
A Missense Variation in PHACTR2 Associates with Impaired Actin Dynamics, Dilated Cardiomyopathy, and Left Ventricular Non-Compaction in Humans

 , , , , , and
, , , , , and
Abstract
1. Introduction
2. Results
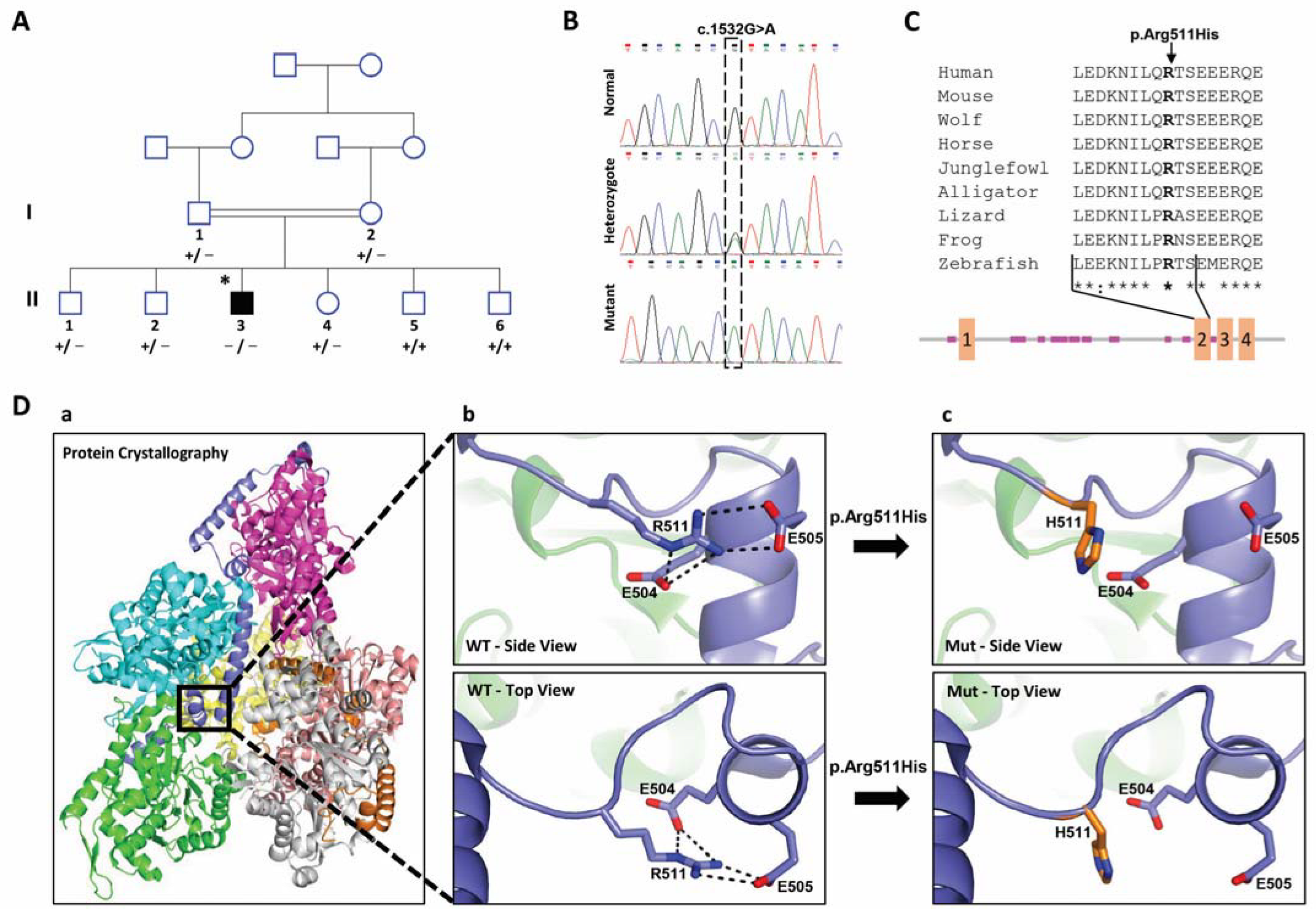
2.1. Clinical Findings
2.2. Identification of the PHACTR2 Variation
2.3. Effect of the Candidate Variation on the Structure of PHACTR2 Protein
2.4. Subcellular Distribution of PHACRT2 Is Not Affected by the Change in Arg 511 to His
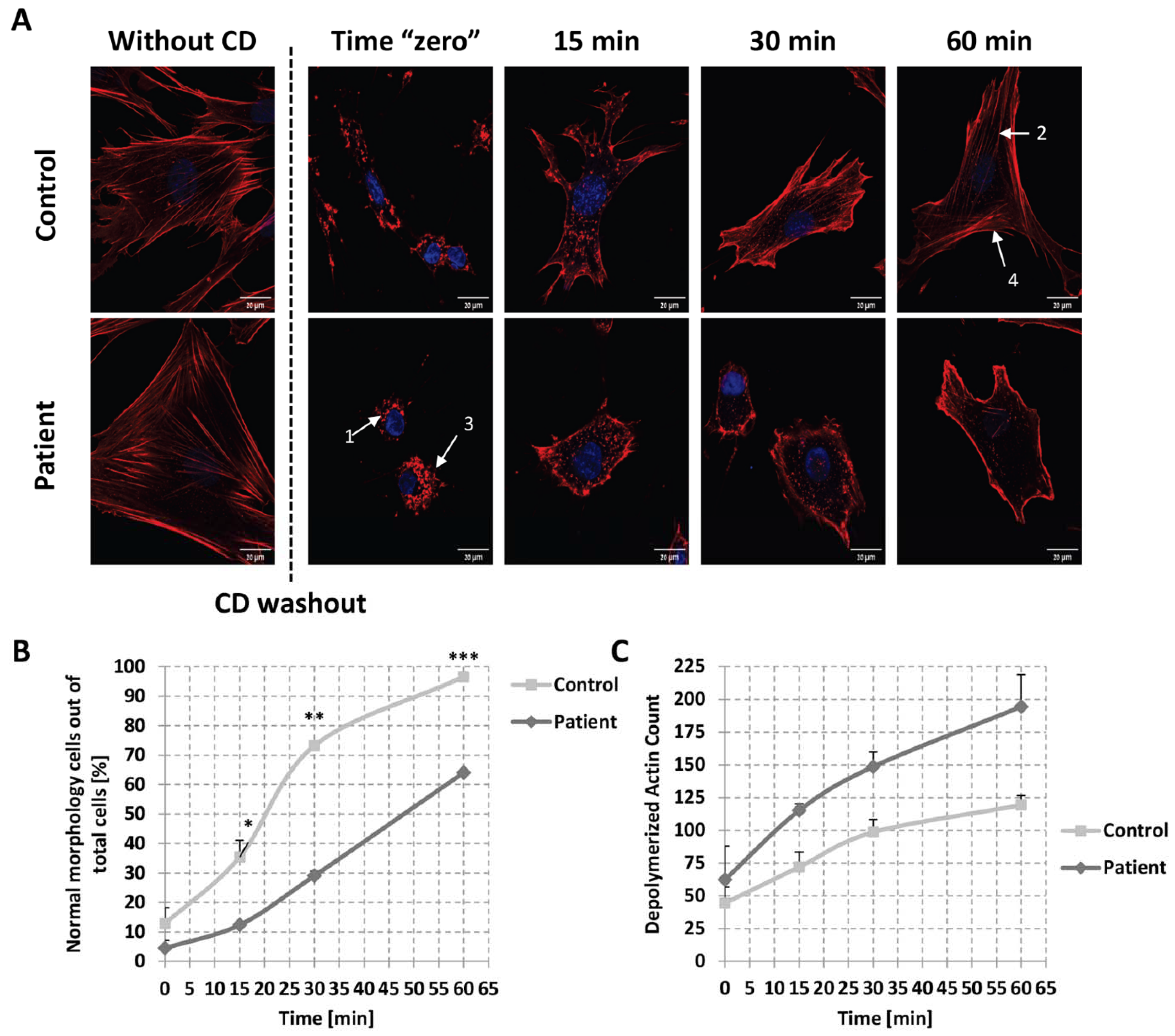
2.5. Globular Actin Pool in Patient’s Fibroblasts
2.6. Kinetics of Actin Repolymerization in Patient’s Fibroblasts
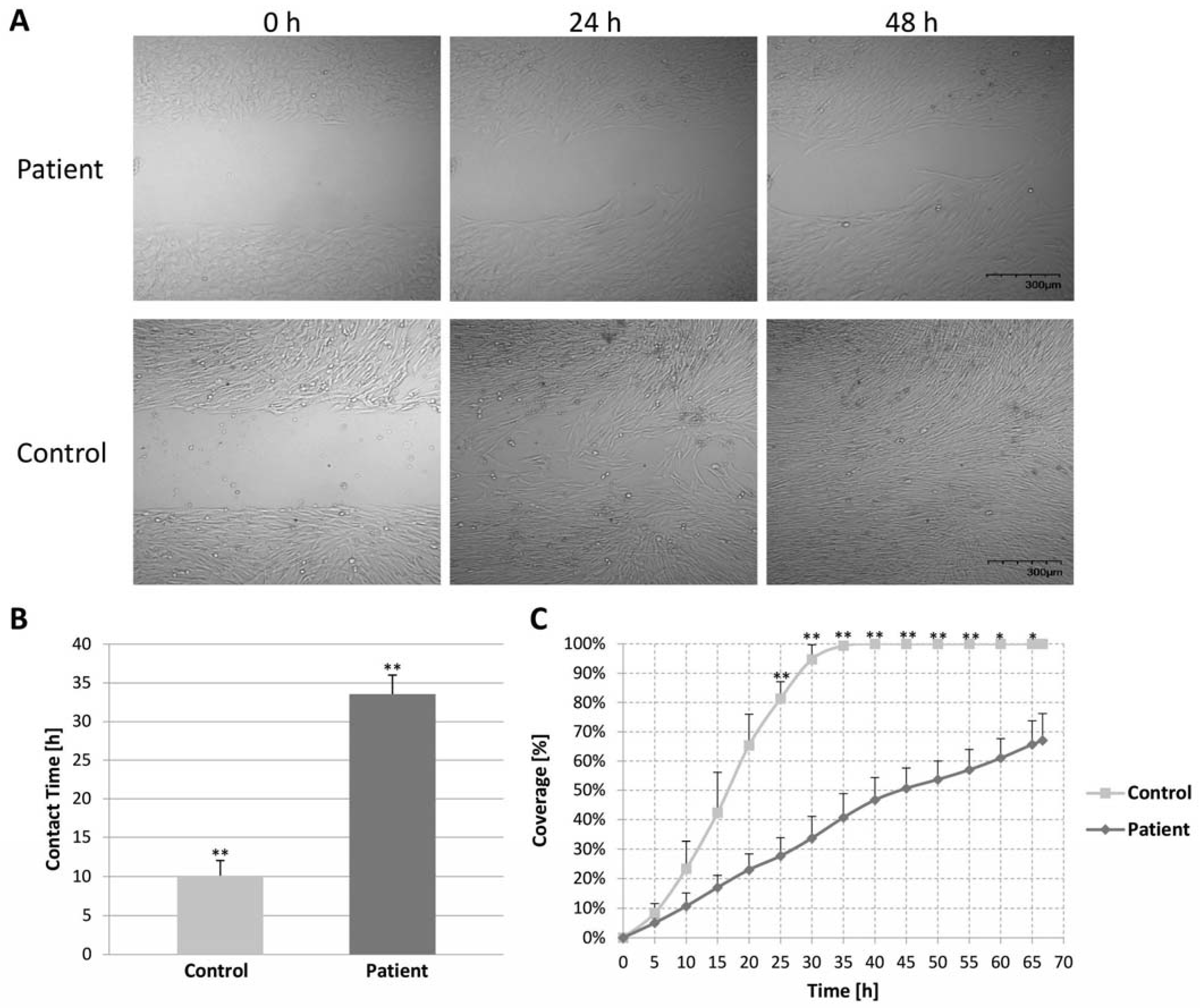
2.7. Migration Assay of Control and Patient Fibroblast Cells (Wound Healing Assay)
3. Discussion
4. Patient and Methods
4.1. Patient
4.2. Genetic Analysis
4.3. Variation Verification
4.4. Protein Structure
4.5. Lentivirus Preparation and Infection
4.6. Immunofluorescence Studies
4.7. Kinetics of Actin Re-Polymerization
4.8. Fractionation of Globular and Filamentous Actin
4.9. Cell Migration Assay
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Web Resources
References
- Hershberger, R.E.; Hedges, D.J.; Morales, A. Dilated cardiomyopathy: The complexity of a diverse genetic architecture. Nat. Rev. Cardiol. 2013, 10, 531–547. [Google Scholar] [CrossRef] [PubMed]
- Pinto, Y.M.; Elliott, P.M.; Arbustini, E.; Adler, Y.; Anastasakis, A.; Bohm, M.; Duboc, D.; Gimeno, J.; de Groote, P.; Imazio, M.; et al. Proposal for a revised definition of dilated cardiomyopathy, hypokinetic non-dilated cardiomyopathy, and its implications for clinical practice: A position statement of the ESC working group on myocardial and pericardial diseases. Eur. Heart J. 2016, 37, 1850–1858. [Google Scholar] [CrossRef] [PubMed]
- Grunig, E.; Tasman, J.A.; Kucherer, H.; Franz, W.; Kubler, W.; Katus, H.A. Frequency and phenotypes of familial dilated cardiomyopathy. J. Am. Coll. Cardiol. 1998, 31, 186–194. [Google Scholar] [CrossRef] [PubMed]
- McNally, E.M.; Golbus, J.R.; Puckelwartz, M.J. Genetic mutations and mechanisms in dilated cardiomyopathy. J. Clin. Investig. 2013, 123, 19–26. [Google Scholar] [CrossRef]
- Mestroni, L.; Rocco, C.; Gregori, D.; Sinagra, G.; Di Lenarda, A.; Miocic, S.; Vatta, M.; Pinamonti, B.; Muntoni, F.; Caforio, A.L.; et al. Familial dilated cardiomyopathy: Evidence for genetic and phenotypic heterogeneity. Heart Muscle Disease Study Group. J. Am. Coll. Cardiol. 1999, 34, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Quiat, D.; Witkowski, L.; Zouk, H.; Daly, K.P.; Roberts, A.E. Retrospective Analysis of Clinical Genetic Testing in Pediatric Primary Dilated Cardiomyopathy: Testing Outcomes and the Effects of Variant Reclassification. J. Am. Heart Assoc. 2020, 9, e016195. [Google Scholar] [CrossRef]
- Parvari, R.; Levitas, A. The mutations associated with dilated cardiomyopathy. Biochem. Res. Int. 2012, 2012, 639250. [Google Scholar] [CrossRef]
- Mazzarotto, F.; Hawley, M.H.; Beltrami, M.; Beekman, L.; de Marvao, A.; McGurk, K.A.; Statton, B.; Boschi, B.; Girolami, F.; Roberts, A.M.; et al. Systematic large-scale assessment of the genetic architecture of left ventricular noncompaction reveals diverse etiologies. Genet. Med. 2021, 23, 856–864. [Google Scholar] [CrossRef]
- Ware, S.M.; Bhatnagar, S.; Dexheimer, P.J.; Wilkinson, J.D.; Sridhar, A.; Fan, X.; Shen, Y.; Tariq, M.; Schubert, J.A.; Colan, S.D.; et al. The genetic architecture of pediatric cardiomyopathy. Am. J. Hum. Genet. 2022, 109, 282–298. [Google Scholar] [CrossRef]
- Schaper, J.; Froede, R.; Hein, S.; Buck, A.; Hashizume, H.; Speiser, B.; Friedl, A.; Bleese, N. Impairment of the myocardial ultrastructure and changes of the cytoskeleton in dilated cardiomyopathy. Circulation 1991, 83, 504–514. [Google Scholar] [CrossRef]
- Peche, V.; Shekar, S.; Leichter, M.; Korte, H.; Schroder, R.; Schleicher, M.; Holak, T.A.; Clemen, C.S.; Ramanath, Y.B.; Pfitzer, G.; et al. CAP2, cyclase-associated protein 2, is a dual compartment protein. Cell Mol. Life Sci. CMLS 2007, 64, 2702–2715. [Google Scholar] [CrossRef] [PubMed]
- Allen, P.B.; Greenfield, A.T.; Svenningsson, P.; Haspeslagh, D.C.; Greengard, P. Phactrs 1-4: A family of protein phosphatase 1 and actin regulatory proteins. Proc. Natl. Acad. Sci. USA 2004, 101, 7187–7192. [Google Scholar] [CrossRef]
- Huttlin, E.L.; Bruckner, R.J.; Paulo, J.A.; Cannon, J.R.; Ting, L.; Baltier, K.; Colby, G.; Gebreab, F.; Gygi, M.P.; Parzen, H.; et al. Architecture of the human interactome defines protein communities and disease networks. Nature 2017, 545, 505–509. [Google Scholar] [CrossRef]
- Kathiresan, S.; Voight, B.F.; Purcell, S.; Musunuru, K.; Ardissino, D.; Mannucci, P.M.; Anand, S.; Engert, J.C.; Samani, N.J.; Schunkert, H.; et al. Genome-wide association of early-onset myocardial infarction with single nucleotide polymorphisms and copy number variants. Nat. Genet. 2009, 41, 334–341. [Google Scholar] [CrossRef] [PubMed]
- Qi, L.; Parast, L.; Cai, T.; Powers, C.; Gervino, E.V.; Hauser, T.H.; Hu, F.B.; Doria, A. Genetic susceptibility to coronary heart disease in type 2 diabetes: 3 independent studies. J. Am. Coll. Cardiol. 2011, 58, 2675–2682. [Google Scholar] [CrossRef] [PubMed]
- Wider, C.; Lincoln, S.J.; Heckman, M.G.; Diehl, N.N.; Stone, J.T.; Haugarvoll, K.; Aasly, J.O.; Gibson, J.M.; Lynch, T.; Rajput, A.; et al. Phactr2 and Parkinson’s disease. Neurosci. Lett. 2009, 453, 9–11. [Google Scholar] [CrossRef]
- Gorski, M.M.; Lecchi, A.; Femia, E.A.; La Marca, S.; Cairo, A.; Pappalardo, E.; Lotta, L.A.; Artoni, A.; Peyvandi, F. Complications of whole-exome sequencing for causal gene discovery in primary platelet secretion defects. Haematologica 2019, 104, 2084–2090. [Google Scholar] [CrossRef]
- Solimini, N.L.; Liang, A.C.; Xu, C.; Pavlova, N.N.; Xu, Q.; Davoli, T.; Li, M.Z.; Wong, K.K.; Elledge, S.J. STOP gene Phactr4 is a tumor suppressor. Proc. Natl. Acad. Sci. USA 2013, 110, E407–E414. [Google Scholar] [CrossRef]
- Kim, T.H.; Goodman, J.; Anderson, K.V.; Niswander, L. Phactr4 regulates neural tube and optic fissure closure by controlling PP1-, Rb-, and E2F1-regulated cell-cycle progression. Dev. Cell 2007, 13, 87–102. [Google Scholar] [CrossRef]
- Mathew, T.; Williams, L.; Navaratnam, G.; Rana, B.; Wheeler, R.; Collins, K.; Harkness, A.; Jones, R.; Knight, D.; O’Gallagher, K.; et al. Diagnosis and assessment of dilated cardiomyopathy: A guideline protocol from the British Society of Echocardiography. Echo Res. Pract. 2017, 4, G1–G13. [Google Scholar] [CrossRef]
- Yamada, T.; Nomura, S. Recent Findings Related to Cardiomyopathy and Genetics. Int. J. Mol. Sci. 2021, 22, 12522. [Google Scholar] [CrossRef] [PubMed]
- Hershberger, R.E. UpToDate—Genetics of Dilated Cardiomyopathy. Available online: https://www.uptodate.com/contents/genetics-of-dilated-cardiomyopathy?search=dilated%20cardiomyopathy%20genetics&source=search_result&selectedTitle=1~150&usage_type=default&display_rank=1 (accessed on 19 January 2022).
- Cossu, C.; Incani, F.; Serra, M.L.; Coiana, A.; Crisponi, G.; Boccone, L.; Rosatelli, M.C. New mutations in DYNC2H1 and WDR60 genes revealed by whole-exome sequencing in two unrelated Sardinian families with Jeune asphyxiating thoracic dystrophy. Clin. Chim. Acta Int. J. Clin. Chem. 2016, 455, 172–180. [Google Scholar] [CrossRef] [PubMed]
- McInerney-Leo, A.M.; Schmidts, M.; Cortés, C.R.; Leo, P.J.; Gener, B.; Courtney, A.D.; Gardiner, B.; Harris, J.A.; Lu, Y.; Marshall, M.; et al. Short-rib polydactyly and Jeune syndromes are caused by mutations in WDR60. Am. J. Hum. Genet. 2013, 93, 515–523. [Google Scholar] [CrossRef] [PubMed]
- Kakar, N.; Horn, D. Expanding the phenotype associated with biallelic WDR60 mutations: Siblings with retinal degeneration and polydactyly lacking other features of short rib thoracic dystrophies. Am. J. Med. Genet. Part A 2018, 176, 438–442. [Google Scholar] [CrossRef]
- Miller, D.T.; Lee, K.; Chung, W.K.; Gordon, A.S.; Herman, G.E.; Klein, T.E.; Stewart, D.R.; Amendola, L.M.; Adelman, K.; Bale, S.J.; et al. ACMG SF v3.0 list for reporting of secondary findings in clinical exome and genome sequencing: A policy statement of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. Off. J. Am. Coll. Med. Genet. 2021, 23, 1381–1390. [Google Scholar] [CrossRef]
- Alsalem, A.B.; Halees, A.S.; Anazi, S.; Alshamekh, S.; Alkuraya, F.S. Autozygome sequencing expands the horizon of human knockout research and provides novel insights into human phenotypic variation. PLoS Genet. 2013, 9, e1004030. [Google Scholar] [CrossRef]
- Fakhro, K.A.; Staudt, M.R.; Ramstetter, M.D.; Robay, A.; Malek, J.A.; Badii, R.; Al-Marri, A.A.; Abi Khalil, C.; Al-Shakaki, A.; Chidiac, O.; et al. The Qatar genome: A population-specific tool for precision medicine in the Middle East. Hum. Genome Var. 2016, 3, 16016. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, E.D.S.; Griffith, S.; Martin, R.; Antonescu, C.; Posey, J.E.; Coban-Akdemir, Z.; Jhangiani, S.N.; Doheny, K.F.; Lupski, J.R.; Valle, D.; et al. Variant-level matching for diagnosis and discovery: Challenges and opportunities. Hum. Mutat. 2022, 43, 782–790. [Google Scholar] [CrossRef] [PubMed]
- Sagara, J.; Arata, T.; Taniguchi, S. Scapinin, the protein phosphatase 1 binding protein, enhances cell spreading and motility by interacting with the actin cytoskeleton. PLoS ONE 2009, 4, e4247. [Google Scholar] [CrossRef]
- Huet, G.; Rajakylä, E.K.; Viita, T.; Skarp, K.P.; Crivaro, M.; Dopie, J.; Vartiainen, M.K. Actin-regulated feedback loop based on Phactr4, PP1 and cofilin maintains the actin monomer pool. J. Cell Sci. 2013, 126, 497–507. [Google Scholar] [CrossRef]
- Mouilleron, S.; Wiezlak, M.; O′Reilly, N.; Treisman, R.; McDonald, N.Q. Structures of the Phactr1 RPEL domain and RPEL motif complexes with G-actin reveal the molecular basis for actin binding cooperativity. Structure 2012, 20, 1960–1970. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Niswander, L. Phactr4: A new integrin modulator required for directional migration of enteric neural crest cells. Cell Adhes. Migr. 2012, 6, 419–423. [Google Scholar] [CrossRef] [PubMed]
- Wiezlak, M.; Diring, J.; Abella, J.; Mouilleron, S.; Way, M.; McDonald, N.Q.; Treisman, R. G-actin regulates the shuttling and PP1 binding of the RPEL protein Phactr1 to control actomyosin assembly. J. Cell Sci. 2012, 125, 5860–5872. [Google Scholar] [CrossRef] [PubMed]
- Itoh, A.; Uchiyama, A.; Taniguchi, S.; Sagara, J. Phactr3/scapinin, a member of protein phosphatase 1 and actin regulator (phactr) family, interacts with the plasma membrane via basic and hydrophobic residues in the N-terminus. PLoS ONE 2014, 9, e113289. [Google Scholar] [CrossRef]
- Favot, L.; Gillingwater, M.; Scott, C.; Kemp, P.R. Overexpression of a family of RPEL proteins modifies cell shape. FEBS Lett. 2005, 579, 100–104. [Google Scholar] [CrossRef]
- Kosmas, K.; Eskandarnaz, A.; Khorsandi, A.B.; Kumar, A.; Ranjan, R.; Eming, S.A.; Noegel, A.A.; Peche, V.S. CAP2 is a regulator of the actin cytoskeleton and its absence changes infiltration of inflammatory cells and contraction of wounds. Eur. J. Cell Biol. 2015, 94, 32–45. [Google Scholar] [CrossRef]
- Fan, Z.; Zhou, S.; Garcia, C.; Fan, L.; Zhou, J. pH-Responsive fluorescent graphene quantum dots for fluorescence-guided cancer surgery and diagnosis. Nanoscale 2017, 9, 4928–4933. [Google Scholar] [CrossRef]
- Jing, Y.; Zhang, L.; Xu, Z.; Chen, H.; Ju, S.; Ding, J.; Guo, Y.; Tian, H. Phosphatase Actin Regulator-1 (PHACTR-1) Knockdown Suppresses Cell Proliferation and Migration and Promotes Cell Apoptosis in the bEnd.3 Mouse Brain Capillary Endothelial Cell Line. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2019, 25, 1291–1300. [Google Scholar] [CrossRef]
- Cao, F.; Liu, M.; Zhang, Q.Z.; Hao, R. PHACTR4 regulates proliferation, migration and invasion of human hepatocellular carcinoma by inhibiting IL-6/Stat3 pathway. Eur. Rev. Med. Pharmacol. Sci. 2016, 20, 3392–3399. [Google Scholar]
- Yang, D.H.; Lee, J.W.; Lee, J.; Moon, E.Y. Dynamic rearrangement of F-actin is required to maintain the antitumor effect of trichostatin A. PLoS ONE 2014, 9, e97352. [Google Scholar] [CrossRef]
- Allain, B.; Jarray, R.; Borriello, L.; Leforban, B.; Dufour, S.; Liu, W.Q.; Pamonsinlapatham, P.; Bianco, S.; Larghero, J.; Hadj-Slimane, R.; et al. Neuropilin-1 regulates a new VEGF-induced gene, Phactr-1, which controls tubulogenesis and modulates lamellipodial dynamics in human endothelial cells. Cell Signal. 2012, 24, 214–223. [Google Scholar] [CrossRef] [PubMed]
- Muhammad, E.; Levitas, A.; Singh, S.R.; Braiman, A.; Ofir, R.; Etzion, S.; Sheffield, V.C.; Etzion, Y.; Carrier, L.; Parvari, R. PLEKHM2 mutation leads to abnormal localization of lysosomes, impaired autophagy flux and associates with recessive dilated cardiomyopathy and left ventricular noncompaction. Hum. Mol. Genet. 2015, 24, 7227–7240. [Google Scholar] [CrossRef] [PubMed]
- Mazzarotto, F.; Tayal, U.; Buchan, R.J.; Midwinter, W.; Wilk, A.; Whiffin, N.; Govind, R.; Mazaika, E.; de Marvao, A.; Dawes, T.J.W.; et al. Reevaluating the Genetic Contribution of Monogenic Dilated Cardiomyopathy. Circulation 2020, 141, 387–398. [Google Scholar] [CrossRef]
- Ceulemans, H.; Bollen, M. Functional diversity of protein phosphatase-1, a cellular economizer and reset button. Physiol. Rev. 2004, 84, 1–39. [Google Scholar] [CrossRef] [PubMed]
- Weber, S.; Meyer-Roxlau, S.; Wagner, M.; Dobrev, D.; El-Armouche, A. Counteracting Protein Kinase Activity in the Heart: The Multiple Roles of Protein Phosphatases. Front. Pharmacol. 2015, 6, 270. [Google Scholar] [CrossRef]
- Aspit, L.; Levitas, A.; Etzion, S.; Krymko, H.; Slanovic, L.; Zarivach, R.; Etzion, Y.; Parvari, R. CAP2 mutation leads to impaired actin dynamics and associates with supraventricular tachycardia and dilated cardiomyopathy. J. Med. Genet. 2019, 56, 228–235. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Age at Onset and Clinical Situation | Age at Follow-Up and Clinical Situation (p/s) | Echo at Follow-Up, 10 Wks | Age at Follow-Up and Clinical Situation (p/s) | Echo at Follow-Up, Teens | CMRI in Teens Cine-SSFP | Repeated Cardiac Holter Monitoring |
|---|---|---|---|---|---|---|
| Prenatal diagnosis at 32 wks Cardiomegaly Increased cardiac/thoracic Circumference ratio: 0.7 (normal: 0.45–0.5) | 10 wks Congestive heart failure Dyspnea Tachypnea Tachycardia Eating difficulty | LVEDD 32 mm z-score- +3.84 LVESD 25 mm z-score- +4.89 EF- 40% | Teens Constant fatigue Exercise intolerance | LVEDD 52 mm z-score- +1.67 LVESD 43 mm z-score- +3.59 EF- 37–40% | LVEDV 101 mL/m2 LVESV 64 mL/m2 EF- 44% Normal origin of coronary artery Non-compaction LV | 22 h Normal sinus rhythm |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Majdalani, P.; Levitas, A.; Krymko, H.; Slanovic, L.; Braiman, A.; Hadad, U.; Dabsan, S.; Horev, A.; Zarivach, R.; Parvari, R. A Missense Variation in PHACTR2 Associates with Impaired Actin Dynamics, Dilated Cardiomyopathy, and Left Ventricular Non-Compaction in Humans. Int. J. Mol. Sci. 2023, 24, 1388. https://doi.org/10.3390/ijms24021388
Majdalani P, Levitas A, Krymko H, Slanovic L, Braiman A, Hadad U, Dabsan S, Horev A, Zarivach R, Parvari R. A Missense Variation in PHACTR2 Associates with Impaired Actin Dynamics, Dilated Cardiomyopathy, and Left Ventricular Non-Compaction in Humans. International Journal of Molecular Sciences. 2023; 24(2):1388. https://doi.org/10.3390/ijms24021388
Chicago/Turabian StyleMajdalani, Pierre, Aviva Levitas, Hanna Krymko, Leonel Slanovic, Alex Braiman, Uzi Hadad, Salam Dabsan, Amir Horev, Raz Zarivach, and Ruti Parvari. 2023. "A Missense Variation in PHACTR2 Associates with Impaired Actin Dynamics, Dilated Cardiomyopathy, and Left Ventricular Non-Compaction in Humans" International Journal of Molecular Sciences 24, no. 2: 1388. https://doi.org/10.3390/ijms24021388
APA StyleMajdalani, P., Levitas, A., Krymko, H., Slanovic, L., Braiman, A., Hadad, U., Dabsan, S., Horev, A., Zarivach, R., & Parvari, R. (2023). A Missense Variation in PHACTR2 Associates with Impaired Actin Dynamics, Dilated Cardiomyopathy, and Left Ventricular Non-Compaction in Humans. International Journal of Molecular Sciences, 24(2), 1388. https://doi.org/10.3390/ijms24021388