyer, R.; Shelton, J.; Lougheed, C.; Breitkopf, T. CCAAT-Enhancer Binding Protein-β Expression and Elevation in Alzheimer’s Disease and Microglial Cell Cultures. PLoS ONE 2014, 9, e86617. [Google Scholar] [CrossRef]
- Figueroa-Romero, C.; Hur, J.; Bender, D.E.; Delaney, C.E.; Cataldo, M.D.; Smith, A.L.; Yung, R.; Ruden, D.M.; Callaghan, B.C.; Feldman, E.L. Identification of Epigenetically Altered Genes in Sporadic Amyotrophic Lateral Sclerosis. PLoS ONE 2012, 7, e52672. [Google Scholar] [CrossRef]
- Valente, T.; Mancera, P.; Tusell, J.M.; Serratosa, J.; Saura, J. C/EBPβ Expression in Activated Microglia in Amyotrophic Lateral Sclerosis. Neurobiol. Aging 2012, 33, 2186–2199. [Google Scholar] [CrossRef]
- Morales-Garcia, J.A.; Gine, E.; Hernandez-Encinas, E.; Aguilar-Morante, D.; Sierra-Magro, A.; Sanz-SanCristobal, M.; Alonso-Gil, S.; Sanchez-Lanzas, R.; Castaño, J.G.; Santos, A.; et al. CCAAT/Enhancer Binding Protein β Silencing Mitigates Glial Activation and Neurodegeneration in a Rat Model of Parkinson’s Disease. Sci. Rep. 2017, 7, 13526. [Google Scholar] [CrossRef]
- Silva, J.P.; Larsson, N.G. Manipulation of Mitochondrial DNA Gene Expression in the Mouse. Biochim. Biophys. Acta Bioenerg. 2002, 1555, 106–110. [Google Scholar] [CrossRef] [PubMed]
- Larsson, N.G.; Wang, J.; Wilhelmsson, H.; Oldfors, A.; Rustin, P.; Lewandoski, M.; Barsh, G.S.; Clayton, D.A. Mitochondrial Transcription Factor A Is Necessary for MtDNA Maintenance and Embryogenesis in Mice. Nat. Genet. 1998, 18, 231–236. [Google Scholar] [CrossRef] [PubMed]
- Kanki, T.; Ohgaki, K.; Gaspari, M.; Gustafsson, C.M.; Fukuoh, A.; Sasaki, N.; Hamasaki, N.; Kang, D. Architectural Role of Mitochondrial Transcription Factor A in Maintenance of Human Mitochondrial DNA. Mol. Cell Biol. 2004, 24, 9823–9834. [Google Scholar] [CrossRef] [PubMed]
- Campbell, C.T.; Kolesar, J.E.; Kaufman, B.A. Mitochondrial Transcription Factor A Regulates Mitochondrial Transcription Initiation, DNA Packaging, and Genome Copy Number. BBA-Gene Regul. Mech. 2012, 1819, 921–929. [Google Scholar] [CrossRef] [PubMed]
- Kang, I.; Chu, C.T.; Kaufman, B.A. The Mitochondrial Transcription Factor TFAM in Neurodegeneration: Emerging Evidence and Mechanisms. FEBS Lett. 2018, 592, 793–811. [Google Scholar] [CrossRef]
- Andreeva, L.; Hiller, B.; Kostrewa, D.; Lässig, C.; de Oliveira Mann, C.C.; Jan Drexler, D.; Maiser, A.; Gaidt, M.; Leonhardt, H.; Hornung, V.; et al. CGAS Senses Long and HMGB/TFAM-Bound U-Turn DNA by Forming Protein-DNA Ladders. Nature 2017, 549, 394–398. [Google Scholar] [CrossRef]
- Bajwa, E.; Pointer, C.B.; Klegeris, A. The Role of Mitochondrial Damage-Associated Molecular Patterns in Chronic Neuroinflammation. Mediat. Inflamm 2019, 2019, 4050796. [Google Scholar] [CrossRef]
- Pulido-Salgado, M.; Vidal-Taboada, J.M.; Saura, J. C/EBPβ and C/EBPδ Transcription Factors: Basic Biology and Roles in the CNS. Prog. Neurobiol. 2015, 132, 1–33. [Google Scholar] [CrossRef]
- Fung, H.C.; Scholz, S.; Matarin, M.; Simón-Sánchez, J.; Hernandez, D.; Britton, A.; Gibbs, J.R.; Langefeld, C.; Stiegert, M.L.; Schymick, J.; et al. Genome-Wide Genotyping in Parkinson’s Disease and Neurologically Normal Controls: First Stage Analysis and Public Release of Data. Lancet Neurol. 2006, 5, 911–916. [Google Scholar] [CrossRef]
- Nalls, M.A.; Pankratz, N.; Lill, C.M.; Do, C.B.; Hernandez, D.G.; Saad, M.; Destefano, A.L.; Kara, E.; Bras, J.; Sharma, M.; et al. Large-Scale Meta-Analysis of Genome-Wide Association Data Identifies Six New Risk Loci for Parkinson’s Disease. Nat. Genet. 2014, 46, 989–993. [Google Scholar] [CrossRef]
- Chang, D.; Nalls, M.A.; Hallgrímsdóttir, I.B.; Hunkapiller, J.; van der Brug, M.; Cai, F.; Kerchner, G.A.; Ayalon, G.; Bingol, B.; Sheng, M.; et al. A Meta-Analysis of Genome-Wide Association Studies Identifies 17 New Parkinson’s Disease Risk Loci. Nat. Genet. 2017, 49, 1511–1516. [Google Scholar] [CrossRef]
- Pickrell, J.K.; Berisa, T.; Liu, J.Z.; Ségurel, L.; Tung, J.Y.; Hinds, D.A. Detection and Interpretation of Shared Genetic Influences on 42 Human Traits. Nat. Genet. 2016, 48, 709–717. [Google Scholar] [CrossRef]
- Dumitriu, A.; Golji, J.; Labadorf, A.T.; Gao, B.; Beach, T.G.; Myers, R.H.; Longo, K.A.; Latourelle, J.C. Integrative Analyses of Proteomics and RNA Transcriptomics Implicate Mitochondrial Processes, Protein Folding Pathways and GWAS Loci in Parkinson Disease. BMC Med. Genom. 2016, 9, 5. [Google Scholar] [CrossRef]
- Lee, C.M.; Barber, G.P.; Casper, J.; Clawson, H.; Diekhans, M.; Gonzalez, J.N.; Hinrichs, A.S.; Lee, B.T.; Nassar, L.R.; Powell, C.C.; et al. UCSC Genome Browser Enters 20th Year. Nucleic Acids Res. 2020, 48, D756–D761. [Google Scholar] [CrossRef]
- Fiorito, V.; Chiabrando, D.; Tolosano, E. Mitochondrial Targeting in Neurodegeneration: A Heme Perspective. Pharmaceuticals 2018, 11, 87. [Google Scholar] [CrossRef]
- Flaskos, J.; McLean, W.G.; Fowler, M.J.; Hargreaves, A.J. Tricresyl Phosphate Inhibits the Formation of Axon-like Processes and Disrupts Neurofilaments in Cultured Mouse N2a and Rat PC12 Cells. Neurosci. Lett. 1998, 242, 101–104. [Google Scholar] [CrossRef]
- Xicoy, H.; Brouwers, J.F.; Kalnytska, O.; Wieringa, B.; Martens, G.J.M. Lipid Analysis of the 6-Hydroxydopamine-Treated SH-SY5Y Cell Model for Parkinson’s Disease. Mol. Neurobiol. 2020, 57, 848–859. [Google Scholar] [CrossRef]
- Glinka, Y.; Gassen, M.; Youdim, M.B.H. Mechanism of 6-Hydroxydopamine Neurotoxicity. J. Neural Transm. Suppl. 1997, 50, 55–66. [Google Scholar] [CrossRef]
- Morales-Garcia, J.A.; Alonso-Gil, S.; Santos, Á.; Perez-Castillo, A. Phosphodiesterase 7 Regulation in Cellular and Rodent Models of Parkinson’s Disease. Mol. Neurobiol. 2020, 57, 806–822. [Google Scholar] [CrossRef]
- Guerrero-Castillo, S.; Baertling, F.; Kownatzki, D.; Wessels, H.J.; Arnold, S.; Brandt, U.; Nijtmans, L. The Assembly Pathway of Mitochondrial Respiratory Chain Complex I. Cell Metab. 2017, 25, 128–139. [Google Scholar] [CrossRef]
- Cardinaux, J.R.; Allaman, I.; Magistretti, P.J. Pro-Inflammatory Cytokines Induce the Transcription Factors C/EBPbeta and C/EBPdelta in Astrocytes. Glia 2000, 29, 91–97. [Google Scholar] [CrossRef]
- Pickles, S.; Vigié, P.; Youle, R.J. Mitophagy and Quality Control Mechanisms in Mitochondrial Maintenance. Curr. Biol. 2018, 28, R170–R185. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of Cells and Tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef]
- Yang, Z.; Klionsky, D.J. Mammalian Autophagy: Core Molecular Machinery and Signaling Regulation. Curr. Opin. Cell Biol. 2010, 22, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Bjørkøy, G.; Lamark, T.; Brech, A.; Outzen, H.; Perander, M.; Øvervatn, A.; Stenmark, H.; Johansen, T. P62/SQSTM1 Forms Protein Aggregates Degraded by Autophagy and Has a Protective Effect on Huntingtin-Induced Cell Death. J. Cell Biol. 2005, 171, 603–614. [Google Scholar] [CrossRef]
- Sánchez-Martın, P.; Komatsu, M. P62/SQSTM1—Steering the Cell through Health and Disease. J. Cell Sci. 2018, 131, jcs222836. [Google Scholar] [CrossRef]
- Mizushima, N.; Murphy, L.O. Autophagy Assays for Biological Discovery and Therapeutic Development. Trends Biochem. Sci. 2020, 45, 1080–1093. [Google Scholar] [CrossRef]
- Pasquier, B. Autophagy Inhibitors. Cell. Mol. Life Sci. 2016, 73, 985–1001. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdel-Aziz, A.K.; Abdelfatah, S.; Abdellatif, M.; Abdoli, A.; Abel, S.; Abeliovich, H.; Abildgaard, M.H.; Abudu, Y.P.; Acevedo-Arozena, A.; et al. Guidelines for the Use and Interpretation of Assays for Monitoring Autophagy (4th Edition). Autophagy 2021, 17, 1–382. [Google Scholar] [CrossRef]
- Obrietan, K.; Hoyt, K.R. CRE-Mediated Transcription Is Increased in Huntington’s Disease Transgenic Mice. J. Neurosci. 2004, 24, 791–796. [Google Scholar] [CrossRef]
- Gómez-Santos, C.; Barrachina, M.; Giménez-Xavier, P.; Dalfó, E.; Ferrer, I.; Ambrosio, S. Induction of C/EBP Beta and GADD153 Expression by Dopamine in Human Neuroblastoma Cells. Relationship with Alpha-Synuclein Increase and Cell Damage. Brain Res. Bull. 2005, 65, 87–95. [Google Scholar] [CrossRef]
- Kfoury, N.; Kapatos, G. Identification of Neuronal Target Genes for CCAAT/Enhancer Binding Proteins. Mol. Cell. Neurosci. 2009, 40, 313–327. [Google Scholar] [CrossRef]
- Wu, Z.; Xia, Y.; Wang, Z.; Su Kang, S.; Lei, K.; Liu, X.; Jin, L.; Wang, X.; Cheng, L.; Ye, K. C/EBPβ/δ-Secretase Signaling Mediates Parkinson’s Disease Pathogenesis via Regulating Transcription and Proteolytic Cleavage of α-Synuclein and MAOB. Mol. Psychiatry 2021, 26, 568–585. [Google Scholar] [CrossRef]
- Ahn, E.H.; Lei, K.; Kang, S.S.; Wang, Z.H.; Liu, X.; Hong, W.; Wang, Y.T.; Edgington-Mitchell, L.E.; Jin, L.; Ye, K. Mitochondrial Dysfunction Triggers the Pathogenesis of Parkinson’s Disease in Neuronal C/EBPβ Transgenic Mice. Mol. Psychiatry 2021, 26, 7838–7850. [Google Scholar] [CrossRef]
- Ahn, E.H.; Kang, S.S.; Liu, X.; Cao, X.; Choi, S.Y.; Musazzi, L.; Mehlen, P.; Ye, K. BDNF and Netrin-1 Repression by C/EBPβ in the Gut Triggers Parkinson’s Disease Pathologies, Associated with Constipation and Motor Dysfunctions. Prog. Neurobiol. 2021, 198, 101905. [Google Scholar] [CrossRef]
- Doxakis, E. Cell-Free MicroRNAs in Parkinson’s Disease: Potential Biomarkers That Provide New Insights into Disease Pathogenesis. Ageing Res. Rev. 2020, 58, 101023. [Google Scholar] [CrossRef]
- Ravanidis, S.; Bougea, A.; Papagiannakis, N.; Koros, C.; Simitsi, A.M.; Pachi, I.; Breza, M.; Stefanis, L.; Doxakis, E. Validation of Differentially Expressed Brain-Enriched MicroRNAs in the Plasma of PD Patients. Ann. Clin. Transl. Neurol. 2020, 7, 1594–1607. [Google Scholar] [CrossRef]
- Szigeti, A.; Bellyei, S.; Gasz, B.; Boronkai, A.; Hocsak, E.; Minik, O.; Bognar, Z.; Varbiro, G.; Sumegi, B.; Gallyas, F. Induction of Necrotic Cell Death and Mitochondrial Permeabilization by Heme Binding Protein 2/SOUL. FEBS Lett. 2006, 580, 6447–6454. [Google Scholar] [CrossRef]
- Paradkar, P.N.; Zumbrennen, K.B.; Paw, B.H.; Ward, D.M.; Kaplan, J. Regulation of Mitochondrial Iron Import through Differential Turnover of Mitoferrin 1 and Mitoferrin 2. Mol. Cell Biol. 2009, 29, 1007–1016. [Google Scholar] [CrossRef]
- Troadec, M.B.; Warner, D.; Wallace, J.; Thomas, K.; Spangrude, G.J.; Phillips, J.; Khalimonchuk, O.; Paw, B.H.; Ward, D.M.V.; Kaplan, J. Targeted Deletion of the Mouse Mitoferrin1 Gene: From Anemia to Protoporphyria. Blood 2011, 117, 5494–5502. [Google Scholar] [CrossRef]
- Rininger, A.; Dejesus, C.; Totten, A.; Wayland, A.; Halterman, M.W. MKP-1 Antagonizes C/EBPβ Activity and Lowers the Apoptotic Threshold after Ischemic Injury. Cell Death Differ. 2012, 19, 1634–1643. [Google Scholar] [CrossRef] [PubMed]
- Rahman, S.M.; Choudhury, M.; Janssen, R.C.; Baquero, K.C.; Miyazaki, M.; Friedman, J.E. CCAAT/Enhancer Binding Protein β Deletion Increases Mitochondrial Function and Protects Mice from LXR-Induced Hepatic Steatosis. Biochem. Biophys. Res. Commun. 2013, 430, 336–339. [Google Scholar] [CrossRef]
- Chen, Y.; Chang, Y.; Zhang, N.; Guo, X.; Sun, G.; Sun, Y. Atorvastatin Attenuates Myocardial Hypertrophy in Spontaneously Hypertensive Rats via the C/EBPβ/PGC-1α/UCP3 Pathway. Cell Physiol. Biochem. 2018, 46, 1009–1018. [Google Scholar] [CrossRef] [PubMed]
- Chiu, C.-H.; Lin, W.-D.; Huang, S.-Y.; Lee, Y.-H. Effect of a C/EBP Gene Replacement on Mitochondrial Biogenesis in Fat Cells. Genes Dev. 2004, 18, 1970–1975. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Bai, F.; Cao, H.; Xu, J.; Fang, L.; Wu, J.; Yuan, Q.; Zhou, Y.; Sun, Q.; He, W.; et al. PDE/CAMP/Epac/C/EBP-β Signaling Cascade Regulates Mitochondria Biogenesis of Tubular Epithelial Cells in Renal Fibrosis. Antioxid. Redox Signal. 2018, 29, 637–652. [Google Scholar] [CrossRef]
- Sun, Q.; Fang, L.; Tang, X.; Lu, S.; Tamm, M.; Stolz, D.; Roth, M. TGF-β Upregulated Mitochondria Mass through the SMAD2/3→C/EBPβ→PRMT1 Signal Pathway in Primary Human Lung Fibroblasts. J. Immunol. 2019, 202, 37–47. [Google Scholar] [CrossRef]
- Aghanoori, M.R.; Agarwal, P.; Gauvin, E.; Nagalingam, R.S.; Bonomo, R.; Yathindranath, V.; Smith, D.R.; Hai, Y.; Lee, S.; Jolivalt, C.G.; et al. CEBPβ Regulation of Endogenous IGF-1 in Adult Sensory Neurons Can Be Mobilized to Overcome Diabetes-Induced Deficits in Bioenergetics and Axonal Outgrowth. Cell Mol. Life Sci. 2022, 79, 193. [Google Scholar] [CrossRef]
- Qin, J.; Yang, Y.; Gao, S.; Liu, Y.; Yu, F.; Zhou, Y.; Lyu, R.; Liu, M.; Liu, X.; Li, D.; et al. Deregulated ALG-2/HEBP2 Axis Alters Microtubule Dynamics and Mitotic Spindle Behavior to Stimulate Cancer Development. J. Cell Physiol. 2017, 232, 3067–3076. [Google Scholar] [CrossRef]
- Szigeti, A.; Hocsak, E.; Rapolti, E.; Racz, B.; Boronkai, A.; Pozsgai, E.; Debreceni, B.; Bognar, Z.; Bellyei, S.; Sumegi, B.; et al. Facilitation of Mitochondrial Outer and Inner Membrane Permeabilization and Cell Death in Oxidative Stress by a Novel Bcl-2 Homology 3 Domain Protein. J. Biol. Chem. 2010, 285, 2140–2151. [Google Scholar] [CrossRef]
- Li, C.; Zhang, Y.; Cheng, X.; Yuan, H.; Zhu, S.; Liu, J.; Wen, Q.; Xie, Y.; Liu, J.; Kroemer, G.; et al. PINK1 and PARK2 Suppress Pancreatic Tumorigenesis through Control of Mitochondrial Iron-Mediated Immunometabolism. Dev. Cell 2018, 46, 441–455.e8. [Google Scholar] [CrossRef]
- Visconte, V.; Avishai, N.; Mahfouz, R.; Tabarroki, A.; Cowen, J.; Sharghi-Moshtaghin, R.; Hitomi, M.; Rogers, H.J.; Hasrouni, E.; Phillips, J.; et al. Distinct Iron Architecture in SF3B1-Mutant Myelodysplastic Syndrome Patients Is Linked to an SLC25A37 Splice Variant with a Retained Intron. Leukemia 2015, 29, 188–195. [Google Scholar] [CrossRef]
- Ni, S.; Kuang, Y.; Yuan, Y.; Yu, B. Mitochondrion-Mediated Iron Accumulation Promotes Carcinogenesis and Warburg Effect through Reactive Oxygen Species in Osteosarcoma. Cancer Cell Int. 2020, 20, 399. [Google Scholar] [CrossRef]
- Huang, R.; Yang, L.; Zhang, Z.; Liu, X.; Fei, Y.; Tong, W.M.; Niu, Y.; Liang, Z. RNA M6A Demethylase ALKBH5 Protects Against Pancreatic Ductal Adenocarcinoma via Targeting Regulators of Iron Metabolism. Front. Cell Dev. Biol. 2021, 9, 2929. [Google Scholar] [CrossRef]
- Zhang, Z.; Jia, H.; Zhang, Q.; Wan, Y.; Zhou, Y.; Jia, Q.; Zhang, W.; Yuan, W.; Cheng, T.; Zhu, X.; et al. Assessment of Hematopoietic Failure Due to Rpl11 Deficiency in a Zebrafish Model of Diamond-Blackfan Anemia by Deep Sequencing. BMC Genom. 2013, 14, 896. [Google Scholar] [CrossRef]
- Del Rey, M.; Benito, R.; Fontanillo, C.; Campos-Laborie, F.J.; Janusz, K.; Velasco-Hernández, T.; Abáigar, M.; Hernández, M.; Cuello, R.; Borrego, D.; et al. Deregulation of Genes Related to Iron and Mitochondrial Metabolism in Refractory Anemia with Ring Sideroblasts. PLoS ONE 2015, 10, e0126555. [Google Scholar] [CrossRef]
- Phillips, J.; Farrell, C.; Wang, Y.; Singal, A.K.; Anderson, K.; Balwani, M.; Bissell, M.; Bonkovsky, H.; Seay, T.; Paw, B.; et al. Strong Correlation of Ferrochelatase Enzymatic Activity with Mitoferrin-1 MRNA in Lymphoblasts of Patients with Protoporphyria. Mol. Genet. Metab. 2019, 128, 391–395. [Google Scholar] [CrossRef]
- Huo, Y.X.; Huang, L.; Zhang, D.F.; Yao, Y.G.; Fang, Y.R.; Zhang, C.; Luo, X.J. Identification of SLC25A37 as a Major Depressive Disorder Risk Gene. J. Psychiatr. Res. 2016, 83, 168–175. [Google Scholar] [CrossRef]
- Dai, X.G.; Li, Q.; Li, T.; Huang, W.B.; Zeng, Z.H.; Yang, Y.; Duan, Z.P.; Wang, Y.J.; Ai, Y.H. The Interaction between C/EBPβ and TFAM Promotes Acute Kidney Injury via Regulating NLRP3 Inflammasome-Mediated Pyroptosis. Mol. Immunol. 2020, 127, 136–145. [Google Scholar] [CrossRef]
- Ma, D.; Panda, S.; Lin, J.D. Temporal Orchestration of Circadian Autophagy Rhythm by C/EBPβ. EMBO J. 2011, 30, 4642–4651. [Google Scholar] [CrossRef]
- Guo, L.; Huang, J.-X.; Liu, Y.; Li, X.; Zhou, S.-R.; Qian, S.-W.; Liu, Y.; Zhu, H.; Huang, H.-Y.; Dang, Y.-J.; et al. Transactivation of Atg4b by C/EBPβ Promotes Autophagy to Facilitate Adipogenesis. Mol. Cell Biol. 2013, 33, 3180–3190. [Google Scholar] [CrossRef]
- Ahmed, M.; Lai, T.H.; Hwang, J.S.; Zada, S.; Pham, T.M.; Kim, D.R. Transcriptional Regulation of Autophagy Genes via Stage-Specific Activation of CEBPB and PPARG during Adipogenesis: A Systematic Study Using Public Gene Expression and Transcription Factor Binding Datasets. Cells 2019, 8, 1321. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.; Lai, T.H.; Pham, T.M.; Zada, S.; Elashkar, O.; Hwang, J.S.; Kim, D.R. Hierarchical Regulation of Autophagy during Adipocyte Differentiation. PLoS ONE 2022, 17, e0250865. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.; Zhang, F.; Ye, Q.; Wang, H. The Circadian Clock Regulates Autophagy Directly through the Nuclear Hormone Receptor Nr1d1/Rev-Erbα and Indirectly via Cebpb/(C/Ebpβ) in Zebrafish. Autophagy 2016, 12, 1292–1309. [Google Scholar] [CrossRef] [PubMed]
- Yuk, J.-M.; Shin, D.-M.; Lee, H.-M.; Yang, C.-S.; Jin, H.S.; Kim, K.-K.; Lee, Z.-W.; Lee, S.-H.; Kim, J.-M.; Jo, E.-K. Vitamin D3 Induces Autophagy in Human Monocytes/Macrophages via Cathelicidin. Cell Host Microbe 2009, 6, 231–243. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.-S.; Kim, J.-J.; Lee, H.-M.; Jin, H.S.; Lee, S.-H.; Park, J.-H.; Kim, S.J.; Kim, J.-M.; Han, Y.-M.; Lee, M.-S.; et al. The AMPK-PPARGC1A Pathway Is Required for Antimicrobial Host Defense through Activation of Autophagy. Autophagy 2014, 10, 785–802. [Google Scholar] [CrossRef]
- Yu, Y.; Zhao, N.; An, J.; Zhang, X. CCAAT/Enhancer-Binding Protein β Mediates the Killing of Toxoplasma Gondii by Inducing Autophagy in Nonhematopoietic Cells. DNA Cell Biol. 2017, 36, 212–218. [Google Scholar] [CrossRef]
- Yang, X.; Xue, P.; Yuan, M.; Xu, X.; Wang, C.; Li, W.; Machens, H.G.; Chen, Z. SESN2 Protects against Denervated Muscle Atrophy through Unfolded Protein Response and Mitophagy. Cell Death Dis 2021, 12, 805. [Google Scholar] [CrossRef]
- Xu, X.; Huang, E.; Luo, B.; Cai, D.; Zhao, X.; Luo, Q.; Jin, Y.; Chen, L.; Wang, Q.; Liu, C.; et al. Methamphetamine Exposure Triggers Apoptosis and Autophagy in Neuronal Cells by Activating the C/EBPb-Related Signaling Pathway. FASEB J. 2018, 32, 6737–6759. [Google Scholar] [CrossRef]
- Huang, E.; Huang, H.; Guan, T.; Liu, C.; Qu, D.; Xu, Y.; Yang, J.; Yan, L.; Xiong, Y.; Liang, T.; et al. Involvement of C/EBPβ-Related Signaling Pathway in Methamphetamine-Induced Neuronal Autophagy and Apoptosis. Toxicol. Lett. 2019, 312, 11–21. [Google Scholar] [CrossRef]
- Liu, X.S.; Chopp, M.; Zhang, R.L.; Hozeska-Solgot, A.; Gregg, S.C.; Buller, B.; Lu, M.; Zhang, Z.G. Angiopoietin 2 Mediates the Differentiation and Migration of Neural Progenitor Cells in the Subventricular Zone after Stroke. J. Biol. Chem. 2009, 284, 22680–22689. [Google Scholar] [CrossRef]
- Cheeran, M.C.J.; Hu, S.; Ni, H.T.; Sheng, W.; Palmquist, J.M.; Peterson, P.K.; Lokensgard, J.R. Neural Precursor Cell Susceptibility to Human Cytomegalovirus Diverges along Glial or Neuronal Differentiation Pathways. J. Neurosci. Res. 2005, 82, 839–850. [Google Scholar] [CrossRef]
- Marshall, J.; Dolan, B.M.; Garcia, E.P.; Sathe, S.; Tang, X.; Mao, Z.; Blair, L.A.C. Calcium Channel and NMDA Receptor Activities Differentially Regulate Nuclear C/EBPβ Levels to Control Neuronal Survival. Neuron 2003, 39, 625–639. [Google Scholar] [CrossRef]
- Peña-Altamira, E.; Polazzi, E.; Moretto, E.; Lauriola, M.; Monti, B. The Transcription Factor CCAAT Enhancer-Binding Protein β Protects Rat Cerebellar Granule Neurons from Apoptosis through Its Transcription-Activating Isoforms. Eur. J. Neurosci. 2014, 39, 176–185. [Google Scholar] [CrossRef]
- Buttery, P.C.; Barker, R.A. Gene and Cell-Based Therapies for Parkinson’s Disease: Where Are We? Neurotherapeutics 2020, 17, 1539–1562. [Google Scholar] [CrossRef]
- Zhang, Y.; Bi, J.; Huang, J.; Tang, Y.; Du, S.; Li, P. Exosome: A Review of Its Classification, Isolation Techniques, Storage, Diagnostic and Targeted Therapy Applications. Int. J. Nanomed. 2020, 15, 6917–6934. [Google Scholar] [CrossRef]
- Screpanti, I.; Romani, L.; Musiani, P.; Modesti, A.; Fattori, E.; Lazzaro, D.; Sellitto, C.; Scarpa, S.; Bellavia, D.; Lattanzio, G. Lymphoproliferative Disorder and Imbalanced T-Helper Response in C/EBP Beta-Deficient Mice. EMBO J. 1995, 14, 1932–1941. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2-ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Morales-Garcia, J.A.; Palomo, V.; Redondo, M.; Alonso-Gil, S.; Gil, C.; Martinez, A.; Perez-Castillo, A. Crosstalk between Phosphodiesterase 7 and Glycogen Synthase Kinase-3: Two Relevant Therapeutic Targets for Neurological Disorders. ACS Chem. Neurosci. 2014, 5, 194–204. [Google Scholar] [CrossRef]
- Dull, T.; Zufferey, R.; Kelly, M.; Mandel, R.J.; Nguyen, M.; Trono, D.; Naldini, L. A Third-Generation Lentivirus Vector with a Conditional Packaging System. J. Virol. 1998, 72, 8463–8471. [Google Scholar] [CrossRef]
- Morales-Garcia, J.A.; Aguilar-Morante, D.; Hernandez-Encinas, E.; Alonso-Gil, S.; Gil, C.; Martinez, A.; Santos, A.; Perez-Castillo, A. Silencing Phosphodiesterase 7B Gene by Lentiviral-ShRNA Interference Attenuates Neurodegeneration and Motor Deficits in Hemiparkinsonian Mice. Neurobiol. Aging 2015, 36, 1160–1173. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 Years of Image Analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Esteras, N.; Adjobo-Hermans, M.J.W.; Abramov, A.Y.; Koopman, W.J.H. Visualization of Mitochondrial Membrane Potential in Mammalian Cells. Methods Cell Biol. 2020, 155, 221–245. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Symbol | Forward Primer (5′ → 3′) | Reverse Primer (5′ → 3′) |
|---|---|---|
| Hebp2 | CCGGCTCAAGTCCTTTCAGT | TCCATCAAAAGACCGCACGA |
| Tfam | CACCCAGATGCAAAAGTTTCA | CCACTCAGCTTTAAAATC CGC |
| Slc25a37 | CAGCTATTGGTGCTTCTGGC | AAAGTGGGAACCTCCTCCCC |
| 18S | CCAGTAAGTGCGGGTCATAAGC | CCTCACTAAACCATCCAATCGG |
| Sequence | Forward Primer (5′ → 3′) | Reverse Primer (5′ → 3′) |
|---|---|---|
| pTFAM/1188 (−1026/+162) | GGCTGTCTCAGAAGGTGGTTAG | CCACATGCTTCGGAGAAACG |
| pTFAM/652 (−490/+162) | TTAAGCTCTGCGGTAAGGCC | CCACATGCTTCGGAGAAACG |
| pTFAM/500 (−338/+162) | CTGGATGCAGGACTGTCTGT | CCACATGCTTCGGAGAAACG |
| pTFAM/352 (−190/+162 | ACAGAGGTGGCTCAACAGC | CCACATGCTTCGGAGAAACG |
| pTFAM/270 (−108/+162) | TCTTATTCCTCCCCCGCAAG | CCACATGCTTCGGAGAAACG |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sierra-Magro, A.; Bartolome, F.; Lozano-Muñoz, D.; Alarcón-Gil, J.; Gine, E.; Sanz-SanCristobal, M.; Alonso-Gil, S.; Cortes-Canteli, M.; Carro, E.; Pérez-Castillo, A.; et al. C/EBPβ Regulates TFAM Expression, Mitochondrial Function and Autophagy in Cellular Models of Parkinson’s Disease. Int. J. Mol. Sci. 2023, 24, 1459. https://doi.org/10.3390/ijms24021459
Sierra-Magro A, Bartolome F, Lozano-Muñoz D, Alarcón-Gil J, Gine E, Sanz-SanCristobal M, Alonso-Gil S, Cortes-Canteli M, Carro E, Pérez-Castillo A, et al. C/EBPβ Regulates TFAM Expression, Mitochondrial Function and Autophagy in Cellular Models of Parkinson’s Disease. International Journal of Molecular Sciences. 2023; 24(2):1459. https://doi.org/10.3390/ijms24021459
Chicago/Turabian StyleSierra-Magro, Ana, Fernando Bartolome, David Lozano-Muñoz, Jesús Alarcón-Gil, Elena Gine, Marina Sanz-SanCristobal, Sandra Alonso-Gil, Marta Cortes-Canteli, Eva Carro, Ana Pérez-Castillo, and et al. 2023. "C/EBPβ Regulates TFAM Expression, Mitochondrial Function and Autophagy in Cellular Models of Parkinson’s Disease" International Journal of Molecular Sciences 24, no. 2: 1459. https://doi.org/10.3390/ijms24021459
APA StyleSierra-Magro, A., Bartolome, F., Lozano-Muñoz, D., Alarcón-Gil, J., Gine, E., Sanz-SanCristobal, M., Alonso-Gil, S., Cortes-Canteli, M., Carro, E., Pérez-Castillo, A., & Morales-García, J. A. (2023). C/EBPβ Regulates TFAM Expression, Mitochondrial Function and Autophagy in Cellular Models of Parkinson’s Disease. International Journal of Molecular Sciences, 24(2), 1459. https://doi.org/10.3390/ijms24021459