
The Advancement of Biodegradable Polyesters as Delivery Systems for Camptothecin and Its Analogues—A Status Report




Abstract
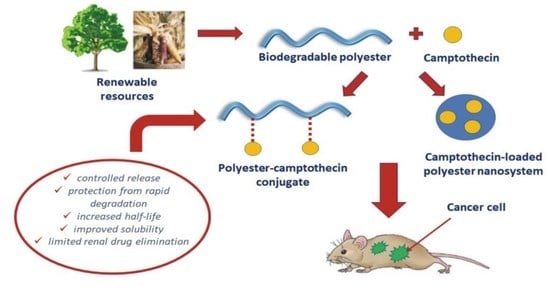
1. Introduction
2. Camptothecin and Its Analogues
2.1. Camptothecin
2.2. Synthetic Analogues of Camptothecin
3. Biodegradable Polyesters in Drug Delivery Systems
4. Polyester Carriers of Camptothecin and Its Analogues in Cancer Therapy
4.1. Encapsulation and/or Adsorption of Camptothecin and Its Analogues onto a Polyester Carrier

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of DDSs | Drug | Polyester Carrier Used for the Formulation | Size, Zeta Potential (ζ) and Entrapment Efficiency (EE) | In Vitro Studies | In Vivo Studies | Additional Studies | Reference | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Release Date | Cytotoxicity | Cellular Uptake | Cell Line | Pharmacokinetics Study | Animal Model | Pharmacodynamics Study | Cell Line | ||||||
| Nanoparticles | Topotecan | PLGA | 243.0 4.0 nm ζ = −2.36 ± 0.6 mV EE = 60.9% 2.2% | Release rate after 1 day: was 25.06% 2.4% at pH 6.5 and 22.45% 2.4% at pH 7.4. | 0.98 (after 48 h) 0.43 (after 72 h) | 2.6% within 1 h | SKOV3 | Noncompartmental method: 17.89 ng/mL AUC0–∞ = 98,978.15 ± 362 ng h/mL 0.3 h | Swiss albino mice | - | SKOV3 | - | [85] |
| 9-CPT | PLGA | 2.6 nm | At pH 7.4, 20% of the drug was released in 20 h | At concentration of 5 µg/mL, after 24 h, percentage of cell viability was equal to approximately 10%. After 24 h, 91% of the cells were killed | Time-dependent cellular uptake, increased with time: about 40 g/mL after 3 h | A2780sn—cytotoxicity study Caco-2—cellular uptake study | Noncompartmental method (total 9-CPT): 868 ng h/mL T1/2 = 2.45 0.27 h MRT = 1.56 0.36 h 59 mL | Male Wistar rats | - | - | Empty NPs were nontoxic | [89,90,91] | |
| CPT-11 | PLGA | 124 12 nm ζ = −20.3 mV EE = 55% 2.7% | At pH 7.4, about 50% of the drug was released within 24 h | IC50 = 36.2 1.2 (after 48 h) | - | HT-29 | - | - | - | - | - | [92] | |
| CPT-11 | PLGA | 169.97 6.29 nm ζ = −0.94 ± 0.6 mV 2.41 % | 3% of the drug was released within 2 h | - | - | - | Compartmental method: AUC0–∞ = 16.8 mg/L·h t1/2β = 3.327 h | Kunming mice | Tumor growth inhibition (TGI) = 86.63% | H22 | ≤5% hemolysis of CPT-11 PLGA NPs at CPT-11 concentration 20–100 µg/mL | [86] | |
| CPT-11 | PCL | 202.1 ± 2.1 nm ζ = ca. − 8.0 mV EE = 65% | 1.5% of the drug was released within 16 days | CPT-11-NPs were significantly more cytotoxic by day 11 compared to day 1 due to the slow release of IRH over the 11 days | - | Primary HGG | - | - | - | - | - | [93] | |
| SN-38 | PLGA–PEG–FOL | 15 nm 0.3 mV 9.2% | At pH 7.4, about 23% of the drug was released within 240 h | 1.1% lower than that of nontargeted NPs (after 48 h) | Higher cellular uptake than PLGA-NPs | HT-29 | - | - | - | - | - | [94] | |
| SN-38 | PLGA | 13 nm mV EE = 77.1% 6.5% | At pH 7.4, about 30% of the drug was released within 240 h | % | - | HT-29 | - | - | - | - | - | [94] | |
| SN-38 | PLGA | 24.5 nm mV 14.9 | At pH 5.0, about 55% of the drug was released within 15 days; at pH 7.4, about 26% of the drug was released within 15 days | IC 50 = 0.874 M (after 48 h) | Active targeting and prolonged circulation properties | CD44 Her2 HGC27 | - | Balb/c nude mice | - | - | In vitro anti-proliferation mechanism revealed downregulated expression of CD44 and Her2 and better inhibition of HGC27 cell growth and invasive activity | [95] | |
| SN-38 | PLGA–PEG–PLGA (70,000:8000:70,000) (HMw) PLGA–PEG–PLGA (6000:10,000:6000) (LMw) | HMw in PBS pH 7,4: 0.25 nm 2.20 mV EE = 6% LMw in PBS pH 7.4: 0.40 nm 1.39 mV EE = 3% | - | Negligible growth inhibition effect. Cell viability approximately 90% | Increasing cellular uptake over time, during 24 h internalization study of fluorescently labeled NPs | SW-480 | - | Wistar rats | - | - | Increased expression of UBD and RGCC genes. Decreased expression of FGF3 and HIST genes. HMw-NPs accumulated rapidly in the liver. LMw-NPs were detected 1 h post injection. After 24 h LMw-NPs were mostly distributed in the liver | [96] | |
| CPT | PLGA | 8.6 nm 0.4 mV 5.5% | 7.4% of the drug was released within 48 h | 4.8% compared with CPT | Concentration-dependent endocytic process: higher cellular uptake after 4 h incubation than free CPT at 150 µg/mL.At 50 and 100 µg/mL, there were no significant differences | HepG2 | - | - | - | - | Functional study of CYP3A4 activity revealed that the activity of the cytochrome P450 may be inhibited by CPT-PLGA NPs | [97] | |
| Micelles | SN-38-BOC | MPEG-P(CL-ran-TMC) | 1 nm ζ = −0.6 mV 1.1% | 1.3% of the drug was released within 24 h | IC50 = 2.1 µg/mL (after 48 h) | - | HCT116 CT26 | - | Female BALB/c mice Female BALB/c nude mice | 2.4% | HCT116 CT26 | SN-38-BOC micelles and free SN-38-BOC inhibited embryonic angiogenesis in transgenic zebrafish embryos. Mice administered with SN-38-BOC micelles maintained body weight | [98] |
| Microspheres | CPT-11 | PLA | 37.2 m EE = 93.4% | About 70% of the drug was released within 1 day | - | - | - | - | - | - | - | - | [99] |
| Nanocapsules | CPT-11 | PLGA | 103.4 nm EE ≈ 65% | At pH 7.4, about 20 % of the drug was released within 5 days. At pH 5.5, about 30% of the drug was released within 5 days | Material inhibited cell survival for both lines. | - | SW1990 Panc-1 | - | BALB/c (nu/nu) nude mice | No significant difference in tumor volume within 5 days after administration. By day 15, the tumor volume was smaller than that of the free drug (p ˂ 0.001) | - | - | [100] |
| Hydrogels | MPEG–CPT | PLGA–PEG–PLGA | - | In PBS about 70% of the drug was released within 35 days. | - | - | - | Mice | Tumor inhibition ratio = 73.1% after 3% (w/w) MPEG-CPT injection (c.a. 56 mg/kg) | Murine S180 sarcoma | - | [101] | |
| CPT-11 | PLGA–PEG–PLGA | - | Sustained release throughout 2 weeks. About 50% of the drug was released within 4 days | In vitro cytotoxicity of PLGA–PEG–PLGA: more than 80% cell viability (10 mg/mL polymer concentration). In vitro hematoxicity of PLGA–PEG–PLGA: (2.8% at 2 mg/mL copolymer concentration). Weak inflammatory response after injection | - | MC3T3 | - | Mice | Tumor inhibition ratio from 86.2% (CPT-11 in thermogel: 1 mg/mL) to 98.2% (CPT-11 in thermogel: 4 mg/mL) | Mice xenografted SW620 human colon tumors | Reduced side-effects in Kunming mice bearing murine solid tumor S180 (drug dose: 45 mg/kg)—lower WBC decrease and rapid WBC recovery in CPT-11 thermogel group than in the CPT-11 group. The body weight of nude mice—no marked difference between the experimental and tumor-free group was observed | [88] | |
| Nanogels | CPT | PLA–PEG–PLA diacrylate | EGDMA 3% 2.49 nm 2.5% EGDMA 6% 8.81 nm 6.9% EGDMA 12% 5.57 nm 4.1% EGDMA 24% 9.68 nm 5.7% EGDMA 50% 8.00 nm 6.1% | In PBS, within 20 days: EGDMA 3%, about 7% of the drug was released. EGDMA 6%, about 4% of the drug was released. EGDMA 12%, about 3% of the drug was released. EGDMA 24%, about 2% of the drug was released. EGDMA 50%, about 1% of the drug was released | - | - | - | - | - | - | - | The size of nanogel did not change within 2 months of storage at 4 °C | [102] |
4.2. Polyester Conjugates of Camptothecin and Its Analogues
| Polymer | Linkage | Drug | In Vitro Studies | In Vivo Studies | Cell Line Tested | Additional Studies | Benefits, Conclusions | References | ||
|---|---|---|---|---|---|---|---|---|---|---|
| Release Kinetics | Cytotoxicity | T1/2 | ||||||||
| (attacic-PLA)100 (PLA-1) and (attacic-PLA)50-b-(isotactic PLA-Pm = 0.79)50 (PLA-2) | Urethane bond | CPT | In buffer solution at pH 1.0, 20% of the drug was released after 35 days from the CPT-PLA-1 and 6% from the CPT-PLA-2 conjugate. In buffer solution at pH 7.4: CPT-PLA-1 led to 7% release, and CPT-PLA-2 led to 4% release after 35 days of incubation | - | - | - | - | PLA matrices were nontoxic according to Spirotox, Protoxkit, and Microtox tests | Drug-release characteristics were strongly influenced by the PLA microstructure chain | [108] |
| PGACL (initiator: PEG-200) PLACL (initiator: PEG-200) poly(CL-co-TMC) (initiator: PEG-200) PLGA (initiator: PEG-200) poly(GL-co-TMC) (initiator: PEG-200) poly(LA-co-TMC) (initiator: PEG-200) PLACL (initiator: PEG-400) PLACL (initiator: PEG-600) | Urethane bond | CPT | In buffer solution, at pH 7.4, after 12 weeks of incubation, 81%, 73%, 32%, 89%, 39%, 35%, 82%, and 89% of CPT were released, respectively | - | - | - | - | Synthesized matrices were nontoxic according to Spirotox, ProtoxkitF, and Microtox tests | The rates of CPT release were shown to be directly dependent on the nature of the synthesized carriers. The kinetic rates of drug release were found to be faster in polymeric conjugates containing CL, rac-LA, or GL units compared to those containing TMC units. The rate of in vitro CPT release from the macromolecular conjugates increased as the Mn of PEG increased | [23] |
| P(EAEP–PPA) | Disulfide bond | CPT | In buffer solution at pH 7.4, about 20% of the drug was released within 1 day. In buffer solution at pH 7.4 + 2 M GSH, about 20% of CPT was released within 1 day. In buffer solution at pH 7.4 + 5 mM GSH, about 35% of drug was released within 1 day. In buffer solution at pH 7.4 + 10 mM GSH, about 50% of the drug was released within 1 day | Synthesized polymers were nontoxic | - | - | 4T1, L929, and HepG2 | Efficient cellular uptake of CPT endocytosis | Efficient inhibition of 4T1 and HepG2 cell proliferation | [109] |
| PEG-3400 | Ester bond | CPT | In PBS, about 90% of CPT was released within 10 days | - | - | - | - | Higher stability of the conjugate at pH 6.0 and 5.5 than at pH 7.4. | - | [112] |
| PEG-40000 | Ester bond, amino acid spacers (alanine, methionine, sarcosine, and glycine) | SN-38 | - | IC50, µM: COLO 205 (colorectal): 9, 0.13 ± 0.022; 13, 0.10 ± 0.024; 18, 0.18 ± 0.013; 23, 0.14 ± 0.032. HT29: 9, 0.21 ± 0.068; 13, 0.21 ± 0.049; 18, 0.34 ± 0.081; 23, 0.52 ± 0.066. OVCAR-3: 9, 0.22 ± 0.01; 13, 0.2 ± 0.047; 18, 0.27 ± 0.11; 23, 0.1 ± 0.032. A549: 9, 3.9 ± 0.97; 13, 2.1 ± 0.28; 18, 5.1 ± 0.95; 23, 3.1 ± 043 | In human plasma: 9–12.5 min, 13–26.8 min, 18–19 min, and 23–12.3 minIn rat plasma: 9–6.3 min, 13–12.4 min, 18–10.5 min, and 23–3.5 min | Female nude mice–human mammary carcinoma (MX1) breast tumor | COLO 205, HT 29, A549, and OVCAR 3 | Much enhanced anticancer activity of SN38 in the MX-1 xenograft mice model compared with CPT-11 | - | [110] |
| CD-123 | Disulfide linker | CPT | Release achieved in the presence of GSH | - | - | - | THP-1 and Hep3B | - | - | [113] |
| PEG2K–PLA4-2K | Ester bond | SN-38 | In PBS at pH 7.4 with addition of 0.2% Tween-80, about 25% of the drug was released within 1 day. In PBS at pH 7.4 with addition of 20% (dialysis medium), about 45% of the drug was released within 1 day | Against BEL-7402: IC50 = 5.33 0.84 g/mL. Against HCT116: IC50 = 0.93 0.18 g/mL | - | - | BEL-7402 and HCT116 | - | - | [111] |
| PEG2K–PLA8-9K | Ester bond | SN-38 | In PBS at pH 7.4 with addition of 0.2% Tween-80, about 25% of the drug was released within 1 day. In PBS at pH 7.4 with addition of 20% dialysis medium, about 45% of the drug was released within 1 day | Against BEL-7402: IC50 = 2.35 0.11 g/mL. Against HCT116: g/mL | - | - | BEL-7402 and HCT116 | - | - | [111] |
| PEG4K–PLA1K | Ester bond | SN-38 | In PBS at pH 7.4 with addition of 0.2% Tween-80, about 25% of the drug was released within 1 day. In PBS at pH 7.4 with addition of 20% dialysis medium, about 50% of the drug was released within 1 day | Against BEL-7402: IC50 = 21.38 2.44 g/mL. Against HCT116: IC50 = 21.06 1.19 g/mL | - | - | BEL-7402 and HCT116 | - | - | [111] |
5. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| 4T1 | Mouse breast cancer cells |
| 9-AC | 9-Aminocamptothecin |
| 9-CPT | Rubitecan |
| 9-NC | 9-Nitrocamptothecin |
| A2780sn | Human ovarian cancer cell line |
| A549 | Human lung carcinoma cell line |
| ALL | Acute lymphocytic leukemia |
| AML | Acute myelogenous leukemia |
| AUC | Area under the curve |
| BAX | Bcl-2-associated X protein |
| BEL-7402 | Human hepatoma cell line |
| Caco-2 | Colorectal adenocarcinoma cell line |
| CD123 | Interleukin-3 receptor alpha chain |
| CD147 mAb | CD147 monoclonal antibody |
| CKD-602 | Belotecan |
| CL | ε-Caprolactone |
| Cmax | Highest concentration of the drug |
| COLO-205 | Human colon carcinoma cell line |
| CPPs | Cell penetrating peptides |
| CPT | Camptothecin |
| CPT-11 | Irinotecan |
| CT26 | Mouse colorectal carcinoma cell line |
| CuAAC | Cu(I)-catalyzed azidealkyne cycloaddition |
| Da | Dalton |
| DDSs | Drug delivery systems |
| DX-8951f | Exatecan |
| EE | Entrapment efficiency |
| EGDMA | Ethylene glycol dimethacrylate |
| FDA | US Food and Drug Administration |
| FGF3 | Fibroblast growth factor 3 gene |
| FOL | Folate |
| GG-211 | Lurtotecan |
| GSH | Glutathione |
| GA | Glycolide monomer |
| H22 | Hepatocellular carcinoma cell line |
| HCT116 | Human colon cancer cell line |
| Hep3B | Human hepatoma cell line |
| HepG2 | Human hepatocellular carcinoma cells |
| HGC27 | Gastric cancer cells |
| HGG | High-grade glioma |
| HIST | Histone cluster 1 gene |
| HMW | High molecular weight |
| HT-29 | Human colorectal adenocarcinoma cell culture |
| IC50 | Drug concentration needed to inhibit 50% of the cell growth |
| IRH LA | Irinotecan hydrochloride trihydrate Lactide monomer |
| LMW | Low molecular weight |
| mAB | Monoclonal antibodies |
| MC3T3 | Mouse osteoblast cell line |
| Mn | Number-averaged molecular weight |
| Mw | Weight-averaged molecular weight |
| mPEG | Monomethoxy-poly(ethylene glycol) |
| MPEG-P(CL-ran-TMC) | Monomethyl poly(ethylene glycol)-poly-(ε-caprolactone)-poly(trimethylene carbonate) |
| mPEG-PLA | Methoxypoly(ethylene glycol)-b-poly(lactide) |
| MRT | Mean residence time |
| MTD | Maximum tolerated dose |
| MX-1 | Human mammary carcinoma cell line |
| NPs | Nanoparticles |
| NX-211 | Liposomal lurtotecan |
| OSI-211 | Liposomal lurtotecan |
| OVCAR 3 | Human ovarian carcinoma cell line |
| P(EAEP-PPA) | Poly(ethyl-bis(2-(acryloxy)ethyl phosphate-co-2-propynylamine) |
| Panc-1 | Human pancreatic cancer cell line |
| PARP | Poly adenosine diphosphate-ribose polymerase |
| PBS | Phosphate buffer solution |
| PCL | Poly(ε-caprolactone) |
| PDCs | Polymer–drug conjugates |
| PDLLA | Poly(D,L-lactide) |
| PEG | Poly(ethylene glycol) |
| PGA | Poly(glycolide), Poly(glycolic acid) |
| PGACL | Poly(glycolide-co-ε-caprolactone) |
| PHB | Poly(3-hydroxybutyrate) |
| PHF | Poly(1-hydroxymethylethylene hydroxy-methyl formal) |
| PLA | Polylactide |
| PLACL | Poly(lactide-co-ε-caprolactone) |
| PLGA | Poly(lactide-co-glycolide) |
| PDLA | Poly(D-lactide) |
| PLLA | Poly(L-lactide) |
| rac-LA | A racemic mixture of L-LA and D-LA |
| RAFT | Reversible addition fragmentation transfer |
| RGCC | Regulator of cell cycle, response gene to complement 32 protein |
| ROMP | Ring-opening metathesis polymerization |
| ROP | Ring-opening polymerization |
| S-180 | Sarcoma bearing Kunming mice |
| SAR | Structure–activity relationship |
| sc-PLA | Stereocomplex PLA |
| SKOV3 | Human ovarian cancer cell line |
| SMPs | SN-38-loaded biodegradable PLGA microparticles |
| SN-38 | 7-Ethyl-10-hydroxycamptothecin |
| SN-38-BOC | 7-Ethyl-10-O-tert-butyl ester-camptothecin |
| SW-480 | Human colon adenocarcinoma cell line |
| SW1990 | Human pancreatic cell line |
| SW620 | Human colon tumor |
| TGI | Tumor growth inhibition |
| THP-1 | Human acute myeloid leukemia cell line |
| Tmax | Time to reach maximum concentration of the drug |
| TMC | Trimethylene carbonate |
| TMC | N-Trimethyl chitosan chloride |
| Top 1 | Topoisomerase I |
| UBD | Ubiquitin D gene |
| UV | Ultraviolet |
| Vss | Volume of distribution at steady stage |
| WBC | White blood cell |
| ζ | Zeta potential |
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef]
- Pérez-Herrero, E.; Fernández-Medarde, A. Advanced targeted therapies in cancer: Drug nanocarriers, the future of chemotherapy. Eur. J. Pharm. Biopharm. 2015, 93, 52–79. [Google Scholar] [CrossRef] [PubMed]
- Hauner, K.; Maisch, P.; Retz, M. Side effects of chemotherapy. Urol. A 2017, 56, 472–479. [Google Scholar] [CrossRef]
- Livshits, Z.; Rao, R.B.; Smith, S.W. An approach to chemotherapy-associated toxicity. Emerg. Med. Clin. N. Am. 2014, 32, 167–203. [Google Scholar] [CrossRef] [PubMed]
- Norouzi, M.; Nazari, B.; Miller, D.W. Injectable hydrogel-based drug delivery systems for local cancer therapy. Drug Discov. Today 2016, 21, 1835–1849. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, T. Side effects of cancer chemotherapy and steps to deal with them. Jpn. J. Cancer Chemother. 1995, 22, 2017–2028. [Google Scholar]
- Zraik, I.M.; Heß-Busch, Y. Management of chemotherapy side effects and their long-term sequelae. Urol. A 2021, 60, 862–871. [Google Scholar] [CrossRef]
- Ramadori, G.; Cameron, S. Effects of systemic chemotherapy on the liver. Ann. Hepatol. 2010, 9, 133–143. [Google Scholar] [CrossRef]
- Chen, Z.; Li, Y.; Tan, B.; Zhao, Q.; Fan, L.; Li, F.; Zhao, X. Progress and current status of molecule-targeted therapy and drug resistance in gastric cancer. Drugs Today 2020, 56, 469–482. [Google Scholar] [CrossRef]
- Senapati, S.; Mahanta, A.K.; Kumar, S.; Maiti, P. Controlled drug delivery vehicles for cancer treatment and their performance. Signal Transduct. Target. Ther. 2018, 3, 7. [Google Scholar] [CrossRef]
- Liu, P.; Chen, G.; Zhang, J. A review of liposomes as a drug delivery system: Current status of approved products, regulatory environments, and future perspectives. Molecules 2022, 27, 1372. [Google Scholar] [CrossRef]
- Zare, H.; Ahmadi, S.; Ghasemi, A.; Ghanbari, M.; Rabiee, N.; Bagherzadeh, M.; Karimi, M.; Webster, T.J.; Hamblin, M.R.; Mostafavi, E. Carbon nanotubes: Smart drug/gene delivery carriers. Int. J. Nanomed. 2021, 16, 1681–1706. [Google Scholar] [CrossRef]
- Chauhan, A.S. Dendrimers for drug delivery. Molecules 2018, 23, 938. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, Z.; Shah, A.; Siddiq, M.; Kraatz, H.B. Polymeric micelles as drug delivery vehicles. RSC Adv. 2014, 4, 17028–17038. [Google Scholar] [CrossRef]
- Larson, N.; Ghandehari, H. Polymeric conjugates for drug delivery. Chem. Mater. 2012, 24, 840–853. [Google Scholar] [CrossRef] [PubMed]
- Bajracharya, R.; Song, J.G.; Patil, B.R.; Lee, S.H.; Noh, H.M.; Kim, D.H.; Kim, G.L.; Seo, S.H.; Park, J.W.; Jeong, S.H.; et al. Functional ligands for improving anticancer drug therapy: Current status and applications to drug delivery systems. Drug Deliv. 2022, 29, 1959–1970. [Google Scholar] [CrossRef] [PubMed]
- Tian, H.R.; Huang, Y.K.; He, J.L.; Zhang, M.Z.; Ni, P.H. CD147 Monoclonal antibody targeted reduction-responsive camptothecin polyphosphoester nanomedicine for drug delivery in hepatocellular carcinoma cells. ACS Appl. Bio Mater. 2021, 4, 4422–4431. [Google Scholar] [CrossRef]
- Berrada, M.; Serreqi, A.; Dabbarh, F.; Owusu, A.; Gupta, A.; Lehnert, S. A novel non-toxic camptothecin formulation for cancer chemotherapy. Biomaterials 2005, 26, 2115–2120. [Google Scholar] [CrossRef] [PubMed]
- Khaiwa, N.; Maarouf, N.R.; Darwish, M.H.; Alhamad, D.W.M.; Sebastian, A.; Hamad, M.; Omar, H.A.; Orive, G.; Al-Tel, T.H. Camptothecin’s journey from discovery to WHO essential medicine: Fifty years of promise. Eur. J. Med. Chem. 2021, 223, 113639. [Google Scholar] [CrossRef]
- Botella, P.; Rivero-Buceta, E. Safe approaches for camptothecin delivery: Structural analogues and nanomedicines. J. Control Release 2017, 247, 28–54. [Google Scholar] [CrossRef]
- Xin, Y.; Yin, M.; Zhao, L.; Meng, F.; Luo, L. Recent progress on nanoparticle-based drug delivery systems for cancer therapy. Cancer Biol. Med. 2017, 14, 228–241. [Google Scholar] [CrossRef] [PubMed]
- Maeda, H.; Greish, K.; Fang, J. The EPR effect and polymeric drugs: A paradigm shift for cancer chemotherapy in the 21st century. Adv. Polym. Sci. 2006, 193, 103–121. [Google Scholar]
- Sobczak, M.; Olędzka, E.; Kwietniewska, M.; Nałęcz-Jawecki, G.; Kołodziejski, W. Promising macromolecular conjugates of camptothecin—the synthesis, characterization and in vitro studies. J. Macromol. Sci. Part A 2014, 51, 254–262. [Google Scholar] [CrossRef]
- Legarza, K.; Yang, L.X. Novel camptothecin derivatives. Vivo 2005, 19, 283–292. [Google Scholar] [PubMed]
- Martino, E.; Della Volpe, S.; Terribile, E.; Benetti, E.; Sakaj, M.; Centamore, A.; Sala, A.; Collina, S. The long story of camptothecin: From traditional medicine to drugs. Bioorg. Med. Chem. Lett. 2017, 27, 701–707. [Google Scholar] [CrossRef] [PubMed]
- Li, F.Z.; Jiang, T.; Li, Q.Y.; Ling, X. Camptothecin (CPT) and its derivatives are known to target topoisomerase I (Top1) as their mechanism of action: Did we miss something in CPT analogue molecular targets for treating human disease such as cancer? Am. J. Cancer Res. 2017, 7, 2350–2394. [Google Scholar]
- Li, Q.Y.; Zu, Y.G.; Shi, R.Z.; Yao, L.P. Review Camptothecin: Current Perspectives. Curr. Med. Chem. 2006, 13, 2021–2039. [Google Scholar] [CrossRef]
- Timothy, L.; MacDonald, M.A.L.; Jetze, J.T. Comprehensive Natural Products Chemistry; Sir Barton, D.K.N., Meth-Cohn, O., Eds.; Elsevier: Amsterdam, The Netherlands, 1999. [Google Scholar]
- Venditto, V.J.; Simanek, E.E. Cancer therapies utilizing the camptothecins: A review of the in vivo literature. Mol. Pharm. 2010, 7, 307–349. [Google Scholar] [CrossRef]
- Amin, S.A.; Adhikari, N.; Jha, T.; Gayen, S. A review on camptothecin analogs with promising cytotoxic profile. Anti-Cancer Agents Med. Chem. 2018, 18, 1796–1814. [Google Scholar] [CrossRef]
- Wani, M.C.; Ronman, P.E.; Lindley, J.T.; Wall, M.E. Plant antitumor agents. 18. Synthesis and biological activity of camptothecin analogs. J. Med. Chem. 1980, 23, 554–560. [Google Scholar] [CrossRef]
- Nicholas, A.W.; Wani, M.C.; Manikumar, G.; Wall, M.E.; Kohn, K.W.; Pommier, Y. Plant antitumor agents. 29. Synthesis and biological activity of ring D and ring E modified analogues of camptothecin. J. Med. Chem. 1990, 33, 972–978. [Google Scholar] [CrossRef] [PubMed]
- Schöffski, P.; Herr, A.; Vermorken, J.B.; Van den Brande, J.; Beijnen, J.H.; Rosing, H.; Volk, J.; Ganser, A.; Adank, S.; Botma, H.J.; et al. Clinical phase II study and pharmacological evaluation of rubitecan in non-pretreated patients with metastatic colorectal cancer-significant effect of food intake on the bioavailability of the oral camptothecin analogue. Eur. J. Cancer 2002, 38, 807–813. [Google Scholar] [CrossRef]
- Clark, J.W. Rubitecan. Expert Opin. Investig. Drugs 2006, 15, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Ho, J.Y.; Liu, J.J.; Lee, H.; Park, J.Y.; Baik, M.; Ko, M.; Lee, S.U.; Choi, Y.J.; Hur, S.Y. CKD-602, a topoisomerase I inhibitor, induces apoptosis and cell-cycle arrest and inhibits invasion in cervical cancer. Mol. Med. 2019, 25, 23. [Google Scholar] [CrossRef] [PubMed]
- MacKenzie, M.J.; Hirte, H.W.; Siu, L.L.; Gelmon, K.; Ptaszynski, M.; Fisher, B.; Eisenhauer, E. A phase I study of OSI-211 and cisplatin as intravenous infusions given on days 1, 2 and 3 every 3 weeks in patients with solid cancers. Ann. Oncol. 2004, 15, 665–670. [Google Scholar] [CrossRef] [PubMed]
- Tomkinson, B.; Bendele, R.; Giles, F.J.; Brown, E.; Gray, A.; Hart, K.; LeRay, J.D.; Meyer, D.; Pelanne, M.; Emerson, D.L. OSI-211, a novel liposomal topoisomerase I inhibitor, is active in SCID mouse models of human AML and ALL. Leuk. Res. 2003, 27, 1039–1050. [Google Scholar] [CrossRef] [PubMed]
- Mitsui, I.; Kumazawa, E.; Hirota, Y.; Aonuma, M.; Sugimori, M.; Ohsuki, S.; Uoto, K.; Ejima, A.; Terasawa, H.; Sato, K. A new water-soluble camptothecin derivative, DX-8951f, exhibits potent antitumor activity against human tumors in vitro and in vivo. Jpn. J. Cancer Res. 1995, 86, 776–782. [Google Scholar] [CrossRef]
- van Hattum, A.H.; Pinedo, H.M.; Schlüper, H.M.; Erkelens, C.A.; Tohgo, A.; Boven, E. The activity profile of the hexacyclic camptothecin derivative DX-8951f in experimental human colon cancer and ovarian cancer. Biochem. Pharm. 2002, 64, 1267–1277. [Google Scholar] [CrossRef]
- Liu, Y.Q.; Li, W.Q.; Morris-Natschke, S.L.; Qian, K.; Yang, L.; Zhu, G.X.; Wu, X.B.; Chen, A.L.; Zhang, S.Y.; Nan, X.; et al. Perspectives on biologically active camptothecin derivatives. Med. Res. Rev. 2015, 35, 753–789. [Google Scholar] [CrossRef]
- Bleiberg, H. CPT-11 in gastrointestinal cancer. Eur. J. Cancer 1999, 35, 371–379. [Google Scholar] [CrossRef]
- Tian, Q.; Zhang, J.; Tan, T.M.; Chan, E.; Duan, W.; Chan, S.Y.; Boelsterli, U.A.; Ho, P.C.; Yang, H.; Bian, J.S.; et al. Human multidrug resistance associated protein 4 confers resistance to camptothecins. Pharm. Res. 2005, 22, 1837–1853. [Google Scholar] [CrossRef]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef]
- Jung, J.Y.; Song, S.H.; Kim, T.Y.; Park, J.H.; Jong, H.S.; Im, S.A.; Kim, T.Y.; Bang, Y.J.; Kim, N.K. The synergism between belotecan and cisplatin in gastric cancer. Cancer Res. Treat. 2006, 38, 159–167. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lim, S.; Cho, B.C.; Jung, J.Y.; Kim, G.M.; Kim, S.H.; Kim, H.R.; Kim, H.S.; Lim, S.M.; Park, J.S.; Lee, J.H.; et al. Phase II study of camtobell inj. (belotecan) in combination with cisplatin in patients with previously untreated, extensive stage small cell lung cancer. Lung Cancer 2013, 80, 313–318. [Google Scholar] [CrossRef]
- Thomas, C.J.; Rahier, N.J.; Hecht, S.M. Camptothecin: Current perspectives. Bioorganic Med. Chem. 2004, 12, 1585–1604. [Google Scholar] [CrossRef] [PubMed]
- Garrison, M.A.; Hammond, L.A.; Geyer, C.E., Jr.; Schwartz, G.; Tolcher, A.W.; Smetzer, L.; Figueroa, J.A.; Ducharme, M.; Coyle, J.; Takimoto, C.H.; et al. A phase I and pharmocokinetic study of exatecan mesylate administered as a protracted 21-day infusion in patients with advanced solid malignancies. Clin. Cancer Res. 2003, 9, 2527–2537. [Google Scholar] [PubMed]
- Zhang, J.A.; Xuan, T.; Parmar, M.; Ma, L.; Ugwu, S.; Ali, S.; Ahmad, I. Development and characterization of a novel liposome-based formulation of SN-38. Int. J. Pharm. 2004, 270, 93–107. [Google Scholar] [CrossRef] [PubMed]
- Kawato, Y.; Aonuma, M.; Hirota, Y.; Kuga, H.; Sato, K. Intracellular roles of SN-38, a metabolite of the camptothecin derivative CPT-11, in the antitumor effect of CPT-11. Cancer Res. 1991, 51, 4187–4191. [Google Scholar]
- Bala, V.; Rao, S.; Boyd, B.J.; Prestidge, C.A. Prodrug and nanomedicine approaches for the delivery of the camptothecin analogue SN38. J. Control Release 2013, 172, 48–61. [Google Scholar] [CrossRef]
- Im, S.H.; Im, D.H.; Park, S.J.; Chung, J.J.; Jung, Y.; Kim, S.H. Stereocomplex polylactide for drug delivery and biomedical applications: A review. Molecules 2021, 26, 2846. [Google Scholar] [CrossRef] [PubMed]
- Luo, F.; Fortenberry, A.; Ren, J.; Qiang, Z. Recent progress in enhancing poly(lactic acid) stereocomplex formation for material property improvement. Front. Chem. 2020, 8, 688. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, H. Poly(lactic acid) stereocomplexes: A decade of progress. Adv. Drug Deliv. Rev. 2016, 107, 97–135. [Google Scholar] [CrossRef]
- Pohlmann, A.R.; Fonseca, F.N.; Paese, K.; Detoni, C.B.; Coradini, K.; Beck, R.C.; Guterres, S.S. Poly(ϵ-caprolactone) microcapsules and nanocapsules in drug delivery. Expert Opin. Drug Deliv. 2013, 10, 623–638. [Google Scholar] [CrossRef]
- Saez-Fernandez, E.; Ruiz, M.A.; Arias, J.L. Drug delivery systems based on poly(epsilon-caprolactone) for cancer treatment. Ars Pharm. 2009, 50, 83–96. [Google Scholar]
- Higgins, N.A. E. I. du Pont de Nemours & Co. Polymers of Hydroxyacetic Acid and Its Ester. US Patent 2676945, 18 October 1950. [Google Scholar]
- Chu, C.C. The in-vitro degradation of poly(glycolic acid) sutures—Effect of pH. J. Biomed. Mater. Res. 1981, 15, 795–804. [Google Scholar] [CrossRef] [PubMed]
- Laufman, H.; Rubel, T. Synthetic absorable sutures. Surg. Gynecol. Obs. 1977, 145, 597–608. [Google Scholar]
- Jain, R.; Shah, N.H.; Malick, A.W.; Rhodes, C.T. Controlled drug delivery by biodegradable poly(ester) devices: Different preparative approaches. Drug Dev. Ind. Pharm. 1998, 24, 703–727. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, D.N.; Bhatia, A.; Kaur, R.; Sharma, R.; Kaur, G.; Dhawan, S. PLGA: A unique polymer for drug delivery. Ther. Deliv. 2015, 6, 41–58. [Google Scholar] [CrossRef] [PubMed]
- Sadat Tabatabaei Mirakabad, F.; Nejati-Koshki, K.; Akbarzadeh, A.; Yamchi, M.R.; Milani, M.; Zarghami, N.; Zeighamian, V.; Rahimzadeh, A.; Alimohammadi, S.; Hanifehpour, Y. PLGA-based nanoparticles as cancer drug delivery systems. Asian Pac. J. Cancer Prev. 2014, 15, 517–535. [Google Scholar] [CrossRef]
- Fernández, J.; Etxeberria, A.; Sarasua, J.R. Synthesis, structure and properties of poly(L-lactide-co-ε-caprolactone) statistical copolymers. J. Mech. Behav. Biomed. Mater. 2012, 9, 100–112. [Google Scholar] [CrossRef]
- Nardo, T.; Chiono, V.; Gentile, P.; Tabrizian, M.; Ciardelli, G. Poly(DL-lactide-co-epsilon-caprolactone) and poly(DL-lactide-co-glycolide) blends for biomedical application: Physical properties, cell compatibility, and in vitro degradation behavior. Int. J. Polym. Mater. Polym. Biomater. 2016, 65, 741–750. [Google Scholar] [CrossRef]
- Meek, M.F.; Jansen, K.; Steendam, R.; van Oeveren, W.; van Wachem, P.B.; van Luyn, M.J. In vitro degradation and biocompatibility of poly(DL-lactide-epsilon-caprolactone) nerve guides. J. Biomed. Mater. Res. A 2004, 68, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Kim, B.S.; Kim, S.H.; Choi, S.W.; Jeong, S.I.; Kwon, I.K.; Kang, S.W.; Nikolovski, J.; Mooney, D.J.; Han, Y.K.; et al. Elastic biodegradable poly(glycolide-co-caprolactone) scaffold for tissue engineering. J. Biomed. Mater. Res. A 2003, 66, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Sharma, U.; Concagh, D.; Core, L.; Kuang, Y.; You, C.; Pham, Q.; Zugates, G.; Busold, R.; Webber, S.; Merlo, J.; et al. The development of bioresorbable composite polymeric implants with high mechanical strength. Nat. Mater. 2018, 17, 96–103. [Google Scholar] [CrossRef]
- Min, C.; Cui, W.; Bei, J.; Wang, S. Biodegradable shape-memory polymer—Polylactide-co-poly(glycolide-co-caprolactone) multiblock copolymer. Polym. Adv. Technol. 2005, 16, 608–615. [Google Scholar] [CrossRef]
- Lin, X.; Yin, M.; Liu, Y.; Li, L.; Ren, X.; Sun, Y.; Huang, T.S. Biodegradable polyhydroxybutyrate/poly-ε-caprolactone fibrous membranes modified by silica composite hydrol for super hydrophobic and outstanding antibacterial application. J. Ind. Eng. Chem. 2018, 63, 303–311. [Google Scholar] [CrossRef]
- Degli Esposti, M.; Chiellini, F.; Bondioli, F.; Morselli, D.; Fabbri, P. Highly porous PHB-based bioactive scaffolds for bone tissue engineering by in situ synthesis of hydroxyapatite. Mater. Sci. Eng. C Mater. Biol. Appl. 2019, 100, 286–296. [Google Scholar] [CrossRef]
- Zhao, J.; Weng, G.; Li, J.; Zhu, J.; Zhao, J. Polyester-based nanoparticles for nucleic acid delivery. Mater. Sci. Eng. C Mater. Biol. Appl. 2018, 92, 983–994. [Google Scholar] [CrossRef] [PubMed]
- Dozier, J.K.; Distefano, M.D. Site-Specific PEGylation of Therapeutic Proteins. Int. J. Mol. Sci. 2015, 16, 25831–25864. [Google Scholar] [CrossRef]
- Babos, G.; Rydz, J.; Kawalec, M.; Klim, M.; Fodor-Kardos, A.; Trif, L.; Feczkó, T. Poly(3-hydroxybutyrate)-based nanoparticles for sorafenib and doxorubicin anticancer drug delivery. Int. J. Mol. Sci. 2020, 21, 7312. [Google Scholar] [CrossRef]
- Mahapatro, A.; Singh, D.K. Biodegradable nanoparticles are excellent vehicle for site directed in-vivo delivery of drugs and vaccines. J. Nanobiotechnol. 2011, 9, 55. [Google Scholar] [CrossRef]
- Gavasane, A.; Pawar, H. Synthetic Biodegradable polymers used in controlled drug delivery system: An overview. Clin. Pharmacol. Biopharm. 2014, 3, 121. [Google Scholar] [CrossRef]
- Tiwari, G.; Tiwari, R.; Sriwastawa, B.; Bhati, L.; Pandey, S.; Pandey, P.; Bannerjee, S.K. Drug delivery systems: An updated review. Int. J. Pharm. Investig. 2012, 2, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Doane, T.; Burda, C. Nanoparticle mediated non-covalent drug delivery. Adv. Drug Deliv. Rev. 2013, 65, 607–621. [Google Scholar] [CrossRef] [PubMed]
- Peer, D.; Karp, J.M.; Hong, S.; Farokhzad, O.C.; Margalit, R.; Langer, R. Nanocarriers as an emerging platform for cancer therapy. Nat. Nanotechnol. 2007, 2, 751–760. [Google Scholar] [CrossRef]
- Kasinski, A.; Zielinska-Pisklak, M.; Kowalczyk, S.; Plichta, A.; Zgadzaj, A.; Oledzka, E.; Sobczak, M. Synthesis and characterization of new biodegradable injectable thermosensitive smart hydrogels for 5-fluorouracil delivery. Int. J. Mol. Sci. 2021, 22, 8330. [Google Scholar] [CrossRef]
- Ding, C.; Li, Z. A review of drug release mechanisms from nanocarrier systems. Mater. Sci. Eng. C 2017, 76. [Google Scholar] [CrossRef]
- Wen, Y.; Wang, Y.Z.; Liu, X.L.; Zhang, W.; Xiong, X.H.; Han, Z.X.; Liang, X.J. Camptothecin-based nanodrug delivery systems. Cancer Biol. Med. 2017, 14, 363–370. [Google Scholar] [CrossRef]
- Hasan, A.S.; Socha, M.; Lamprecht, A.; El Ghazouani, F.; Sapin, A.; Hoffman, A.; Maincent, P.; Ubrich, N. Effect of the microencapsulation of nanoparticles on the reduction of burst release. Int. J. Pharm. 2007, 344, 53–61. [Google Scholar] [CrossRef]
- Yeo, Y.; Park, K.N. Control of encapsulation efficiency and initial burst in polymeric microparticle systems. Arch. Pharmacal Res. 2004, 27, 1–12. [Google Scholar] [CrossRef]
- Ahmed, A.; Sarwar, S.; Hu, Y.; Munir, M.U.; Nisar, M.F.; Ikram, F.; Asif, A.; Rahman, S.U.; Chaudhry, A.A.; Rehman, I.U. Surface-modified polymeric nanoparticles for drug delivery to cancer cells. Expert Opin. Drug Deliv. 2021, 18, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Caraglia, M.; Marra, M.; Misso, G.; Lamberti, M.; Salzano, G.; De Rosa, G.; Abbruzzese, A. Tumour-specific uptake of anti-cancer drugs: The future is here. Curr. Drug Metab. 2012, 13, 4–21. [Google Scholar] [CrossRef] [PubMed]
- Padhi, S.; Kapoor, R.; Verma, D.; Panda, A.K.; Iqbal, Z. Formulation and optimization of topotecan nanoparticles: In vitro characterization, cytotoxicity, cellular uptake and pharmacokinetic outcomes. J. Photochem. Photobiol. B-Biol. 2018, 183, 222–232. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Yang, Y.; Jia, Q.; Hao, Y.; Liu, J.; Huang, G. Preparation and evaluation of irinotecan poly(lactic-co-glycolic acid) nanoparticles for enhanced anti-tumor therapy. AAPS PharmSciTech 2019, 20, 133. [Google Scholar] [CrossRef] [PubMed]
- Tseng, Y.Y.; Yang, T.C.; Chen, S.M.; Yang, S.T.; Tang, Y.L.; Liu, S.J. Injectable SN-38-embedded polymeric microparticles promote antitumor efficacy against malignant glioma in an animal model. Pharmaceutics 2020, 12, 479. [Google Scholar] [CrossRef] [PubMed]
- Ci, T.Y.; Chen, L.; Yu, L.; Ding, J.D. Tumor regression achieved by encapsulating a moderately soluble drug into a polymeric thermogel. Sci. Rep. 2014, 4, 5473. [Google Scholar] [CrossRef]
- Derakhshandeh, K.; Erfan, M.; Dadashzadeh, S. Encapsulation of 9-nitrocamptothecin, a novel anticancer drug, in biodegradable nanoparticles: Factorial design, characterization and release kinetics. Eur. J. Pharm. Biopharm. 2007, 66, 34–41. [Google Scholar] [CrossRef]
- Dadashzadeh, S.; Derakhshandeh, K.; Shirazi, F.H. 9-nitrocamptothecin polymeric nanoparticles: Cytotoxicity and pharmacokinetic studies of lactone and total forms of drug in rats. Anticancer Drugs 2008, 19, 805–811. [Google Scholar] [CrossRef]
- Derakhshandeh, K.; Hochhaus, G.; Dadashzadeh, S. In-vitro cellular uptake and transport study of 9-nitrocamptothecin PLGA nanoparticles across Caco-2 cell monolayer model. Iran. J. Pharm. Res. 2011, 10, 425–434. [Google Scholar]
- Mohammady, H.; Dinarvand, R.; Manesh, M.E.; Ebrahimnejad, P. Encapsulation of irinotecan in polymeric nanoparticles: Characterization, release kinetic and cytotoxicity evaluation. Nanomed. J. 2016, 3, 159–168. [Google Scholar] [CrossRef]
- Mahmoud, B.S.; McConville, C. Development and Optimization of irinotecan-loaded PCL nanoparticles and their cytotoxicity against primary high-grade glioma cells. Pharmaceutics 2021, 13, 541. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimnejad, P.; Dinarvand, R.; Sajadi, A.; Jaafari, M.R.; Nomani, A.R.; Azizi, E.; Rad-Malekshahi, M.; Atyabi, F. Preparation and in vitro evaluation of actively targetable nanoparticles for SN-38 delivery against HT-29 cell lines. Nanomedicine 2010, 6, 478–485. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Luo, H.Y.; Cao, Z.; Chen, Y.; Gao, J.B.; Li, Y.Q.; Jiang, Q.; Xu, R.H.; Liu, J. Dual-targeting hybrid nanoparticles for the delivery of SN38 to Her2 and CD44 overexpressed human gastric cancer. Nanoscale 2016, 8, 11543–11558. [Google Scholar] [CrossRef]
- Dimchevska, S.; Geskovski, N.; Koliqi, R.; Matevska-Geskovska, N.; Gomez Vallejo, V.; Szczupak, B.; Sebastian, E.S.; Llop, J.; Hristov, D.R.; Monopoli, M.P.; et al. Efficacy assessment of self-assembled PLGA-PEG-PLGA nanoparticles: Correlation of nano-bio interface interactions, biodistribution, internalization and gene expression studies. Int. J. Pharm. 2017, 533, 389–401. [Google Scholar] [CrossRef]
- Bao, H.; Zhang, Q.; Yan, Z. The impact of camptothecin-encapsulated poly(lactic-co-glycolic acid) nanoparticles on the activity of cytochrome P450 in vitro. Int. J. Nanomed. 2019, 14, 383–391. [Google Scholar] [CrossRef] [PubMed]
- Song, L.J.; Zeng, A.Q.; Hu, M.X.; Lin, Y.C.; Shu, Y.Q.; Huang, X.Z.; Gong, C.Y.; Xie, Y.M.; Wu, Q.J. Biodegradable polymeric micelle-mediated delivery of a pH-activatable prodrug of 7-ethyl-10-hydroxy-camptothecin (SN-38) to enhance anti-angiogenesis and anti-tumor activity. J. Biomed. Nanotechnol. 2018, 14, 267–280. [Google Scholar] [CrossRef]
- Nishino, S.; Kishida, A.; Yoshizawa, H. Morphology control of polylactide microspheres enclosing irinotecan hydrochloride with polylactide based polymer surfactant for reduction of initial burst. Int. J. Pharm. 2007, 330, 32–36. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Hu, B.R.; Xu, J.; Wu, J.Z.; Ye, B.L. Preparation of biodegradable polymeric nanocapsules for treatment of malignant tumor using coaxial capillary microfluidic device. Cancer Biother. Radiopharm. 2020, 35, 570–580. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Chang, G.T.; Zhang, H.; Ding, J.D. Injectable block copolymer hydrogels for sustained release of a PEGylated drug. Int. J. Pharm. 2008, 348, 95–106. [Google Scholar] [CrossRef]
- Lee, W.C.; Li, Y.C.; Chu, I.M. Amphiphilic poly(D,L-lactic acid)/poly(ethylene glycol)/poly(D,L-lactic acid) nanogels for controlled release of hydrophobic drugs. Macromol. Biosci. 2006, 6, 846–854. [Google Scholar] [CrossRef]
- Manandhar, S.; Sjöholm, E.; Bobacka, J.; Rosenholm, J.M.; Bansal, K.K. Polymer-drug conjugates as nanotheranostic agents. J. Nanotheranostics 2021, 2, 63–81. [Google Scholar] [CrossRef]
- Marasini, N.; Haque, S.; Kaminskas, L.M. Polymer-drug conjugates as inhalable drug delivery systems: A review. Curr. Opin. Colloid Interface Sci. 2017, 31, 18–29. [Google Scholar] [CrossRef]
- Muluneh, F.S.; Nath, L.K. Polymer-drug conjugates: Novel carriers for cancer chemotherapy. Polym. Plast. Technol. Eng. 2018, 58, 158–171. [Google Scholar] [CrossRef]
- Elvira, C.; Gallardo, A.; San Roman, J.; Cifuentes, A. Covalent polymer-drug conjugates. Molecules 2005, 10, 114. [Google Scholar] [CrossRef]
- Mulas, K.; Stefanowicz, Z.; Oledzka, E.; Sobczak, M. Current state of the polymeric delivery systems of fluoroquinolones—A review. J. Control Release 2019, 294, 195–215. [Google Scholar] [CrossRef]
- Oledzka, E.; Horeglad, P.; Gruszczyńska, Z.; Plichta, A.; Nałęcz-Jawecki, G.; Sobczak, M. Polylactide conjugates of camptothecin with different drug release abilities. Molecules 2014, 19, 19460. [Google Scholar] [CrossRef]
- Du, X.; Sun, Y.; Zhang, M.; He, J.; Ni, P. Polyphosphoester-camptothecin prodrug with reduction-response prepared via Michael addition polymerization and click reaction. ACS Appl. Mater. Interfaces 2017, 9, 13939–13949. [Google Scholar] [CrossRef]
- Zhao, H.; Rubio, B.; Sapra, P.; Wu, D.C.; Reddy, P.; Sai, P.; Martinez, A.; Gao, Y.; Lozanguiez, Y.; Longley, C.; et al. Novel prodrugs of SN38 using multiarm poly(ethylene glycol) linkers. Bioconjugate Chem. 2008, 19, 849–859. [Google Scholar] [CrossRef]
- Lu, L.; Zheng, Y.; Weng, S.Q.; Zhu, W.W.; Chen, J.H.; Zhang, X.M.; Lee, R.J.; Bo, Y.; Jia, H.L.; Qin, L.X. Complete regression of xenograft tumors using biodegradable mPEG-PLA-SN38 block copolymer micelles. Colloids Surf. B Biointerfaces 2016, 142, 417–423. [Google Scholar] [CrossRef]
- Fleming, A.B.; Haverstick, K.; Saltzman, W.M. In vitro cytotoxicity and in vivo distribution after direct delivery of PEG-camptothecin conjugates to the rat brain. Bioconjugate Chem. 2004, 15, 1364–1375. [Google Scholar] [CrossRef]
- Li, B.; Zhao, W.Y.; Zhang, X.F.; Wang, J.F.; Luo, X.; Baker, S.D.; Jordan, C.T.; Dong, Y.Z. Design, synthesis and evaluation of anti-CD123 antibody drug conjugates. Bioorganic Med. Chem. 2016, 24, 5855–5860. [Google Scholar] [CrossRef] [PubMed]







| Drug | Solubility in Water | MTD (mg/m2/d) | Clinical Trial Status | Reference | |
|---|---|---|---|---|---|
| CPT | 2.9 mM | 0.046 | - | - | [29] |
| Topotecan | 100 mM | 0.1 | 1.5 | Approved | [29,40] |
| Irinotecan (CPT-11) | 25 mM | 1.14 | 290–320 | Approved | [40,41] |
| Rubitecan (9-CPT) | 239 g/mL | 0.085 | 1.5 | Phase III | [29,42,43] |
| Belotecan (CKD-602) | 77.9 g/mL | 0.094 | 0.5 | Phase II | [43,44,45] |
| Lurtotecan (GG-211) | 713 g/mL | 0.006 | 1.2 | Phase II | [43,46,47] |
| Exatecan (DX-8951f) | 221 g/mL | 0.008 | 0.3 for heavily pretreated patients 0.5 for minimally pretreated patients | Phase III | [43,46,47] |
| SN-38 | 11–38 g/mL | 0.09 | - | - | [42,48,49,50] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Strzelecka, K.; Piotrowska, U.; Sobczak, M.; Oledzka, E. The Advancement of Biodegradable Polyesters as Delivery Systems for Camptothecin and Its Analogues—A Status Report. Int. J. Mol. Sci. 2023, 24, 1053. https://doi.org/10.3390/ijms24021053
Strzelecka K, Piotrowska U, Sobczak M, Oledzka E. The Advancement of Biodegradable Polyesters as Delivery Systems for Camptothecin and Its Analogues—A Status Report. International Journal of Molecular Sciences. 2023; 24(2):1053. https://doi.org/10.3390/ijms24021053
Chicago/Turabian StyleStrzelecka, Katarzyna, Urszula Piotrowska, Marcin Sobczak, and Ewa Oledzka. 2023. "The Advancement of Biodegradable Polyesters as Delivery Systems for Camptothecin and Its Analogues—A Status Report" International Journal of Molecular Sciences 24, no. 2: 1053. https://doi.org/10.3390/ijms24021053
APA StyleStrzelecka, K., Piotrowska, U., Sobczak, M., & Oledzka, E. (2023). The Advancement of Biodegradable Polyesters as Delivery Systems for Camptothecin and Its Analogues—A Status Report. International Journal of Molecular Sciences, 24(2), 1053. https://doi.org/10.3390/ijms24021053