

Development of a Cytotoxic Antibody–Drug Conjugate Targeting Membrane Immunoglobulin E-Positive Cells
 ,
,  and
and
Abstract
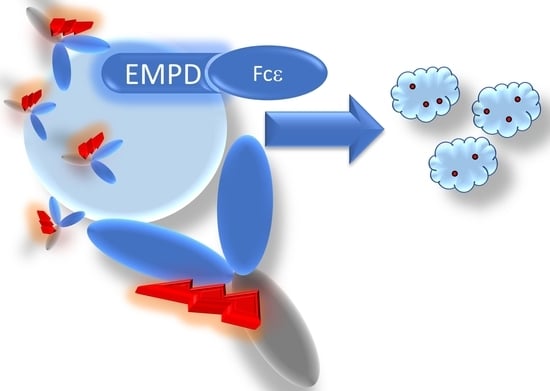
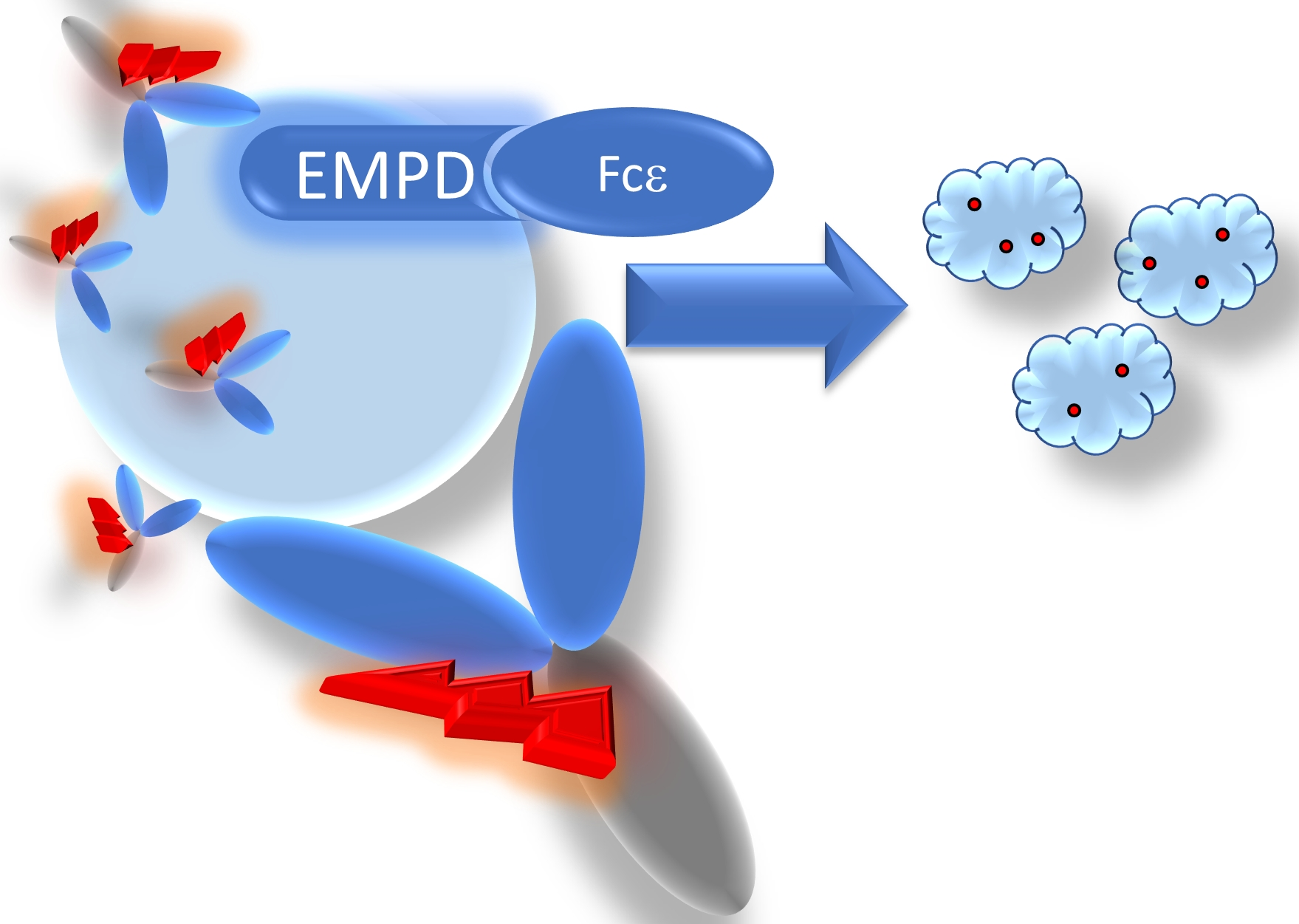{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
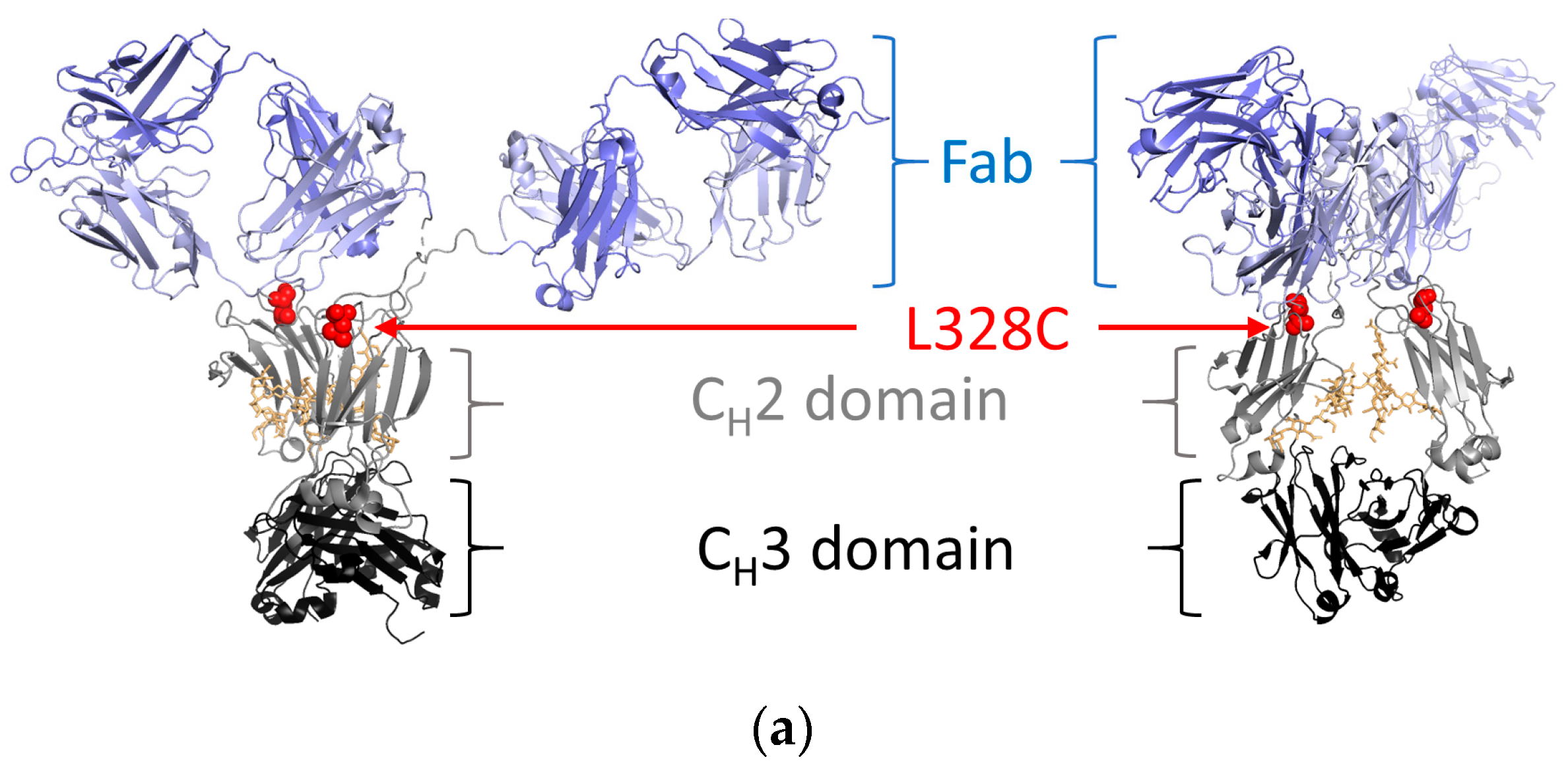
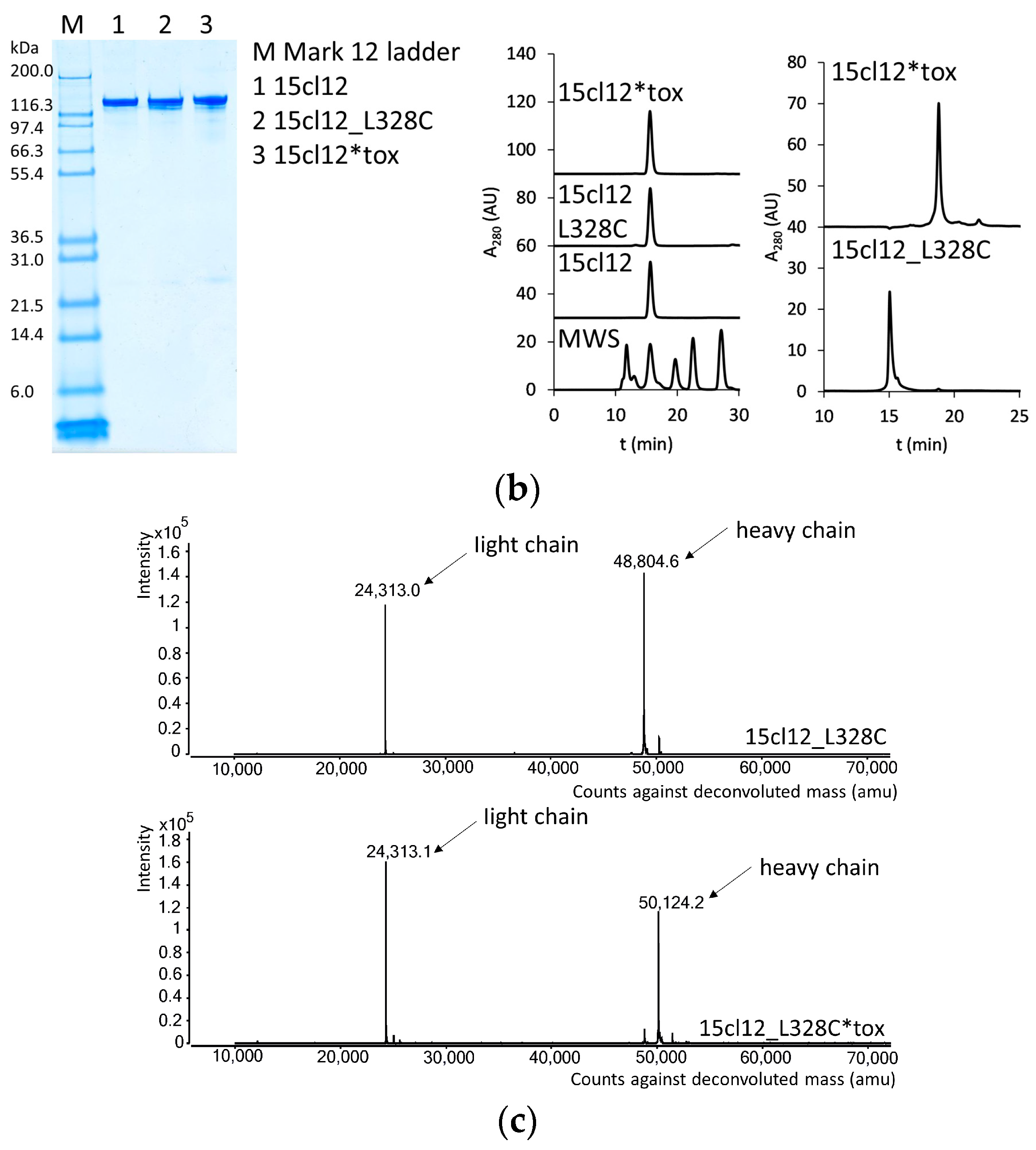
2.1. Antibody Production and Characterization
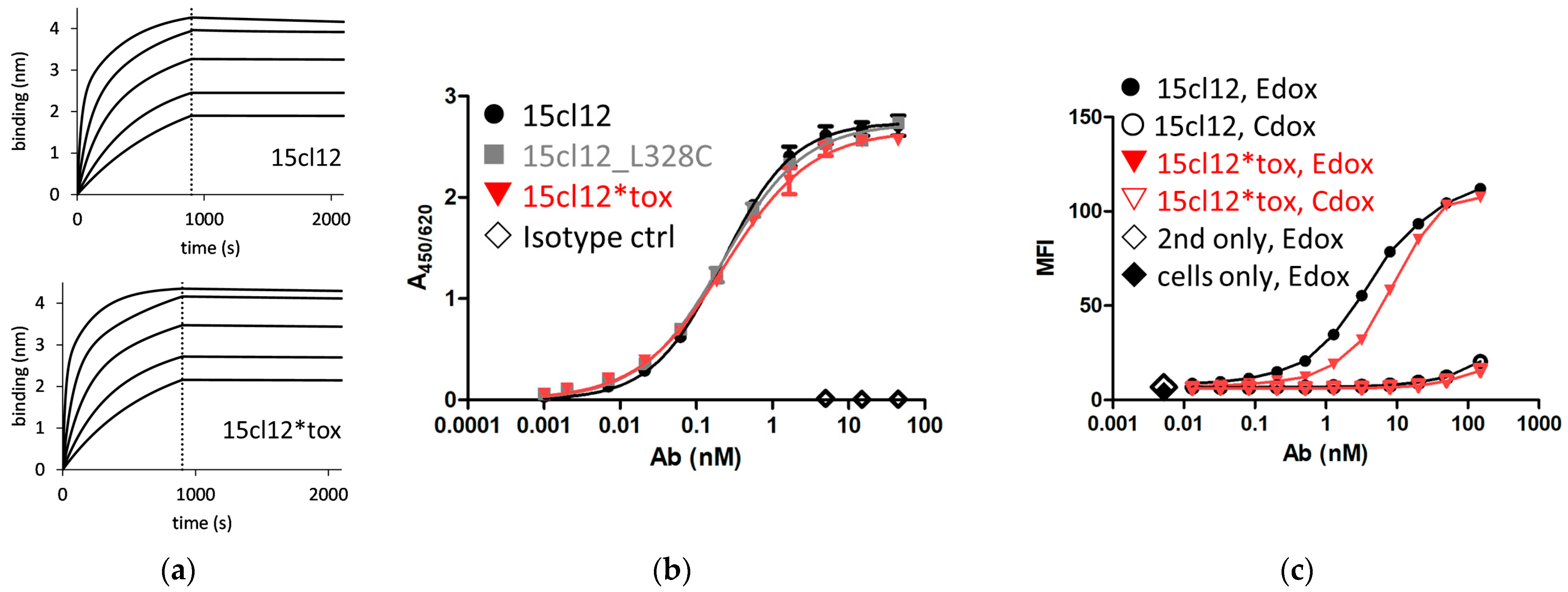
2.2. Binding Properties of 15cl12 and the ADC Derivate
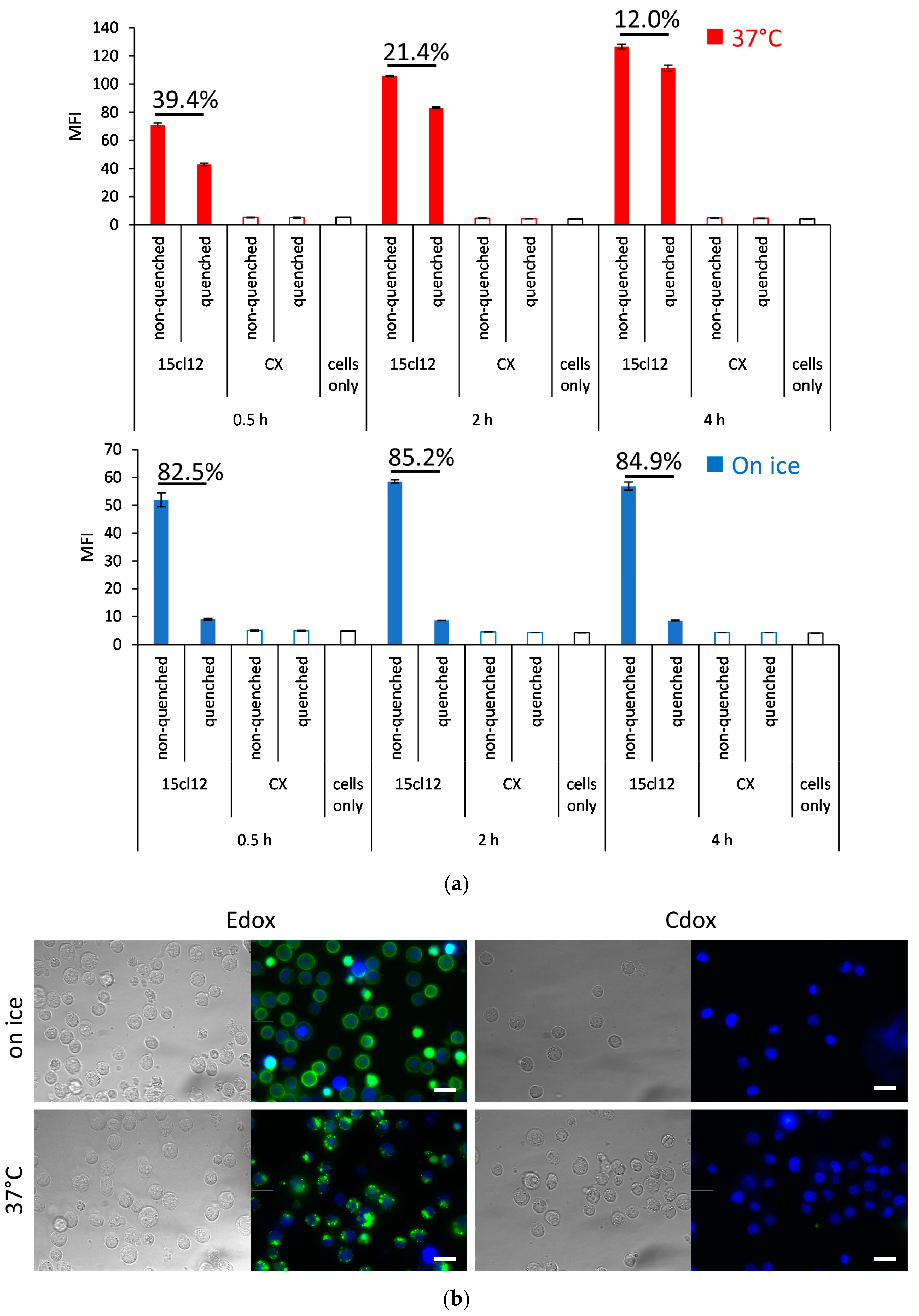
2.3. Internalization Experiments
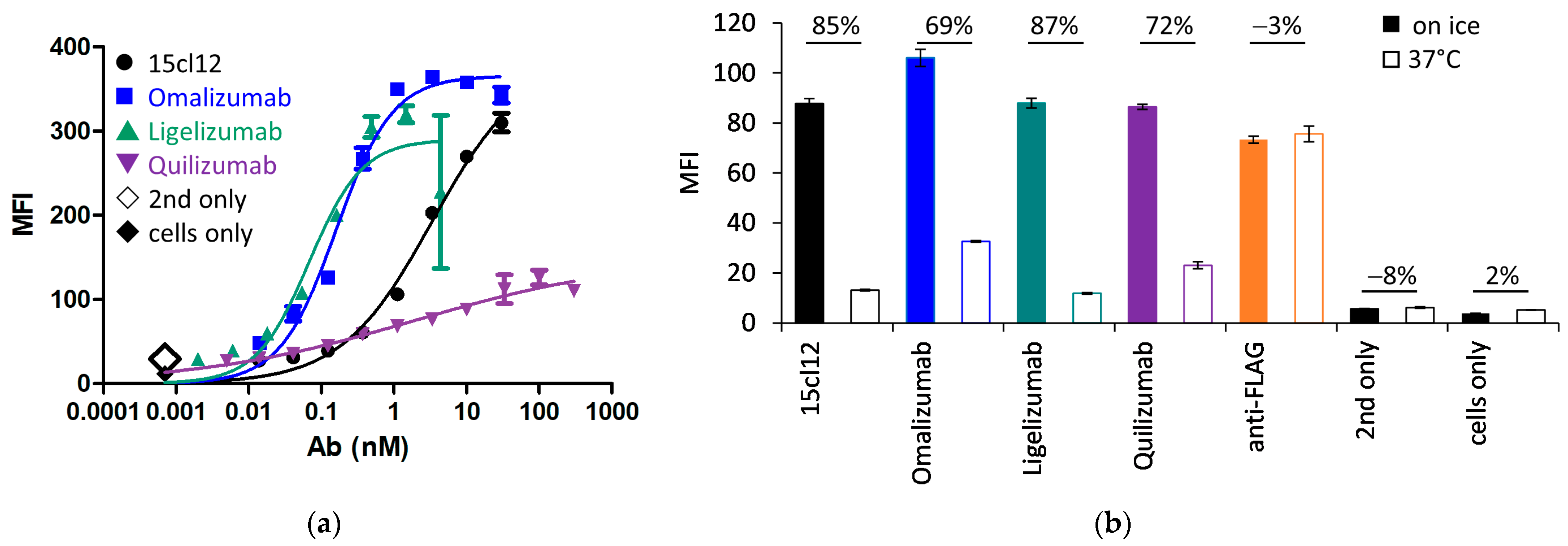
2.4. Comparison of Binding and Internalization Properties with Other Anti-IgE Antibodies
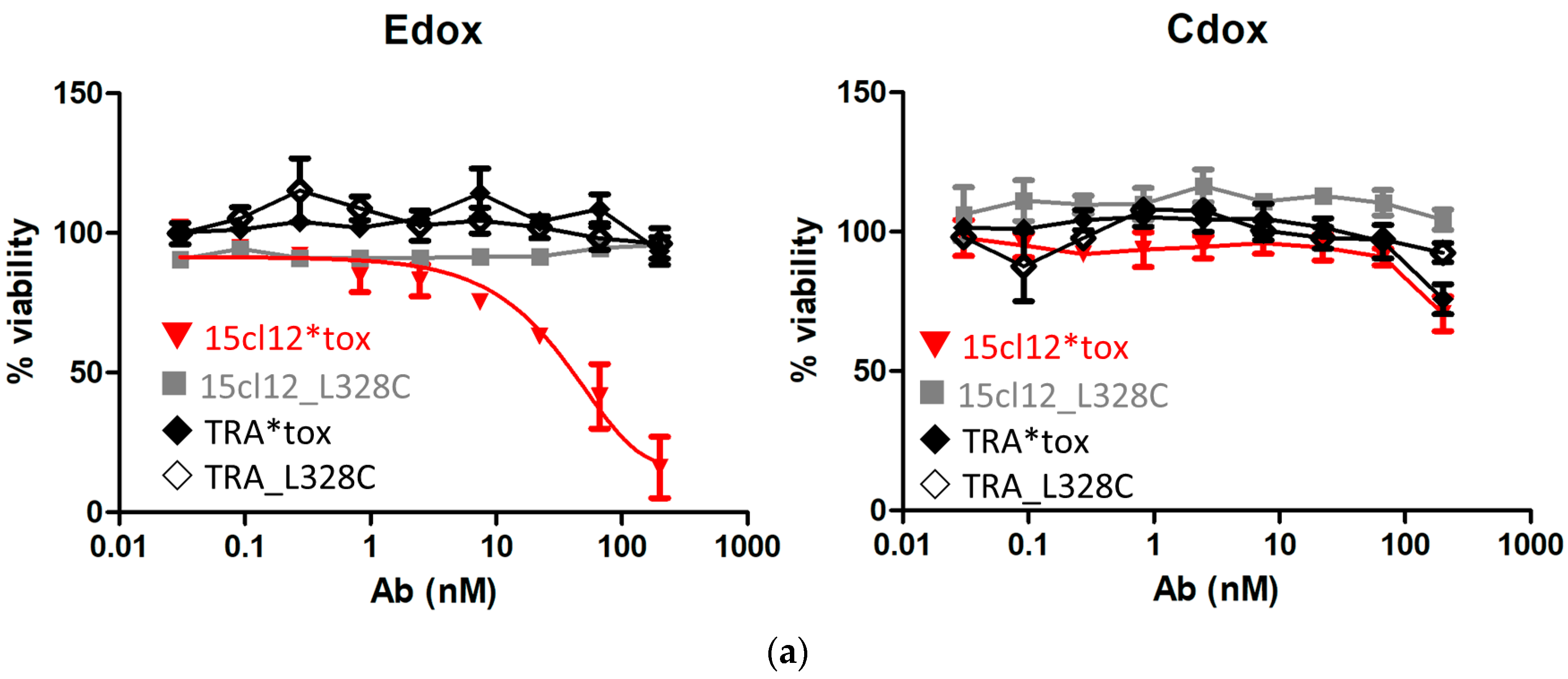
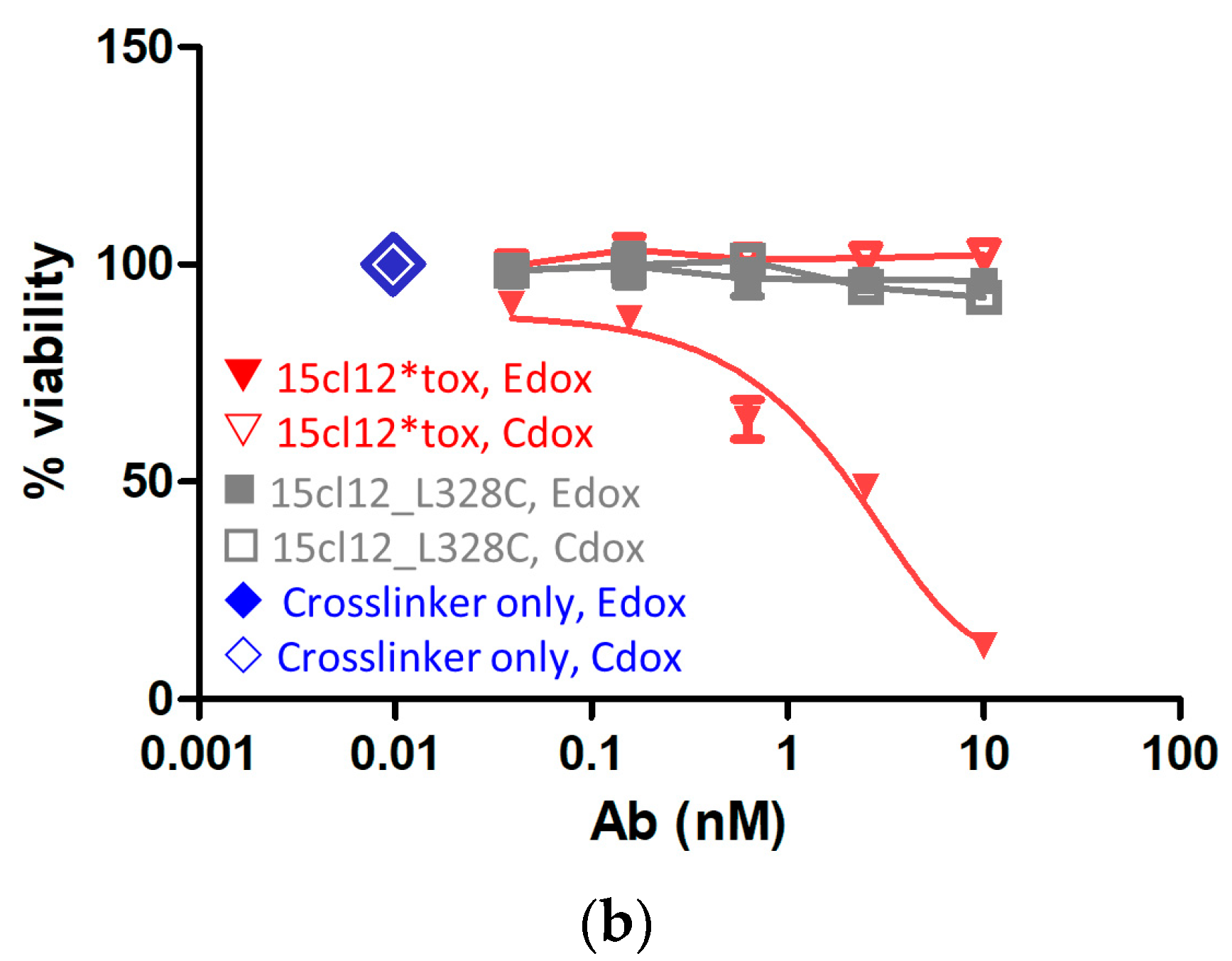
2.5. Specific Cytotoxic Activity of the ADC
3. Discussion
4. Materials and Methods
4.1. Protein Expression, Purification, and Preparation of Toxin Conjugates
4.2. Protein Characterization
4.2.1. SDS-PAGE
4.2.2. SEC-HPLC
4.2.3. HIC
4.2.4. Mass Spectrometry Analysis
4.3. Target Antigen Binding
4.3.1. Biolayer Interferometry
4.3.2. ELISA
4.3.3. Cell Culture
4.3.4. Cell Staining
4.3.5. Competition between EMPD-Binding Antibodies 15cl12 and Quilizumab
4.4. Internalization Assays
4.4.1. Quenching of Surface Fluorescence
4.4.2. Microscopy
4.4.3. Internalization of Fcε
4.5. Cytotoxicity Experiments
4.5.1. Direct Specific Cytotoxic Activity of the ADC
4.5.2. Cytotoxicity after Cross-Linking of Anti-IgE Antibodies
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Holgate, S.T. The epidemic of allergy and asthma. Nature 1999, 402 (Suppl. 6760), 2–4. [Google Scholar] [CrossRef] [PubMed]
- Gould, H.J.; Sutton, B.J. IgE in allergy and asthma today. Nat. Rev. Immunol. 2008, 8, 205–217. [Google Scholar] [CrossRef]
- Gasser, P.; Tarchevskaya, S.S.; Guntern, P.; Brigger, D.; Ruppli, R.; Zbären, N.; Kleinboelting, S.; Heusser, C.; Jardetzky, T.S.; Eggel, A. The mechanistic and functional profile of the therapeutic anti-IgE antibody ligelizumab differs from omalizumab. Nat. Commun. 2020, 11, 165. [Google Scholar] [CrossRef] [PubMed]
- Guntern, P.; Eggel, A. Past, present and future of anti-IgE biologicals. Allergy 2020, 75, 2491. [Google Scholar] [CrossRef]
- Fahy, J.V. Reducing IgE levels as a strategy for the treatment of asthma. Clin. Exp. Allergy 2003, 30, 16–21. [Google Scholar] [CrossRef]
- Wade-Vallance, A.K.; Yang, Z.; Libang, J.B.; Robinson, M.J.; Tarlinton, D.M.; Allen, C.D.C. B cell receptor ligation induces IgE plasma cell elimination. J. Exp. Med. 2023, 220, e20220964. [Google Scholar] [CrossRef]
- Chen, J.-B.; Wu, P.C.; Hung, A.F.-H.; Chu, C.-Y.; Tsai, T.-F.; Yu, H.-M.; Chang, H.-Y.; Chang, T.W. Unique Epitopes on CεmX in IgE–B Cell Receptors Are Potentially Applicable for Targeting IgE-Committed B Cells. J. Immunol. 2010, 184, 1748–1756. [Google Scholar] [CrossRef]
- Chowdhury, P.S.; Chen, Y.; Yang, C.; Cook, K.E.; Nyborg, A.C.; Ettinger, R.; Herbst, R.; Kiener, P.A.; Wu, H. Targeting the junction of CɛmX and ɛ-migis for the specific depletion of mIgE-expressing B cells. Mol. Immunol. 2012, 52, 279–288. [Google Scholar] [CrossRef]
- Gauvreau, G.M.; Arm, J.P.; Boulet, L.P.; Leigh, R.; Cockcroft, D.W.; Davis, B.E.; Mayers, I.; FitzGerald, J.M.; Dahlen, B.; Killian, K.J.; et al. Efficacy and safety of multiple doses of QGE031 (ligelizumab) versus omalizumab and placebo in inhibiting allergen-induced early asthmatic responses. J. Allergy Clin. Immunol. 2016, 138, 1051–1059. [Google Scholar] [CrossRef]
- Goodnow, C.C.; Cyster, J.G.; Hartley, S.B.; Bell, S.E.; Cooke, M.P.; Healy, J.I.; Akkaraju, S.; Rathmell, J.C.; Pogue, S.L.; Shokat, K.P. Self-tolerance checkpoints in B lymphocyte development. Adv. Immunol. 1995, 59, 279–368. [Google Scholar] [CrossRef]
- Nemazee, D.; Buerki, K. Clonal deletion of autoreactive B lymphocytes in bone marrow chimeras. Proc. Natl. Acad. Sci. USA 1989, 86, 8039–8043. [Google Scholar] [CrossRef] [PubMed]
- Brightbill, H.D.; Jeet, S.; Lin, Z.; Yan, D.; Zhou, M.; Tan, M.; Nguyen, A.; Yeh, S.; Delarosa, D.; Leong, S.R.; et al. Antibodies specific for a segment of human membrane IgE deplete IgE-producing B cells in humanized mice. J. Clin. Investig. 2010, 120, 2218–2229. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.J.; Chen, N.Y.; Chen, J.B.; Lu, C.S.; Hung, A.F.H.; Shiung, Y.Y.; Wu, P.C.; Pan, R.L.; Chang, T.W. Ce{open}mX peptide-carrying HBcAg virus-like particles induced antibodies that down-regulate mIgE-B lymphocytes. Mol. Immunol. 2012, 52, 190–199. [Google Scholar] [CrossRef]
- Brightbill, H.; Lin, Y.; Lin, Z.; Tan, M.; Meng, Y.; Balazs, M.; Chung, S.; Wu, L. Quilizumab is an Afucosylated Humanized Anti-M1 Prime Therapeutic Antibody. Clin. Anti-inflamm. Anti-Allergy Drugs 2013, 1, 24–31. [Google Scholar] [CrossRef]
- Harris, J.M.; Maciuca, R.; Bradley, M.S.; Cabanski, C.R.; Scheerens, H.; Lim, J.; Cai, F.; Kishnani, M.; Liao, X.C.; Samineni, D.; et al. A randomized trial of the efficacy and safety of quilizumab in adults with inadequately controlled allergic asthma. Respir. Res. 2016, 17, 29. [Google Scholar] [CrossRef] [PubMed]
- Kirak, O.; Riethmüller, G. A novel, nonanaphylactogenic, bispecific IgE-CD3 antibody eliminates IgE+ B cells. J. Allergy Clin. Immunol. 2015, 136, 800–802.e3. [Google Scholar] [CrossRef]
- Rodak, A.; Stadlmayr, G.; Stadlbauer, K.; Lichtscheidl, D.; Bobbili, M.R.; Rüker, F.; Wozniak-Knopp, G. Bispecific t-cell engagers targeting membrane-bound ige. Biomedicines 2021, 9, 1568. [Google Scholar] [CrossRef]
- Chu, S.Y.; Horton, H.M.; Pong, E.; Leung, I.W.L.; Chen, H.; Nguyen, D.H.; Bautista, C.; Muchhal, U.S.; Bernett, M.J.; Moore, G.L.; et al. Reduction of total IgE by targeted coengagement of IgE B-cell receptor and FcγRIIb with Fc-engineered antibody. J. Allergy Clin. Immunol. 2012, 129, 1102–1115. [Google Scholar] [CrossRef]
- Macro, M.; André, I.; Comby, E.; Chèze, S.; Chapon, F.; Ballet, J.J.; Reman, O.; Leporrier, M.; Troussard, X. IgE multiple myeloma. Leuk. Lymphoma 1999, 32, 597–603. [Google Scholar] [CrossRef]
- Hejl, C.; Mestiri, R.; Carmoi, T.; Bugier, S.; Chianea, D.; Renard, C.; Vest, P. IgE monoclonal gammopathy: A case report and literature review. Clin. Biochem. 2018, 51, 103–109. [Google Scholar] [CrossRef]
- Nakahara, W.; Ogawa, T.; Matsunaga, H.; Iwasa, Y.; Horita, M.; Ikeda, M.; Asako, M.; Iio, S.; Iwama, Y.; Oka, K.; et al. IgE Plasma Cell Leukemia Harboring t(11;14) and 1q Amplification. Case Rep. Hematol. 2023, 2023, 4747989. [Google Scholar] [CrossRef] [PubMed]
- Johansson, S.G.O.; Bennich, H. Immunological studies of an atypical (myeloma) immunoglobulin. Immunology 1967, 13, 381–394. [Google Scholar]
- West, N.C.; Smith, A.M.; Ward, R. IgE myeloma associated with plasma cell leukaemia. Postgrad. Med. J. 1983, 59, 784–785. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Avet-Loiseau, H.; Garand, R.; Lodé, L.; Harousseau, J.L.; Bataille, R. Translocation t(11;14)(q13;q32) is the hallmark of IgM, IgE, and nonsecretory multiple myeloma variants. Blood 2003, 101, 1570–1571. [Google Scholar] [CrossRef] [PubMed]
- Kehl, N.; Kilian, M.; Michel, J.; Wagner, T.R.; Uhrig, S.; Brobeil, A.; Sester, L.S.; Blobner, S.; Steiger, S.; Hundemer, M.; et al. IgE type multiple myeloma exhibits hypermutated phenotype and tumor reactive T cells. J. Immunother. Cancer 2022, 10, e005815. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Dees, S.; Grewal, I.S. Overcoming the challenges associated with CD3+ T-cell redirection in cancer. Br. J. Cancer 2021, 124, 1037–1048. [Google Scholar] [CrossRef]
- Cosenza, M.; Sacchi, S.; Pozzi, S. Cytokine Release Syndrome Associated with T-Cell-Based Therapies for Hematological Malignancies: Pathophysiology, Clinical Presentation, and Treatment. Int. J. Mol. Sci. 2021, 22, 7652. [Google Scholar] [CrossRef]
- Fu, Z.; Li, S.; Han, S.; Shi, C.; Zhang, Y. Antibody drug conjugate: The “biological missile” for targeted cancer therapy. Signal Transduct. Target. Ther. 2022, 7, 93. [Google Scholar] [CrossRef]
- Kaplon, H.; Chenoweth, A.; Crescioli, S.; Reichert, J.M. Antibodies to watch in 2022. MAbs 2022, 14, 2014296. [Google Scholar] [CrossRef]
- Kaplon, H.; Crescioli, S.; Chenoweth, A.; Visweswaraiah, J.; Reichert, J.M. Antibodies to watch in 2023. MAbs 2023, 15, 2153410. [Google Scholar] [CrossRef]
- Vigl, B.; Salhat, N.; Parth, M.; Pankevych, H.; Mairhofer, A.; Bartl, S.; Smrzka, O.W. Quantitative in vitro and in vivo models to assess human IgE B cell receptor crosslinking by IgE and EMPD IgE targeting antibodies. J. Immunol. Methods 2017, 449, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Fager Ferrari, M.; Lemonakis, K.; Förnvik Jonsson, M. A rare case of IgE kappa monoclonal gammopathy of undetermined significance identified in a Swedish female. Scand. J. Clin. Lab. Investig. 2021, 81, 385–388. [Google Scholar] [CrossRef]
- Nafría Jiménez, B.; Oliveros Conejero, R. IgE multiple myeloma: Detection and follow-up. Adv. Lab. Med. 2022, 3, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Hamblett, K.J.; Senter, P.D.; Chace, D.F.; Sun, M.M.C.; Lenox, J.; Cerveny, C.G.; Kissler, K.M.; Bernhardt, S.X.; Kopcha, A.K.; Zabinski, R.F.; et al. Effects of drug loading on the antitumor activity of a monoclonal antibody drug conjugate. Clin. Cancer Res. 2004, 10, 7063–7070. [Google Scholar] [CrossRef] [PubMed]
- Junutula, J.R.; Raab, H.; Clark, S.; Bhakta, S.; Leipold, D.D.; Weir, S.; Chen, Y.; Simpson, M.; Tsai, S.P.; Dennis, M.S.; et al. Site-specific conjugation of a cytotoxic drug to an antibody improves the therapeutic index. Nat. Biotechnol. 2008, 26, 925–932. [Google Scholar] [CrossRef] [PubMed]
- Edelman, G.M.; Cunningham, B.A.; Gall, W.E.; Gottlieb, P.D.; Rutishauser, U.; Waxdal, M.J. The covalent structure of an entire gammaG immunoglobulin molecule. Proc. Natl. Acad. Sci. USA 1969, 63, 78–85. [Google Scholar] [CrossRef]
- Stadlmayr, G.; Stracke, F.; Stadlbauer, K.; Rybka, J.; Dickgiesser, S.; Rasche, N.; Becker, S.; Toleikis, L.; Rüker, F.; Knopp, G.W. Efficient spontaneous site-selective cysteine-mediated toxin attachment within a structural loop of antibodies. Biochim. Biophys. Acta Gen. Subj. 2022, 1866, 130155. [Google Scholar] [CrossRef]
- Kaempffe, A.; Dickgiesser, S.; Rasche, N.; Paoletti, A.; Bertotti, E.; De Salve, I.; Sirtori, F.R.; Kellner, R.; Könning, D.; Hecht, S.; et al. Effect of Conjugation Site and Technique on the Stability and Pharmacokinetics of Antibody-Drug Conjugates. J. Pharm. Sci. 2021, 110, 3776–3785. [Google Scholar] [CrossRef]
- Dickgiesser, S.; Kellner, R.; Kolmar, H.; Rasche, N. Site-Specific Conjugation of Thiol-Reactive Cytotoxic Agents to Nonnative Cysteines of Engineered Monoclonal Antibodies. Methods Mol. Biol. 2019, 2033, 1–14. [Google Scholar]
- Kuo, B.S.; Li, C.H.; Chen, J.B.; Shiung, Y.Y.; Chu, C.Y.; Lee, C.H.; Liu, Y.J.; Kuo, J.H.; Hsu, C.; Su, H.W.; et al. IgE-neutralizing UB-221 mAb, distinct from omalizumab and ligelizumab, exhibits CD23-mediated IgE downregulation and relieves urticaria symptoms. J. Clin. Investig. 2022, 132, 1–16. [Google Scholar] [CrossRef]
- Xiao, D.; Luo, L.; Li, J.; Wang, Z.; Liu, L.; Xie, F.; Feng, J.; Zhou, X. Development of bifunctional anti-PD-L1 antibody MMAE conjugate with cytotoxicity and immunostimulation. Bioorg. Chem. 2021, 116, 105366. [Google Scholar] [CrossRef]
- Wang, Z.; Li, H.; Gou, L.; Li, W.; Wang, Y. Antibody–drug conjugates: Recent advances in payloads. Acta Pharm. Sin. B 2023, 13, 4025–4059. [Google Scholar] [CrossRef] [PubMed]
- Collins, D.M.; Bossenmaier, B.; Kollmorgen, G.; Niederfellner, G. Acquired resistance to antibody-drug conjugates. Cancers 2019, 11, 394. [Google Scholar] [CrossRef] [PubMed]
- Bergler, W.; Petroiami, G.; Schadel, A. Feasibility of Proliferation Studies Using the BrdU and MTT Assays with a Head and Neck Carcinoma Cell Line. ORL 1993, 55, 230–235. [Google Scholar] [CrossRef] [PubMed]
- Stengl, A.; Hörl, D.; Leonhardt, H.; Helma, J. A simple and sensitive high-content Assay for the characterization of antiproliferative therapeutic antibodies. SLAS Discov. 2017, 22, 309–315. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hartley, J.A.; Flynn, M.J.; Bingham, J.P.; Corbett, S.; Reinert, H.; Tiberghien, A.; Masterson, L.A.; Antonow, D.; Adams, L.; Chowdhury, S.; et al. Pre-clinical pharmacology and mechanism of action of SG3199, the pyrrolobenzodiazepine (PBD) dimer warhead component of antibody-drug conjugate (ADC) payload tesirine. Sci. Rep. 2018, 8, 10479. [Google Scholar] [CrossRef]
- Lian, X.; Kats, D.; Rasmussen, S.; Martin, L.R.; Karki, A.; Keller, C.; Berlow, N.E. Design considerations of an IL13Rα2 antibody–drug conjugate for diffuse intrinsic pontine glioma. Acta Neuropathol. Commun. 2021, 9, 88. [Google Scholar] [CrossRef]
- Perrino, E.; Steiner, M.; Krall, N.; Bernardes, G.J.L.; Pretto, F.; Casi, G.; Neri, D. Curative properties of noninternalizing antibody-drug conjugates based on maytansinoids. Cancer Res. 2014, 74, 2569–2578. [Google Scholar] [CrossRef]
- Ritchie, M.; Tchistiakova, L.; Scott, N. Implications of receptor-mediated endocytosis and intracellular trafficking dynamics in the development of antibody drug conjugates. MAbs 2013, 5, 13–21. [Google Scholar] [CrossRef]
- Du, X.; Beers, R.; FitzGerald, D.J.; Pastan, I. Differential cellular internalization of anti-CD19 and -CD22 immunotoxins results in different cytotoxic activity. Cancer Res. 2008, 68, 6300–6305. [Google Scholar] [CrossRef]
- Goldenberg, D.M.; Sharkey, R.M. Antibody-drug conjugates targeting TROP-2 and incorporating SN-38: A case study of anti-TROP-2 sacituzumab govitecan. MAbs 2019, 11, 987–995. [Google Scholar] [CrossRef] [PubMed]
- Richman, L.P.; Vonderheide, R.H. Role of crosslinking for agonistic CD40 monoclonal antibodies as immune therapy of cancer. Cancer Immunol. Res. 2014, 2, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Eggel, A.; Baravalle, G.; Hobi, G.; Kim, B.; Buschor, P.; Forrer, P.; Shin, J.S.; Vogel, M.; Stadler, B.M.; Dahinden, C.A.; et al. Accelerated dissociation of IgE-FcεRI complexes by disruptive inhibitors actively desensitizes allergic effector cells. J. Allergy Clin. Immunol. 2014, 133, 1709–1719.e8. [Google Scholar] [CrossRef] [PubMed]
- Jabs, F.; Plum, M.; Laursen, N.S.; Jensen, R.K.; Mølgaard, B.; Miehe, M.; Mandolesi, M.; Rauber, M.M.; Pfützner, W.; Jakob, T.; et al. Trapping IgE in a closed conformation by mimicking CD23 binding prevents and disrupts FcϵRI interaction. Nat. Commun. 2018, 9, 7. [Google Scholar] [CrossRef] [PubMed]
- Bailly, C.; Bodet-Milin, C.; Rousseau, C.; Faivre-Chauvet, A.; Kraeber-Bodéré, F.; Barbet, J. Pretargeting for imaging and therapy in oncological nuclear medicine. EJNMMI Radiopharm. Chem. 2017, 2, 6. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodak, A.; Stadlbauer, K.; Bobbili, M.R.; Smrzka, O.; Rüker, F.; Wozniak Knopp, G. Development of a Cytotoxic Antibody–Drug Conjugate Targeting Membrane Immunoglobulin E-Positive Cells. Int. J. Mol. Sci. 2023, 24, 14997. https://doi.org/10.3390/ijms241914997
Rodak A, Stadlbauer K, Bobbili MR, Smrzka O, Rüker F, Wozniak Knopp G. Development of a Cytotoxic Antibody–Drug Conjugate Targeting Membrane Immunoglobulin E-Positive Cells. International Journal of Molecular Sciences. 2023; 24(19):14997. https://doi.org/10.3390/ijms241914997
Chicago/Turabian StyleRodak, Aleksandra, Katharina Stadlbauer, Madhusudhan Reddy Bobbili, Oskar Smrzka, Florian Rüker, and Gordana Wozniak Knopp. 2023. "Development of a Cytotoxic Antibody–Drug Conjugate Targeting Membrane Immunoglobulin E-Positive Cells" International Journal of Molecular Sciences 24, no. 19: 14997. https://doi.org/10.3390/ijms241914997
APA StyleRodak, A., Stadlbauer, K., Bobbili, M. R., Smrzka, O., Rüker, F., & Wozniak Knopp, G. (2023). Development of a Cytotoxic Antibody–Drug Conjugate Targeting Membrane Immunoglobulin E-Positive Cells. International Journal of Molecular Sciences, 24(19), 14997. https://doi.org/10.3390/ijms241914997