

Unveiling Novel Avenues in mTOR-Targeted Therapeutics: Advancements in Glioblastoma Treatment


 and
and
Abstract
:1. Introduction
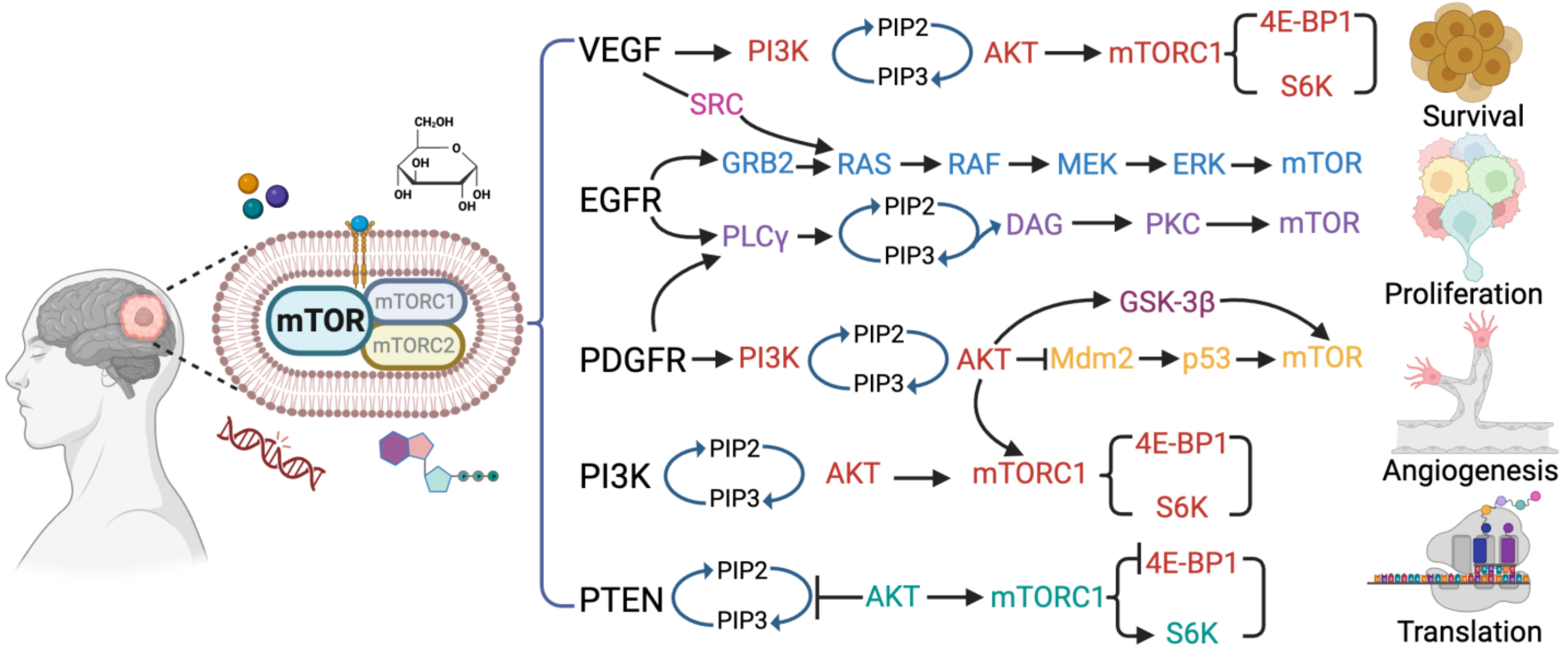
2. The mTOR Signaling Pathway in Glioblastoma
3. The mTOR Inhibitor and Its Limitations
4. Potential Biomarkers for mTOR Inhibitor Response
5. Strategies for Targeting mTOR in Glioblastoma
5.1. Combination Therapies
5.2. Personalized Medicines
5.3. Nanotechnology-Based Drug Delivery
5.4. Extracellular Vesicle as Drug Delivery Vehicle
6. Preclinical and Clinical Studies
6.1. Preclinical Studies and Outcome
6.2. Ongoing and Completed Clinical Trials
7. Challenges for Targeting mTOR in Glioblastoma
8. Future Perspectives and New Therapeutic Approaches
8.1. Potential of Novel mTOR Inhibitors
8.2. Targeting Specific Downstream Effectors of mTOR
8.3. Potential of Immunotherapy in Combination with mTOR Inhibitor
8.4. Targeting Crosstalk of mTOR with Other Signaling Pathways
9. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Schaff, L.R.; Mellinghoff, I.K. Glioblastoma and Other Primary Brain Malignancies in Adults: A Review. JAMA 2023, 329, 574–587. [Google Scholar] [CrossRef] [PubMed]
- Grochans, S.; Cybulska, A.M.; Simińska, D.; Korbecki, J.; Kojder, K.; Chlubek, D.; Baranowska-Bosiacka, I. Epidemiology of Glioblastoma Multiforme—Literature Review. Cancers 2022, 14, 2412. [Google Scholar] [CrossRef] [PubMed]
- Oronsky, B.; Reid, T.R.; Oronsky, A.; Sandhu, N.; Knox, S.J. A Review of Newly Diagnosed Glioblastoma. Front. Oncol. 2021, 10, 574012. [Google Scholar] [CrossRef] [PubMed]
- Rong, L.; Li, N.; Zhang, Z. Emerging Therapies for Glioblastoma: Current State and Future Directions. J. Exp. Clin. Cancer Res. 2022, 41, 142. [Google Scholar] [CrossRef] [PubMed]
- Medikonda, R.; Dunn, G.; Rahman, M.; Fecci, P.; Lim, M. A Review of Glioblastoma Immunotherapy. J. Neurooncol. 2021, 151, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Shergalis, A.; Bankhead, A.; Luesakul, U.; Muangsin, N.; Neamati, N. Current Challenges and Opportunities in Treating Glioblastoma. Pharmacol. Rev. 2018, 70, 412–445. [Google Scholar] [CrossRef]
- Pineda, E.; Domenech, M.; Hernández, A.; Comas, S.; Balaña, C. Recurrent Glioblastoma: Ongoing Clinical Challenges and Future Prospects. OncoTargets Ther. 2023, 16, 71–86. [Google Scholar] [CrossRef]
- Bikfalvi, A.; da Costa, C.A.; Avril, T.; Barnier, J.-V.; Bauchet, L.; Brisson, L.; Cartron, P.F.; Castel, H.; Chevet, E.; Chneiweiss, H.; et al. Challenges in Glioblastoma Research: Focus on the Tumor Microenvironment. Trends Cancer 2023, 9, 9–27. [Google Scholar] [CrossRef]
- Noch, E.K.; Ramakrishna, R.; Magge, R. Challenges in the Treatment of Glioblastoma: Multisystem Mechanisms of Therapeutic Resistance. World Neurosurg. 2018, 116, 505–517. [Google Scholar] [CrossRef] [PubMed]
- Singh, G.; Fritz, S.E.; Seufzer, B.; Boris-Lawrie, K. The mRNA Encoding the JUND Tumor Suppressor Detains Nuclear RNA-Binding Proteins to Assemble Polysomes That Are Unaffected by mTOR. J. Biol. Chem. 2020, 295, 7763–7773. [Google Scholar] [CrossRef] [PubMed]
- Singh, G.; Seufzer, B.; Song, Z.; Zucko, D.; Heng, X.; Boris-Lawrie, K. HIV-1 Hypermethylated Guanosine Cap Licenses Specialized Translation Unaffected by mTOR. Proc. Natl. Acad. Sci. USA 2022, 119, e2105153118. [Google Scholar] [CrossRef] [PubMed]
- Mathieu, J.; Detraux, D.; Kuppers, D.; Wang, Y.; Cavanaugh, C.; Sidhu, S.; Levy, S.; Robitaille, A.M.; Ferreccio, A.; Bottorff, T.; et al. Folliculin Regulates mTORC1/2 and WNT Pathways in Early Human Pluripotency. Nat. Commun. 2019, 10, 632. [Google Scholar] [CrossRef]
- Yan, J.; Wang, R.; Horng, T. mTOR Is Key to T Cell Transdifferentiation. Cell Metab. 2019, 29, 241–242. [Google Scholar] [CrossRef]
- Kim, J.; Guan, K.-L. mTOR as a Central Hub of Nutrient Signalling and Cell Growth. Nat. Cell Biol. 2019, 21, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Colardo, M.; Segatto, M.; Di Bartolomeo, S. Targeting RTK-PI3K-mTOR Axis in Gliomas: An Update. Int. J. Mol. Sci. 2021, 22, 4899. [Google Scholar] [CrossRef]
- Divé, I.; Klann, K.; Michaelis, J.B.; Heinzen, D.; Steinbach, J.P.; Münch, C.; Ronellenfitsch, M.W. Inhibition of mTOR Signaling Protects Human Glioma Cells from Hypoxia-Induced Cell Death in an Autophagy-Independent Manner. Cell Death Discov. 2022, 8, 409. [Google Scholar] [CrossRef]
- Mecca, C.; Giambanco, I.; Donato, R.; Arcuri, C. Targeting mTOR in Glioblastoma: Rationale and Preclinical/Clinical Evidence. Dis. Markers 2018, 2018, 9230479. [Google Scholar] [CrossRef]
- Papavassiliou, K.A.; Papavassiliou, A.G. The Bumpy Road towards mTOR Inhibition in Glioblastoma: Quo Vadis? Biomedicines 2021, 9, 1809. [Google Scholar] [CrossRef] [PubMed]
- Laplante, M.; Sabatini, D.M. mTOR Signaling in Growth Control and Disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef]
- Liu, G.Y.; Sabatini, D.M. mTOR at the Nexus of Nutrition, Growth, Ageing and Disease. Nat. Rev. Mol. Cell Biol. 2020, 21, 183–203. [Google Scholar] [CrossRef]
- Wälchli, M.; Berneiser, K.; Mangia, F.; Imseng, S.; Craigie, L.-M.; Stuttfeld, E.; Hall, M.N.; Maier, T. Regulation of Human mTOR Complexes by DEPTOR. eLife 2021, 10, e70871. [Google Scholar] [CrossRef]
- Marques-Ramos, A.; Cervantes, R. Expression of mTOR in Normal and Pathological Conditions. Mol. Cancer 2023, 22, 112. [Google Scholar] [CrossRef] [PubMed]
- Zou, Z.; Tao, T.; Li, H.; Zhu, X. mTOR Signaling Pathway and mTOR Inhibitors in Cancer: Progress and Challenges. Cell Biosci. 2020, 10, 31. [Google Scholar] [CrossRef] [PubMed]
- Jhanwar-Uniyal, M.; Wainwright, J.V.; Mohan, A.L.; Tobias, M.E.; Murali, R.; Gandhi, C.D.; Schmidt, M.H. Diverse Signaling Mechanisms of mTOR Complexes: mTORC1 and mTORC2 in Forming a Formidable Relationship. Adv. Biol. Regul. 2019, 72, 51–62. [Google Scholar] [CrossRef]
- Duzgun, Z.; Eroglu, Z.; Biray Avci, C. Role of mTOR in Glioblastoma. Gene 2016, 575, 187–190. [Google Scholar] [CrossRef] [PubMed]
- Torrisi, F.; Alberghina, C.; D’Aprile, S.; Pavone, A.M.; Longhitano, L.; Giallongo, S.; Tibullo, D.; Di Rosa, M.; Zappalà, A.; Cammarata, F.P.; et al. The Hallmarks of Glioblastoma: Heterogeneity, Intercellular Crosstalk and Molecular Signature of Invasiveness and Progression. Biomedicines 2022, 10, 806. [Google Scholar] [CrossRef] [PubMed]
- Dumas, A.A.; Pomella, N.; Rosser, G.; Guglielmi, L.; Vinel, C.; Millner, T.O.; Rees, J.; Aley, N.; Sheer, D.; Wei, J.; et al. Microglia Promote Glioblastoma via mTOR-Mediated Immunosuppression of the Tumour Microenvironment. EMBO J. 2020, 39, e103790. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Wang, H.; Jiang, X. mTORC1 beyond Anabolic Metabolism: Regulation of Cell Death. J. Cell Biol. 2022, 221, e202208103. [Google Scholar] [CrossRef] [PubMed]
- Masui, K.; Harachi, M.; Ikegami, S.; Yang, H.; Onizuka, H.; Yong, W.H.; Cloughesy, T.F.; Muragaki, Y.; Kawamata, T.; Arai, N.; et al. mTORC2 Links Growth Factor Signaling with Epigenetic Regulation of Iron Metabolism in Glioblastoma. J. Biol. Chem. 2019, 294, 19740–19751. [Google Scholar] [CrossRef]
- Popova, N.V.; Jücker, M. The Role of mTOR Signaling as a Therapeutic Target in Cancer. Int. J. Mol. Sci. 2021, 22, 1743. [Google Scholar] [CrossRef]
- Sandoval, J.A.; Tomilov, A.; Datta, S.; Allen, S.; O’Donnell, R.; Sears, T.; Woolard, K.; Kovalskyy, D.; Angelastro, J.M.; Cortopassi, G. Novel mTORC1 Inhibitors Kill Glioblastoma Stem Cells. Pharmaceuticals 2020, 13, 419. [Google Scholar] [CrossRef]
- Eckerdt, F.D.; Bell, J.B.; Gonzalez, C.; Oh, M.S.; Perez, R.E.; Mazewski, C.; Fischietti, M.; Goldman, S.; Nakano, I.; Platanias, L.C. Combined PI3Kα-mTOR Targeting of Glioma Stem Cells. Sci. Rep. 2020, 10, 21873. [Google Scholar] [CrossRef]
- Dominguez, J.F.; Rosberger, H.; Garell, P.; Gandhi, C.D.; Jhanwar-Uniyal, M. Abstract 5410: First, Second, and Third Generation mTOR Pathways Inhibitors for Treatment of Glioblastoma. Cancer Res. 2022, 82, 5410. [Google Scholar] [CrossRef]
- Babak, S.; Mason, W.P. mTOR Inhibition in Glioblastoma: Requiem for a Dream? Neuro-Oncology 2018, 20, 584–585. [Google Scholar] [CrossRef]
- Blagosklonny, M.V. Rapamycin for Longevity: Opinion Article. Aging 2019, 11, 8048–8067. [Google Scholar] [CrossRef]
- Kwitkowski, V.E.; Prowell, T.M.; Ibrahim, A.; Farrell, A.T.; Justice, R.; Mitchell, S.S.; Sridhara, R.; Pazdur, R. FDA Approval Summary: Temsirolimus as Treatment for Advanced Renal Cell Carcinoma. Oncologist 2010, 15, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Liang, Y.; Wu, M.; Wang, X.; Fu, H.; Chen, Y.; Wang, Z. The Novel mTOR Inhibitor CCI-779 (Temsirolimus) Induces Antiproliferative Effects through Inhibition of mTOR in Bel-7402 Liver Cancer Cells. Cancer Cell Int. 2013, 13, 30. [Google Scholar] [CrossRef] [PubMed]
- Raymond, E.; Alexandre, J.; Faivre, S.; Vera, K.; Materman, E.; Boni, J.; Leister, C.; Korth-Bradley, J.; Hanauske, A.; Armand, J.-P. Safety and Pharmacokinetics of Escalated Doses of Weekly Intravenous Infusion of CCI-779, a Novel mTOR Inhibitor, in Patients with Cancer. J. Clin. Oncol. 2004, 22, 2336–2347. [Google Scholar] [CrossRef] [PubMed]
- Hartford, C.M.; Desai, A.A.; Janisch, L.; Karrison, T.; Rivera, V.M.; Berk, L.; Loewy, J.W.; Kindler, H.; Stadler, W.M.; Knowles, H.L.; et al. A Phase I Trial to Determine the Safety, Tolerability, and Maximum Tolerated Dose of Deforolimus in Patients with Advanced Malignancies. Clin. Cancer Res. 2009, 15, 1428–1434. [Google Scholar] [CrossRef]
- Apsel, B.; Blair, J.A.; Gonzalez, B.; Nazif, T.M.; Feldman, M.E.; Aizenstein, B.; Hoffman, R.; Williams, R.L.; Shokat, K.M.; Knight, Z.A. Targeted Polypharmacology: Discovery of Dual Inhibitors of Tyrosine and Phosphoinositide Kinases. Nat. Chem. Biol. 2008, 4, 691–699. [Google Scholar] [CrossRef]
- Yu, K.; Toral-Barza, L.; Shi, C.; Zhang, W.-G.; Lucas, J.; Shor, B.; Kim, J.; Verheijen, J.; Curran, K.; Malwitz, D.J.; et al. Biochemical, Cellular, and in Vivo Activity of Novel ATP-Competitive and Selective Inhibitors of the Mammalian Target of Rapamycin. Cancer Res. 2009, 69, 6232–6240. [Google Scholar] [CrossRef]
- Cheng, N.-T.; Guo, A.; Cui, Y.-P. Intra-Articular Injection of Torin 1 Reduces Degeneration of Articular Cartilage in a Rabbit Osteoarthritis Model. Bone Jt. Res. 2016, 5, 218–224. [Google Scholar] [CrossRef]
- Thoreen, C.C.; Kang, S.A.; Chang, J.W.; Liu, Q.; Zhang, J.; Gao, Y.; Reichling, L.J.; Sim, T.; Sabatini, D.M.; Gray, N.S. An ATP-Competitive Mammalian Target of Rapamycin Inhibitor Reveals Rapamycin-Resistant Functions of mTORC1. J. Biol. Chem. 2009, 284, 8023–8032. [Google Scholar] [CrossRef] [PubMed]
- García-Martínez, J.M.; Moran, J.; Clarke, R.G.; Gray, A.; Cosulich, S.C.; Chresta, C.M.; Alessi, D.R. Ku-0063794 Is a Specific Inhibitor of the Mammalian Target of Rapamycin (mTOR). Biochem. J. 2009, 421, 29–42. [Google Scholar] [CrossRef] [PubMed]
- Naing, A.; Aghajanian, C.; Raymond, E.; Olmos, D.; Schwartz, G.; Oelmann, E.; Grinsted, L.; Burke, W.; Taylor, R.; Kaye, S.; et al. Safety, Tolerability, Pharmacokinetics and Pharmacodynamics of AZD8055 in Advanced Solid Tumours and Lymphoma. Br. J. Cancer 2012, 107, 1093–1099. [Google Scholar] [CrossRef] [PubMed]
- Miller, N. Abstract B146: XL388: A Novel, Selective, Orally Bioavailable mTORC1 and mTORC2 Inhibitor That Demonstrates Pharmacodynamic and Antitumor Activity in Multiple Human Cancer Xenograft Models. Mol. Cancer Ther. 2009, 8, B146. [Google Scholar] [CrossRef]
- Netland, I.A.; Førde, H.E.; Sleire, L.; Leiss, L.; Rahman, M.A.; Skeie, B.S.; Gjerde, C.H.; Enger, P.Ø.; Goplen, D. Dactolisib (NVP-BEZ235) Toxicity in Murine Brain Tumour Models. BMC Cancer 2016, 16, 657. [Google Scholar] [CrossRef] [PubMed]
- Wicki, A.; Brown, N.; Xyrafas, A.; Bize, V.; Hawle, H.; Berardi, S.; Cmiljanović, N.; Cmiljanović, V.; Stumm, M.; Dimitrijević, S.; et al. First-in Human, Phase 1, Dose-Escalation Pharmacokinetic and Pharmacodynamic Study of the Oral Dual PI3K and mTORC1/2 Inhibitor PQR309 in Patients with Advanced Solid Tumors (SAKK 67/13). Eur. J. Cancer 2018, 96, 6–16. [Google Scholar] [CrossRef]
- Shapiro, G.I.; Bell-McGuinn, K.M.; Molina, J.R.; Bendell, J.; Spicer, J.; Kwak, E.L.; Pandya, S.S.; Millham, R.; Borzillo, G.; Pierce, K.J.; et al. First-in-Human Study of PF-05212384 (PKI-587), a Small-Molecule, Intravenous, Dual Inhibitor of PI3K and mTOR in Patients with Advanced Cancer. Clin. Cancer Res. 2015, 21, 1888–1895. [Google Scholar] [CrossRef]
- Guenzle, J.; Akasaka, H.; Joechle, K.; Reichardt, W.; Venkatasamy, A.; Hoeppner, J.; Hellerbrand, C.; Fichtner-Feigl, S.; Lang, S.A. Pharmacological Inhibition of mTORC2 Reduces Migration and Metastasis in Melanoma. Int. J. Mol. Sci. 2020, 22, 30. [Google Scholar] [CrossRef]
- Kuroshima, K.; Yoshino, H.; Okamura, S.; Tsuruda, M.; Osako, Y.; Sakaguchi, T.; Sugita, S.; Tatarano, S.; Nakagawa, M.; Enokida, H. Potential New Therapy of Rapalink-1, a New Generation Mammalian Target of Rapamycin Inhibitor, against Sunitinib-resistant Renal Cell Carcinoma. Cancer Sci. 2020, 111, 1607–1618. [Google Scholar] [CrossRef]
- Jhanwar-Uniyal, M. Mighty RapaLink-1 Vanquishes Undruggable Mutant mTOR in Glioblastoma. Transl. Cancer Res. 2017, 6, S143. [Google Scholar] [CrossRef]
- Fan, C.; Lin, X.; Wang, E. Clinicopathological Significance of Cathepsin D Expression in Non-Small Cell Lung Cancer Is Conditional on Apoptosis-Associated Protein Phenotype: An Immunohistochemistry Study. Tumour Biol. 2012, 33, 1045–1052. [Google Scholar] [CrossRef] [PubMed]
- Westhoff, M.-A.; Kandenwein, J.A.; Karl, S.; Vellanki, S.H.K.; Braun, V.; Eramo, A.; Antoniadis, G.; Debatin, K.-M.; Fulda, S. The Pyridinylfuranopyrimidine Inhibitor, PI-103, Chemosensitizes Glioblastoma Cells for Apoptosis by Inhibiting DNA Repair. Oncogene 2009, 28, 3586–3596. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Wang, J.; Shao, W.; Wu, C.; Chen, Z.; To, S.T.; Li, W. Recent Advances in the Use of PI3K Inhibitors for Glioblastoma Multiforme: Current Preclinical and Clinical Development. Mol. Cancer 2017, 16, 100. [Google Scholar] [CrossRef] [PubMed]
- Fan, Q.-W.; Weiss, W.A. Inhibition of PI3K-Akt-mTOR Signaling in Glioblastoma by mTORC1/2 Inhibitors. Methods Mol. Biol. 2012, 821, 349–359. [Google Scholar] [CrossRef]
- Bagci-Onder, T.; Wakimoto, H.; Anderegg, M.; Cameron, C.; Shah, K. A Dual PI3K/mTOR Inhibitor, PI-103, Cooperates with Stem Cell–Delivered TRAIL in Experimental Glioma Models. Cancer Res. 2011, 71, 154–163. [Google Scholar] [CrossRef] [PubMed]
- Del Alcazar, C.R.G.; Gillam, M.C.; Mukherjee, B.; Tomimatsu, N.; Gao, X.; Yan, J.; Xie, X.-J.; Bachoo, R.; Li, L.; Habib, A.A.; et al. Inhibition of DNA Double-Strand Break Repair by the Dual PI3K/mTOR Inhibitor NVP-BEZ235 as a Strategy for Radiosensitization of Glioblastoma. Clin. Cancer Res. 2014, 20, 1235–1248. [Google Scholar] [CrossRef] [PubMed]
- Serra, V.; Markman, B.; Scaltriti, M.; Eichhorn, P.J.A.; Valero, V.; Guzman, M.; Botero, M.L.; Llonch, E.; Atzori, F.; Di Cosimo, S.; et al. NVP-BEZ235, a Dual PI3K/mTOR Inhibitor, Prevents PI3K Signaling and Inhibits the Growth of Cancer Cells with Activating PI3K Mutations. Cancer Res. 2008, 68, 8022–8030. [Google Scholar] [CrossRef] [PubMed]
- Ali, E.S.; Mitra, K.; Akter, S.; Ramproshad, S.; Mondal, B.; Khan, I.N.; Islam, M.T.; Sharifi-Rad, J.; Calina, D.; Cho, W.C. Recent Advances and Limitations of mTOR Inhibitors in the Treatment of Cancer. Cancer Cell Int. 2022, 22, 284. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Qiu, W.; Sun, T.; Wang, L.; Du, C.; Hu, Y.; Liu, W.; Feng, F.; Chen, Y.; Sun, H. Therapeutic Strategies of Glioblastoma (GBM): The Current Advances in the Molecular Targets and Bioactive Small Molecule Compounds. Acta Pharm. Sin. B 2022, 12, 1781–1804. [Google Scholar] [CrossRef]
- Hua, H.; Kong, Q.; Zhang, H.; Wang, J.; Luo, T.; Jiang, Y. Targeting mTOR for Cancer Therapy. J. Hematol. Oncol. 2019, 12, 71. [Google Scholar] [CrossRef]
- Singh, S.; Barik, D.; Arukha, A.P.; Prasad, S.; Mohapatra, I.; Singh, A.; Singh, G. Small Molecule Targeting Immune Cells: A Novel Approach for Cancer Treatment. Biomedicines 2023, 11, 2621. [Google Scholar] [CrossRef]
- Akers, J.C.; Ramakrishnan, V.; Kim, R.; Phillips, S.; Kaimal, V.; Mao, Y.; Hua, W.; Yang, I.; Fu, C.-C.; Nolan, J.; et al. miRNA Contents of Cerebrospinal Fluid Extracellular Vesicles in Glioblastoma Patients. J. Neurooncol. 2015, 123, 205–216. [Google Scholar] [CrossRef] [PubMed]
- Schreck, K.C.; Allen, A.N.; Wang, J.; Pratilas, C.A. Combination MEK and mTOR Inhibitor Therapy Is Active in Models of Glioblastoma. Neurooncol. Adv. 2020, 2, vdaa138. [Google Scholar] [CrossRef]
- Olmez, I.; Brenneman, B.; Xiao, A.; Serbulea, V.; Benamar, M.; Zhang, Y.; Manigat, L.; Abbas, T.; Lee, J.; Nakano, I.; et al. Combined CDK4/6 and mTOR Inhibition Is Synergistic against Glioblastoma via Multiple Mechanisms. Clin. Cancer Res. 2017, 23, 6958–6968. [Google Scholar] [CrossRef]
- Su, Y.-K.; Bamodu, O.A.; Su, I.-C.; Pikatan, N.W.; Fong, I.-H.; Lee, W.-H.; Yeh, C.-T.; Chiu, H.-Y.; Lin, C.-M. Combined Treatment with Acalabrutinib and Rapamycin Inhibits Glioma Stem Cells and Promotes Vascular Normalization by Downregulating BTK/mTOR/VEGF Signaling. Pharmaceuticals 2021, 14, 876. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Peng, X.; Li, X.; Liu, H.; Zhao, B.; Elkabets, M.; Liu, Y.; Wang, W.; Wang, R.; Zhong, Y.; et al. BKM120 Sensitizes Glioblastoma to the PARP Inhibitor Rucaparib by Suppressing Homologous Recombination Repair. Cell Death Dis. 2021, 12, 546. [Google Scholar] [CrossRef]
- El Hage, A.; Dormond, O. Combining mTOR Inhibitors and T Cell-Based Immunotherapies in Cancer Treatment. Cancers 2021, 13, 1359. [Google Scholar] [CrossRef] [PubMed]
- Ronellenfitsch, M.W.; Luger, A.-L.; Steinbach, J.P. EGFR and mTOR as Therapeutic Targets in Glioblastoma. Oncotarget 2019, 10, 4721–4723. [Google Scholar] [CrossRef] [PubMed]
- Xia, Q.; Xu, M.; Zhang, P.; Liu, L.; Meng, X.; Dong, L. Therapeutic Potential of Autophagy in Glioblastoma Treatment with Phosphoinositide 3-Kinase/Protein Kinase B/Mammalian Target of Rapamycin Signaling Pathway Inhibitors. Front. Oncol. 2020, 10, 572904. [Google Scholar] [CrossRef] [PubMed]
- Haas, N.B.; Appleman, L.J.; Stein, M.; Redlinger, M.; Wilks, M.; Xu, X.; Onorati, A.; Kalavacharla, A.; Kim, T.; Zhen, C.J.; et al. Autophagy Inhibition to Augment mTOR Inhibition: A Phase I/II Trial of Everolimus and Hydroxychloroquine in Patients with Previously Treated Renal Cell Carcinoma. Clin. Cancer Res. 2019, 25, 2080–2087. [Google Scholar] [CrossRef] [PubMed]
- Hau, A.M.; Greenwood, J.A.; Löhr, C.V.; Serrill, J.D.; Proteau, P.J.; Ganley, I.G.; McPhail, K.L.; Ishmael, J.E. Coibamide A Induces mTOR-Independent Autophagy and Cell Death in Human Glioblastoma Cells. PLoS ONE 2013, 8, e65250. [Google Scholar] [CrossRef] [PubMed]
- Khabibov, M.; Garifullin, A.; Boumber, Y.; Khaddour, K.; Fernandez, M.; Khamitov, F.; Khalikova, L.; Kuznetsova, N.; Kit, O.; Kharin, L. Signaling Pathways and Therapeutic Approaches in Glioblastoma Multiforme (Review). Int. J. Oncol. 2022, 60, 69. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, E.; Kumar, A.; Zhang, Y.; Wang, A.S.; Chen, K.; Lim, Y.; Shai, A.; Taylor, J.W.; Clarke, J.; Hilz, S.; et al. PI3K/AKT/mTOR Signaling Pathway Activity in IDH-Mutant Diffuse Glioma and Clinical Implications. Neuro-Oncology 2022, 24, 1471–1481. [Google Scholar] [CrossRef]
- Mowforth, O.D.; Brannigan, J.; El Khoury, M.; Sarathi, C.I.P.; Bestwick, H.; Bhatti, F.; Mair, R. Personalised Therapeutic Approaches to Glioblastoma: A Systematic Review. Front. Med. 2023, 10, 1166104. [Google Scholar] [CrossRef]
- Rončević, A.; Koruga, N.; Soldo Koruga, A.; Rončević, R.; Rotim, T.; Šimundić, T.; Kretić, D.; Perić, M.; Turk, T.; Štimac, D. Personalized Treatment of Glioblastoma: Current State and Future Perspective. Biomedicines 2023, 11, 1579. [Google Scholar] [CrossRef]
- Ghiaseddin, A.; Hoang Minh, L.B.; Janiszewska, M.; Shin, D.; Wick, W.; Mitchell, D.A.; Wen, P.Y.; Grossman, S.A. Adult Precision Medicine: Learning from the Past to Enhance the Future. Neuro-Oncol. Adv. 2021, 3, vdaa145. [Google Scholar] [CrossRef]
- Tang, L.; Feng, Y.; Gao, S.; Mu, Q.; Liu, C. Nanotherapeutics Overcoming the Blood-Brain Barrier for Glioblastoma Treatment. Front. Pharmacol. 2021, 12, 786700. [Google Scholar] [CrossRef]
- Séhédic, D.; Roncali, L.; Djoudi, A.; Buchtova, N.; Avril, S.; Chérel, M.; Boury, F.; Lacoeuille, F.; Hindré, F.; Garcion, E. Rapamycin-Loaded Lipid Nanocapsules Induce Selective Inhibition of the mTORC1-Signaling Pathway in Glioblastoma Cells. Front. Bioeng. Biotechnol. 2021, 8, 602998. [Google Scholar] [CrossRef]
- Ferreira, N.N.; Granja, S.; Boni, F.I.; Ferreira, L.M.B.; Reis, R.M.; Baltazar, F.; Gremião, M.P.D. A Novel Strategy for Glioblastoma Treatment Combining Alpha-Cyano-4-Hydroxycinnamic Acid with Cetuximab Using Nanotechnology-Based Delivery Systems. Drug Deliv. Transl. Res. 2020, 10, 594–609. [Google Scholar] [CrossRef] [PubMed]
- Vanza, J.; Jani, P.; Pandya, N.; Tandel, H. Formulation and Statistical Optimization of Intravenous Temozolomide-Loaded PEGylated Liposomes to Treat Glioblastoma Multiforme by Three-Level Factorial Design. Drug Dev. Ind. Pharm. 2018, 44, 923–933. [Google Scholar] [CrossRef] [PubMed]
- Householder, K.T.; DiPerna, D.M.; Chung, E.P.; Wohlleb, G.M.; Dhruv, H.D.; Berens, M.E.; Sirianni, R.W. Intravenous Delivery of Camptothecin-Loaded PLGA Nanoparticles for the Treatment of Intracranial Glioma. Int. J. Pharm. 2015, 479, 374–380. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.-H.; Lin, X.-N.; Wei, F.; Feng, W.; Huang, Z.-C.; Wang, P.; Ren, L.; Diao, Y. Enhanced Brain Targeting of Temozolomide in Polysorbate-80 Coated Polybutylcyanoacrylate Nanoparticles. Int. J. Nanomed. 2011, 6, 445–452. [Google Scholar] [CrossRef]
- Craparo, E.F.; Drago, S.E.; Quaglia, F.; Ungaro, F.; Cavallaro, G. Development of a Novel Rapamycin Loaded Nano- into Micro-Formulation for Treatment of Lung Inflammation. Drug Deliv. Transl. Res. 2022, 12, 1859–1872. [Google Scholar] [CrossRef]
- Deshpande, N.; Ramesh, A.; Nandi, D.; Nguyen, A.; Brouillard, A.; Kulkarni, A. Supramolecular Polysaccharide Nanotheranostics That Inhibit Cancer Cells Growth and Monitor Targeted Therapy Response. Nanotheranostics 2020, 4, 156–172. [Google Scholar] [CrossRef]
- Boada, C.; Zinger, A.; Tsao, C.; Zhao, P.; Martinez, J.O.; Hartman, K.; Naoi, T.; Sukhoveshin, R.; Sushnitha, M.; Molinaro, R.; et al. Rapamycin-Loaded Biomimetic Nanoparticles Reverse Vascular Inflammation. Circ. Res. 2020, 126, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Escalona-Rayo, O.; Fuentes-Vázquez, P.; Jardon-Xicotencatl, S.; García-Tovar, C.G.; Mendoza-Elvira, S.; Quintanar-Guerrero, D. Rapamycin-Loaded Polysorbate 80-Coated PLGA Nanoparticles: Optimization of Formulation Variables and in Vitro Anti-Glioma Assessment. J. Drug Deliv. Sci. Technol. 2019, 52, 488–499. [Google Scholar] [CrossRef]
- Bai, H.; Lee, J.S.; Chen, E.; Wang, M.; Xing, Y.; Fahmy, T.M.; Dardik, A. Covalent Modification of Pericardial Patches for Sustained Rapamycin Delivery Inhibits Venous Neointimal Hyperplasia. Sci. Rep. 2017, 7, 40142. [Google Scholar] [CrossRef]
- Khondee, S.; Rabinsky, E.F.; Owens, S.R.; Joshi, B.P.; Qiu, Z.; Duan, X.; Zhao, L.; Wang, T.D. Targeted Therapy of Colorectal Neoplasia with Rapamycin in Peptide-Labeled Pegylated Octadecyl Lithocholate Micelles. J. Control. Release 2015, 199, 114–121. [Google Scholar] [CrossRef]
- Elsaid, N.; Somavarapu, S.; Jackson, T.L. Cholesterol-Poly(Ethylene) Glycol Nanocarriers for the Transscleral Delivery of Sirolimus. Exp. Eye Res. 2014, 121, 121–129. [Google Scholar] [CrossRef]
- Kim, M.-S.; Kim, J.-S.; Cho, W.K.; Hwang, S.-J. Enhanced Solubility and Oral Absorption of Sirolimus Using D-α-Tocopheryl Polyethylene Glycol Succinate Micelles. Artif. Cells Nanomed. Biotechnol. 2013, 41, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Elsaid, N.; Jackson, T.L.; Gunic, M.; Somavarapu, S. Positively Charged Amphiphilic Chitosan Derivative for the Transscleral Delivery of Rapamycin. Investig. Ophthalmol. Vis. Sci. 2012, 53, 8105–8111. [Google Scholar] [CrossRef] [PubMed]
- Comas, M.; Toshkov, I.; Kuropatwinski, K.K.; Chernova, O.B.; Polinsky, A.; Blagosklonny, M.V.; Gudkov, A.V.; Antoch, M.P. New Nanoformulation of Rapamycin Rapatar Extends Lifespan in Homozygous P53−/− Mice by Delaying Carcinogenesis. Aging 2012, 4, 715–722. [Google Scholar] [CrossRef]
- Woo, H.N.; Chung, H.K.; Ju, E.J.; Jung, J.; Kang, H.-W.; Lee, S.-W.; Seo, M.-H.; Lee, J.S.; Lee, J.S.; Park, H.J.; et al. Preclinical Evaluation of Injectable Sirolimus Formulated with Polymeric Nanoparticle for Cancer Therapy. Int. J. Nanomed. 2012, 7, 2197–2208. [Google Scholar] [CrossRef]
- Dane, K.Y.; Nembrini, C.; Tomei, A.A.; Eby, J.K.; O’Neil, C.P.; Velluto, D.; Swartz, M.A.; Inverardi, L.; Hubbell, J.A. Nano-Sized Drug-Loaded Micelles Deliver Payload to Lymph Node Immune Cells and Prolong Allograft Survival. J. Control. Release 2011, 156, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Li, F.; Mahato, R.I. Poly(Ethylene Glycol)-Block-Poly(2-Methyl-2-Benzoxycarbonyl-Propylene Carbonate) Micelles for Rapamycin Delivery: In Vitro Characterization and Biodistribution. J. Pharm. Sci. 2011, 100, 2418–2429. [Google Scholar] [CrossRef] [PubMed]
- Zou, J.; Zhang, X.; Yang, H.; Zhu, Y.; Ma, H.; Wang, S. Rapamycin-Loaded Nanoparticles for Inhibition of Neointimal Hyperplasia in Experimental Vein Grafts. Ann. Vasc. Surg. 2011, 25, 538–546. [Google Scholar] [CrossRef]
- Tian, W.; Liu, J.; Guo, Y.; Shen, Y.; Zhou, D.; Guo, S. Self-Assembled Micelles of Amphiphilic PEGylated Rapamycin for Loading Paclitaxel and Resisting Multidrug Resistant Cancer Cells. J. Mater. Chem. B Mater. Biol. Med. 2015, 3, 1204–1207. [Google Scholar] [CrossRef]
- Katiyar, S.S.; Muntimadugu, E.; Rafeeqi, T.A.; Domb, A.J.; Khan, W. Co-Delivery of Rapamycin- and Piperine-Loaded Polymeric Nanoparticles for Breast Cancer Treatment. Drug Deliv. 2016, 23, 2608–2616. [Google Scholar] [CrossRef]
- Guo, S.; Lin, C.M.; Xu, Z.; Miao, L.; Wang, Y.; Huang, L. Co-Delivery of Cisplatin and Rapamycin for Enhanced Anticancer Therapy through Synergistic Effects and Microenvironment Modulation. ACS Nano 2014, 8, 4996–5009. [Google Scholar] [CrossRef]
- Hasenstein, J.R.; Shin, H.-C.; Kasmerchak, K.; Buehler, D.; Kwon, G.S.; Kozak, K.R. Antitumor Activity of Triolimus: A Novel Multidrug-Loaded Micelle Containing Paclitaxel, Rapamycin, and 17-AAG. Mol. Cancer Ther. 2012, 11, 2233–2242. [Google Scholar] [CrossRef]
- Wang, R.; Liang, Q.; Zhang, X.; Di, Z.; Wang, X.; Di, L. Tumor-Derived Exosomes Reversing TMZ Resistance by Synergistic Drug Delivery for Glioma-Targeting Treatment. Colloids Surf. B Biointerfaces 2022, 215, 112505. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, J.; Gao, W.; Xie, N. Exosomes as Anticancer Drug Delivery Vehicles: Prospects and Challenges. Front. Biosci. Landmark 2022, 27, 293. [Google Scholar] [CrossRef] [PubMed]
- Di, C.; Zhang, Q.; Wang, Y.; Wang, F.; Chen, Y.; Gan, L.; Zhou, R.; Sun, C.; Li, H.; Zhang, X.; et al. Exosomes as Drug Carriers for Clinical Application. Artif. Cells Nanomed. Biotechnol. 2018, 46, 564–570. [Google Scholar] [CrossRef]
- Ghalavand, M.; Moradi-Chaleshtori, M.; Dorostkar, R.; Mohammadi-Yeganeh, S.; Hashemi, S.M. Exosomes Derived from Rapamycin-Treated 4T1 Breast Cancer Cells Induced Polarization of Macrophages to M1 Phenotype. Biotechnol. Appl. Biochem. 2023, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Wang, X.; Guo, X.; Wan, Q.; Teng, Y.; Liu, J. Development of Rapamycin-Encapsulated Exosome-Mimetic Nanoparticles-in-PLGA Microspheres for Treatment of Hemangiomas. Biomed. Pharmacother. 2022, 148, 112737. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhang, Z.; Li, Y.; Su, L.; Duan, Y.; Zhang, H.; An, J.; Ni, T.; Li, X.; Zhang, X. Therapeutic Effect of Rapamycin-Loaded Small Extracellular Vesicles Derived from Mesenchymal Stem Cells on Experimental Autoimmune Uveitis. Front. Immunol. 2022, 13, 864956. [Google Scholar] [CrossRef]
- Mehryab, F.; Rabbani, S.; Shekari, F.; Nazari, A.; Goshtasbi, N.; Haeri, A. Sirolimus-Loaded Exosomes as a Promising Vascular Delivery System for the Prevention of Post-Angioplasty Restenosis. Drug Deliv. Transl. Res. 2023, 1–19. [Google Scholar] [CrossRef]
- Bagheri, S.; Rahban, M.; Bostanian, F.; Esmaeilzadeh, F.; Bagherabadi, A.; Zolghadri, S.; Stanek, A. Targeting Protein Kinases and Epigenetic Control as Combinatorial Therapy Options for Advanced Prostate Cancer Treatment. Pharmaceutics 2022, 14, 515. [Google Scholar] [CrossRef] [PubMed]
- Saari, H.; Lázaro-Ibáñez, E.; Viitala, T.; Vuorimaa-Laukkanen, E.; Siljander, P.; Yliperttula, M. Microvesicle- and Exosome-Mediated Drug Delivery Enhances the Cytotoxicity of Paclitaxel in Autologous Prostate Cancer Cells. J. Control. Release 2015, 220, 727–737. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, L.; Tang, M.; Li, H.; Guo, X.; Yang, X. The Effects of Umbilical Cord-Derived Macrophage Exosomes Loaded with Cisplatin on the Growth and Drug Resistance of Ovarian Cancer Cells. Drug Dev. Ind. Pharm. 2020, 46, 1150–1162. [Google Scholar] [CrossRef]
- Osterman, C.J.D.; Lynch, J.C.; Leaf, P.; Gonda, A.; Ferguson Bennit, H.R.; Griffiths, D.; Wall, N.R. Curcumin Modulates Pancreatic Adenocarcinoma Cell-Derived Exosomal Function. PLoS ONE 2015, 10, e0132845. [Google Scholar] [CrossRef]
- Zhao, D.; Tao, W.; Li, S.; Chen, Y.; Sun, Y.; He, Z.; Sun, B.; Sun, J. Apoptotic Body–Mediated Intercellular Delivery for Enhanced Drug Penetration and Whole Tumor Destruction. Sci. Adv. 2021, 7, eabg0880. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Song, J.; Kim, I.G.; You, G.; Kim, H.; Ahn, J.-H.; Mok, H. Exosome-Mediated Delivery of Transforming Growth Factor-β Receptor 1 Kinase Inhibitors and Toll-like Receptor 7/8 Agonists for Combination Therapy of Tumors. Acta Biomater. 2022, 141, 354–363. [Google Scholar] [CrossRef]
- Değirmenci, N.S.; Uslu, M.; Kırbaş, O.K.; Şahin, F.; Önay Uçar, E. Lapatinib Loaded Exosomes as a Drug Delivery System in Breast Cancer. J. Drug Deliv. Sci. Technol. 2022, 75, 103584. [Google Scholar] [CrossRef]
- Bai, L.; Liu, Y.; Guo, K.; Zhang, K.; Liu, Q.; Wang, P.; Wang, X. Ultrasound Facilitates Naturally Equipped Exosomes Derived from Macrophages and Blood Serum for Orthotopic Glioma Treatment. ACS Appl. Mater. Interfaces 2019, 11, 14576–14587. [Google Scholar] [CrossRef]
- Bao, P.; Zheng, Z.-T.; Ye, J.-J.; Zhang, X.-Z. Apoptotic Body-Mediated Intracellular Delivery Strategy for Enhanced STING Activation and Improved Tumor Immunogenicity. Nano Lett. 2022, 22, 2217–2227. [Google Scholar] [CrossRef]
- Li, Y.; Gao, Y.; Gong, C.; Wang, Z.; Xia, Q.; Gu, F.; Hu, C.; Zhang, L.; Guo, H.; Gao, S. A33 Antibody-Functionalized Exosomes for Targeted Delivery of Doxorubicin against Colorectal Cancer. Nanomedicine 2018, 14, 1973–1985. [Google Scholar] [CrossRef]
- Fan, M.; Liu, H.; Yan, H.; Che, R.; Jin, Y.; Yang, X.; Zhou, X.; Yang, H.; Ge, K.; Liang, X.-J.; et al. A CAR T-Inspiring Platform Based on Antibody-Engineered Exosomes from Antigen-Feeding Dendritic Cells for Precise Solid Tumor Therapy. Biomaterials 2022, 282, 121424. [Google Scholar] [CrossRef] [PubMed]
- Si, K.; Dai, Z.; Li, Z.; Ye, Z.; Ding, B.; Feng, S.; Sun, B.; Shen, Y.; Xiao, Z. Engineered Exosome-Mediated Messenger RNA and Single-Chain Variable Fragment Delivery for Human Chimeric Antigen Receptor T-Cell Engineering. Cytotherapy 2023, 25, 615–624. [Google Scholar] [CrossRef]
- Liu, T.; Li, T.; Zheng, Y.; Xu, X.; Sun, R.; Zhan, S.; Guo, X.; Zhao, Z.; Zhu, W.; Feng, B.; et al. Evaluating Adipose-Derived Stem Cell Exosomes as miRNA Drug Delivery Systems for the Treatment of Bladder Cancer. Cancer Med. 2022, 11, 3687–3699. [Google Scholar] [CrossRef]
- Jeong, K.; Yu, Y.J.; You, J.Y.; Rhee, W.J.; Kim, J.A. Exosome-Mediated microRNA-497 Delivery for Anti-Cancer Therapy in a Microfluidic 3D Lung Cancer Model. Lab Chip 2020, 20, 548–557. [Google Scholar] [CrossRef]
- Lou, G.; Chen, L.; Xia, C.; Wang, W.; Qi, J.; Li, A.; Zhao, L.; Chen, Z.; Zheng, M.; Liu, Y. MiR-199a-Modified Exosomes from Adipose Tissue-Derived Mesenchymal Stem Cells Improve Hepatocellular Carcinoma Chemosensitivity through mTOR Pathway. J. Exp. Clin. Cancer Res. 2020, 39, 4. [Google Scholar] [CrossRef]
- Katakowski, M.; Buller, B.; Zheng, X.; Lu, Y.; Rogers, T.; Osobamiro, O.; Shu, W.; Jiang, F.; Chopp, M. Exosomes from Marrow Stromal Cells Expressing miR-146b Inhibit Glioma Growth. Cancer Lett. 2013, 335, 201–204. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.; Kim, M.; Lee, Y.; Byun, J.W.; Hwang, D.W.; Lee, M. Systemic Delivery of microRNA-21 Antisense Oligonucleotides to the Brain Using T7-Peptide Decorated Exosomes. J. Control. Release 2020, 317, 273–281. [Google Scholar] [CrossRef]
- Zhao, L.; Gu, C.; Gan, Y.; Shao, L.; Chen, H.; Zhu, H. Exosome-Mediated siRNA Delivery to Suppress Postoperative Breast Cancer Metastasis. J. Control. Release 2020, 318, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.-F.; Zuo, F.-F.; Yin, B.-C.; Ye, B.-C. Delivery of siRNA Based on Engineered Exosomes for Glioblastoma Therapy by Targeting STAT3. Biomater. Sci. 2022, 10, 1582–1590. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Miao, Y.; Wang, Y.; He, S.; Guo, L.; Mao, J.; Chen, M.; Yang, Y.; Zhang, X.; Gan, Y. Tumour-Derived Extracellular Vesicle Membrane Hybrid Lipid Nanovesicles Enhance siRNA Delivery by Tumour-Homing and Intracellular Freeway Transportation. J. Extracell. Vesicles 2022, 11, e12198. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Shi, J.; Xie, J.; Wang, Y.; Sun, J.; Liu, T.; Zhao, Y.; Zhao, X.; Wang, X.; Ma, Y.; et al. Large-Scale Generation of Functional mRNA-Encapsulating Exosomes via Cellular Nanoporation. Nat. Biomed. Eng. 2020, 4, 69–83. [Google Scholar] [CrossRef]
- Mizrak, A.; Bolukbasi, M.F.; Ozdener, G.B.; Brenner, G.J.; Madlener, S.; Erkan, E.P.; Ströbel, T.; Breakefield, X.O.; Saydam, O. Genetically Engineered Microvesicles Carrying Suicide mRNA/Protein Inhibit Schwannoma Tumor Growth. Mol. Ther. 2013, 21, 101–108. [Google Scholar] [CrossRef]
- Kamerkar, S.; Leng, C.; Burenkova, O.; Jang, S.C.; McCoy, C.; Zhang, K.; Dooley, K.; Kasera, S.; Zi, T.; Sisó, S.; et al. Exosome-Mediated Genetic Reprogramming of Tumor-Associated Macrophages by exoASO-STAT6 Leads to Potent Monotherapy Antitumor Activity. Sci. Adv. 2022, 8, eabj7002. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.M.; Yang, Y.; Oh, S.J.; Hong, Y.; Seo, M.; Jang, M. Cancer-Derived Exosomes as a Delivery Platform of CRISPR/Cas9 Confer Cancer Cell Tropism-Dependent Targeting. J. Control. Release 2017, 266, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, J.; Tan, J.; Wu, C.; Zhang, J.; Liu, T.; Fan, C.; Li, J.; Zhang, Y. Extracellular Vesicles Engineered with Valency-Controlled DNA Nanostructures Deliver CRISPR/Cas9 System for Gene Therapy. Nucleic Acids Res. 2020, 48, 8870–8882. [Google Scholar] [CrossRef] [PubMed]
- Bai, J.; Duan, J.; Liu, R.; Du, Y.; Luo, Q.; Cui, Y.; Su, Z.; Xu, J.; Xie, Y.; Lu, W. Engineered Targeting tLyp-1 Exosomes as Gene Therapy Vectors for Efficient Delivery of siRNA into Lung Cancer Cells. Asian J. Pharm. Sci. 2020, 15, 461–471. [Google Scholar] [CrossRef]
- Cheng, Q.; Shi, X.; Han, M.; Smbatyan, G.; Lenz, H.-J.; Zhang, Y. Reprogramming Exosomes as Nanoscale Controllers of Cellular Immunity. J. Am. Chem. Soc. 2018, 140, 16413–16417. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Lu, G.; Nie, W.; Huang, L.-L.; Zhang, Y.; Fan, W.; Wu, G.; Liu, H.; Xie, H.-Y. Self-Activatable Photo-Extracellular Vesicle for Synergistic Trimodal Anticancer Therapy. Adv. Mater. 2021, 33, 2005562. [Google Scholar] [CrossRef] [PubMed]
- Gong, C.; Tian, J.; Wang, Z.; Gao, Y.; Wu, X.; Ding, X.; Qiang, L.; Li, G.; Han, Z.; Yuan, Y.; et al. Functional Exosome-Mediated Co-Delivery of Doxorubicin and Hydrophobically Modified microRNA 159 for Triple-Negative Breast Cancer Therapy. J. Nanobiotechnol. 2019, 17, 93. [Google Scholar] [CrossRef]
- Liang, G.; Zhu, Y.; Ali, D.J.; Tian, T.; Xu, H.; Si, K.; Sun, B.; Chen, B.; Xiao, Z. Engineered Exosomes for Targeted Co-Delivery of miR-21 Inhibitor and Chemotherapeutics to Reverse Drug Resistance in Colon Cancer. J. Nanobiotechnol. 2020, 18, 10. [Google Scholar] [CrossRef]
- Li, B.; Chen, X.; Qiu, W.; Zhao, R.; Duan, J.; Zhang, S.; Pan, Z.; Zhao, S.; Guo, Q.; Qi, Y.; et al. Synchronous Disintegration of Ferroptosis Defense Axis via Engineered Exosome-Conjugated Magnetic Nanoparticles for Glioblastoma Therapy. Adv. Sci. 2022, 9, e2105451. [Google Scholar] [CrossRef]
- Pham, T.C.; Jayasinghe, M.K.; Pham, T.T.; Yang, Y.; Wei, L.; Usman, W.M.; Chen, H.; Pirisinu, M.; Gong, J.; Kim, S.; et al. Covalent Conjugation of Extracellular Vesicles with Peptides and Nanobodies for Targeted Therapeutic Delivery. J. Extracell. Vesicles 2021, 10, e12057. [Google Scholar] [CrossRef] [PubMed]
- Butreddy, A.; Kommineni, N.; Dudhipala, N. Exosomes as Naturally Occurring Vehicles for Delivery of Biopharmaceuticals: Insights from Drug Delivery to Clinical Perspectives. Nanomaterials 2021, 11, 1481. [Google Scholar] [CrossRef]
- Zhou, Y.; Wang, L.; Chen, L.; Wu, W.; Yang, Z.; Wang, Y.; Wang, A.; Jiang, S.; Qin, X.; Ye, Z.; et al. Glioblastoma Cell-Derived Exosomes Functionalized with Peptides as Efficient Nanocarriers for Synergistic Chemotherapy of Glioblastoma with Improved Biosafety. Nano Res. 2023, 1–11. [Google Scholar] [CrossRef]
- Garofalo, M.; Villa, A.; Rizzi, N.; Kuryk, L.; Rinner, B.; Cerullo, V.; Yliperttula, M.; Mazzaferro, V.; Ciana, P. Extracellular Vesicles Enhance the Targeted Delivery of Immunogenic Oncolytic Adenovirus and Paclitaxel in Immunocompetent Mice. J. Control. Release 2019, 294, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Rackles, E.; Lopez, P.H.; Falcon-Perez, J.M. Extracellular Vesicles as Source for the Identification of Minimally Invasive Molecular Signatures in Glioblastoma. Semin. Cancer Biol. 2022, 87, 148–159. [Google Scholar] [CrossRef] [PubMed]
- Khatami, S.H.; Karami, N.; Taheri-Anganeh, M.; Taghvimi, S.; Tondro, G.; Khorsand, M.; Soltani Fard, E.; Sedighimehr, N.; Kazemi, M.; Rahimi Jaberi, K.; et al. Exosomes: Promising Delivery Tools for Overcoming Blood-Brain Barrier and Glioblastoma Therapy. Mol. Neurobiol. 2023, 60, 4659–4678. [Google Scholar] [CrossRef]
- Feldman, M.E.; Apsel, B.; Uotila, A.; Loewith, R.; Knight, Z.A.; Ruggero, D.; Shokat, K.M. Active-Site Inhibitors of mTOR Target Rapamycin-Resistant Outputs of mTORC1 and mTORC2. PLoS Biol. 2009, 7, e38. [Google Scholar] [CrossRef]
- Fan, Q.; Aksoy, O.; Wong, R.A.; Ilkhanizadeh, S.; Novotny, C.J.; Gustafson, W.C.; Truong, A.Y.-Q.; Cayanan, G.; Simonds, E.F.; Haas-Kogan, D.; et al. A Kinase Inhibitor Targeted to mTORC1 Drives Regression in Glioblastoma. Cancer Cell 2017, 31, 424–435. [Google Scholar] [CrossRef] [PubMed]
- Fan, Q.-W.; Weiss, W.A. Targeting the RTK-PI3K-mTOR Axis in Malignant Glioma: Overcoming Resistance. Curr. Top. Microbiol. Immunol. 2010, 347, 279–296. [Google Scholar] [CrossRef]
- Yu, Z.; Xie, G.; Zhou, G.; Cheng, Y.; Zhang, G.; Yao, G.; Chen, Y.; Li, Y.; Zhao, G. NVP-BEZ235, a Novel Dual PI3K-mTOR Inhibitor Displays Anti-Glioma Activity and Reduces Chemoresistance to Temozolomide in Human Glioma Cells. Cancer Lett. 2015, 367, 58–68. [Google Scholar] [CrossRef]
- Mukherjee, B.; Tomimatsu, N.; Amancherla, K.; Camacho, C.V.; Pichamoorthy, N.; Burma, S. The Dual PI3K/mTOR Inhibitor NVP-BEZ235 Is a Potent Inhibitor of ATM- and DNA-PKCs-Mediated DNA Damage Responses. Neoplasia 2012, 14, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Prasad, G.; Sottero, T.; Yang, X.; Mueller, S.; James, C.D.; Weiss, W.A.; Polley, M.-Y.; Ozawa, T.; Berger, M.S.; Aftab, D.T.; et al. Inhibition of PI3K/mTOR Pathways in Glioblastoma and Implications for Combination Therapy with Temozolomide. Neuro-Oncology 2011, 13, 384–392. [Google Scholar] [CrossRef] [PubMed]
- Mallon, R.; Feldberg, L.R.; Lucas, J.; Chaudhary, I.; Dehnhardt, C.; Santos, E.D.; Chen, Z.; dos Santos, O.; Ayral-Kaloustian, S.; Venkatesan, A.; et al. Antitumor Efficacy of PKI-587, a Highly Potent Dual PI3K/mTOR Kinase Inhibitor. Clin. Cancer Res. 2011, 17, 3193–3203. [Google Scholar] [CrossRef] [PubMed]
- Heffron, T.P.; Ndubaku, C.O.; Salphati, L.; Alicke, B.; Cheong, J.; Drobnick, J.; Edgar, K.; Gould, S.E.; Lee, L.B.; Lesnick, J.D.; et al. Discovery of Clinical Development Candidate GDC-0084, a Brain Penetrant Inhibitor of PI3K and mTOR. ACS Med. Chem. Lett. 2016, 7, 351–356. [Google Scholar] [CrossRef] [PubMed]
- Sunayama, J.; Sato, A.; Matsuda, K.; Tachibana, K.; Suzuki, K.; Narita, Y.; Shibui, S.; Sakurada, K.; Kayama, T.; Tomiyama, A.; et al. Dual Blocking of mTor and PI3K Elicits a Prodifferentiation Effect on Glioblastoma Stem-like Cells. Neuro-Oncology 2010, 12, 1205–1219. [Google Scholar] [CrossRef] [PubMed]
- Cerniglia, G.J.; Karar, J.; Tyagi, S.; Christofidou-Solomidou, M.; Rengan, R.; Koumenis, C.; Maity, A. Inhibition of Autophagy as a Strategy to Augment Radiosensitization by the Dual Phosphatidylinositol 3-Kinase/Mammalian Target of Rapamycin Inhibitor NVP-BEZ235. Mol. Pharmacol. 2012, 82, 1230–1240. [Google Scholar] [CrossRef] [PubMed]
- Kahn, J.; Hayman, T.J.; Jamal, M.; Rath, B.H.; Kramp, T.; Camphausen, K.; Tofilon, P.J. The mTORC1/mTORC2 Inhibitor AZD2014 Enhances the Radiosensitivity of Glioblastoma Stem-like Cells. Neuro-Oncology 2014, 16, 29–37. [Google Scholar] [CrossRef]
- Liu, T.-J.; Koul, D.; LaFortune, T.; Tiao, N.; Shen, R.J.; Maira, S.-M.; Garcia-Echevrria, C.; Yung, W.K.A. NVP-BEZ235, a Novel Dual Phosphatidylinositol 3-Kinase/Mammalian Target of Rapamycin Inhibitor, Elicits Multifaceted Antitumor Activities in Human Gliomas. Mol. Cancer Ther. 2009, 8, 2204–2210. [Google Scholar] [CrossRef]
- Wise-Draper, T.M.; Moorthy, G.; Salkeni, M.A.; Karim, N.A.; Thomas, H.E.; Mercer, C.A.; Beg, M.S.; O’Gara, S.; Olowokure, O.; Fathallah, H.; et al. A Phase Ib Study of the Dual PI3K/mTOR Inhibitor Dactolisib (BEZ235) Combined with Everolimus in Patients with Advanced Solid Malignancies. Target. Oncol. 2017, 12, 323–332. [Google Scholar] [CrossRef]
- Cloughesy, T.F.; Mischel, P.S.; Omuro, A.M.P.; Prados, M.; Wen, P.Y.; Wu, B.; Rockich, K.; Xu, Y.; Lager, J.J.; Mellinghoff, I.K. Tumor Pharmacokinetics (PK) and Pharmacodynamics (PD) of SAR245409 (XL765) and SAR245408 (XL147) Administered as Single Agents to Patients with Recurrent Glioblastoma (GBM): An Ivy Foundation Early-Phase Clinical Trials Consortium Study. J. Clin. Oncol. 2013, 31, 2012. [Google Scholar] [CrossRef]
- Wen, P.Y.; Chang, S.M.; Lamborn, K.R.; Kuhn, J.G.; Norden, A.D.; Cloughesy, T.F.; Robins, H.I.; Lieberman, F.S.; Gilbert, M.R.; Mehta, M.P.; et al. Phase I/II Study of Erlotinib and Temsirolimus for Patients with Recurrent Malignant Gliomas: North American Brain Tumor Consortium Trial 04-02. Neuro-Oncology 2014, 16, 567–578. [Google Scholar] [CrossRef] [PubMed]
- Mooney, J.; Bernstock, J.D.; Ilyas, A.; Ibrahim, A.; Yamashita, D.; Markert, J.M.; Nakano, I. Current Approaches and Challenges in the Molecular Therapeutic Targeting of Glioblastoma. World Neurosurg. 2019, 129, 90–100. [Google Scholar] [CrossRef]
- Yeini, E.; Ofek, P.; Albeck, N.; Rodriguez Ajamil, D.; Neufeld, L.; Eldar-Boock, A.; Kleiner, R.; Vaskovich, D.; Koshrovski-Michael, S.; Dangoor, S.I.; et al. Targeting Glioblastoma: Advances in Drug Delivery and Novel Therapeutic Approaches. Adv. Ther. 2021, 4, 2000124. [Google Scholar] [CrossRef]
- Khan, F.; Pang, L.; Dunterman, M.; Lesniak, M.S.; Heimberger, A.B.; Chen, P. Macrophages and Microglia in Glioblastoma: Heterogeneity, Plasticity, and Therapy. J. Clin. Investig. 2023, 133, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Dou, J.; Yu, Y.; Zhao, Y.; Fan, Y.; Cheng, J.; Xu, X.; Liu, W.; Guan, S.; Chen, Z.; et al. mTOR ATP-Competitive Inhibitor INK128 Inhibits Neuroblastoma Growth via Blocking mTORC Signaling. Apoptosis 2015, 20, 50–62. [Google Scholar] [CrossRef]
- Guichard, S.M.; Curwen, J.; Bihani, T.; D’Cruz, C.M.; Yates, J.W.T.; Grondine, M.; Howard, Z.; Davies, B.R.; Bigley, G.; Klinowska, T.; et al. AZD2014, an Inhibitor of mTORC1 and mTORC2, Is Highly Effective in ER+ Breast Cancer When Administered Using Intermittent or Continuous Schedules. Mol. Cancer Ther. 2015, 14, 2508–2518. [Google Scholar] [CrossRef] [PubMed]
- Qin, X.; Jiang, B.; Zhang, Y. 4E-BP1, a Multifactor Regulated Multifunctional Protein. Cell Cycle 2016, 15, 781–786. [Google Scholar] [CrossRef]
- Yi, W.; Gupta, S.; Ricker, E.; Manni, M.; Jessberger, R.; Chinenov, Y.; Molina, H.; Pernis, A.B. The mTORC1-4E-BP-eIF4E Axis Controls de Novo Bcl6 Protein Synthesis in T Cells and Systemic Autoimmunity. Nat. Commun. 2017, 8, 254. [Google Scholar] [CrossRef]
- Ruoff, R.; Katsara, O.; Kolupaeva, V. Cell Type-Specific Control of Protein Synthesis and Proliferation by FGF-Dependent Signaling to the Translation Repressor 4E-BP. Proc. Natl. Acad. Sci. USA 2016, 113, 7545–7550. [Google Scholar] [CrossRef]
- Fu, W.; Hall, M.N. Regulation of mTORC2 Signaling. Genes 2020, 11, 1045. [Google Scholar] [CrossRef]
- Chen, S.; Sang, N. Hypoxia-Inducible Factor-1: A Critical Player in the Survival Strategy of Stressed Cells. J. Cell Biochem. 2016, 117, 267–278. [Google Scholar] [CrossRef]
- Abd El-Fattah, E.E.; Saber, S.; Youssef, M.E.; Eissa, H.; El-Ahwany, E.; Amin, N.A.; Alqarni, M.; Batiha, G.E.-S.; Obaidullah, A.J.; Kaddah, M.M.Y.; et al. AKT-AMPKα-mTOR-Dependent HIF-1α Activation Is a New Therapeutic Target for Cancer Treatment: A Novel Approach to Repositioning the Antidiabetic Drug Sitagliptin for the Management of Hepatocellular Carcinoma. Front. Pharmacol. 2022, 12, 720173. [Google Scholar] [CrossRef]
- Himes, B.T.; Geiger, P.A.; Ayasoufi, K.; Bhargav, A.G.; Brown, D.A.; Parney, I.F. Immunosuppression in Glioblastoma: Current Understanding and Therapeutic Implications. Front. Oncol. 2021, 11, 770561. [Google Scholar] [CrossRef] [PubMed]
- Salehi, S.; Sosa, R.A.; Jin, Y.-P.; Kageyama, S.; Fishbein, M.C.; Rozengurt, E.; Kupiec-Weglinski, J.W.; Reed, E.F. Outside-in HLA Class I Signaling Regulates ICAM-1 Clustering and Endothelial Cell-Monocyte Interactions via mTOR in Transplant Antibody-Mediated Rejection. Am. J. Transplant. 2018, 18, 1096–1109. [Google Scholar] [CrossRef] [PubMed]
- Muntjewerff, E.M.; Meesters, L.D.; van den Bogaart, G.; Revelo, N.H. Reverse Signaling by MHC-I Molecules in Immune and Non-Immune Cell Types. Front. Immunol. 2020, 11, 605958. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; Liu, B.; Sun, Q.; Xiong, X.; Chen, Q. Immune Checkpoint Targeted Therapy in Glioma: Status and Hopes. Front. Immunol. 2020, 11, 578877. [Google Scholar] [CrossRef]
- Mafi, S.; Mansoori, B.; Taeb, S.; Sadeghi, H.; Abbasi, R.; Cho, W.C.; Rostamzadeh, D. mTOR-Mediated Regulation of Immune Responses in Cancer and Tumor Microenvironment. Front. Immunol. 2022, 12, 774103. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Zhao, Q. Emerging Role of mTOR in Tumor Immune Contexture: Impact on Chemokine-Related Immune Cells Migration. Theranostics 2020, 10, 6231–6244. [Google Scholar] [CrossRef]
- Conciatori, F.; Ciuffreda, L.; Bazzichetto, C.; Falcone, I.; Pilotto, S.; Bria, E.; Cognetti, F.; Milella, M. mTOR Cross-Talk in Cancer and Potential for Combination Therapy. Cancers 2018, 10, 23. [Google Scholar] [CrossRef]
- Barzegar Behrooz, A.; Talaie, Z.; Jusheghani, F.; Łos, M.J.; Klonisch, T.; Ghavami, S. Wnt and PI3K/Akt/mTOR Survival Pathways as Therapeutic Targets in Glioblastoma. Int. J. Mol. Sci. 2022, 23, 1353. [Google Scholar] [CrossRef] [PubMed]
- Bazzoni, R.; Bentivegna, A. Role of Notch Signaling Pathway in Glioblastoma Pathogenesis. Cancers 2019, 11, 292. [Google Scholar] [CrossRef] [PubMed]
- Gersey, Z.; Osiason, A.D.; Bloom, L.; Shah, S.; Thompson, J.W.; Bregy, A.; Agarwal, N.; Komotar, R.J. Therapeutic Targeting of the Notch Pathway in Glioblastoma Multiforme. World Neurosurg. 2019, 131, 252–263.e2. [Google Scholar] [CrossRef] [PubMed]
- Sidorov, M.; Dighe, P.; Woo, R.W.L.; Rodriguez-Brotons, A.; Chen, M.; Ice, R.J.; Vaquero, E.; Jian, D.; Desprez, P.-Y.; Nosrati, M.; et al. Dual Targeting of EGFR and MTOR Pathways Inhibits Glioblastoma Growth by Modulating the Tumor Microenvironment. Cells 2023, 12, 547. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| mTOR Inhibitors | Target | Activity | Limitations | References |
|---|---|---|---|---|
| Rapamycin | FKPB12/mTORC1 | Inhibits lymphocyte activation and induces cell cycle arrest |
| [35] |
| RAD001 (Everolimus) | mTORC1 | Reduces VEGF expression and inhibits glycolysis | [36] | |
| CCI-779 (Temsirolimus) | Inhibits mTOR activity and regulates cell division | [37,38] | ||
| AP23573 (Ridaforolimus) | Inhibits PTEN-independent tumor cell proliferation and AKT activation |
| [39] | |
| PP242 PP30 | mTORC1 and mTORC2 in an ATP-competitive manner | Affects cell cycle, cell proliferation, and cap-dependent translation | [40] | |
| WYE-354 WAY-600 WYE-687 | mTOR/PI3K | Reduces mTORC1 and mTORC2 substrate phosphorylation in response to amino acids, and growth factors and induces PI3K-AKT |
| [41] |
| Torin-1 | mTOR | Inhibits mTORC1 and mTORC2 complex, impairs cell proliferation, suppresses rapamycin-resistant functions of mTORC1 | [42,43] | |
| Ku-0063794 | mTOR in an ATP-competitive manner | Inhibits AKT activation and hydrophobic motif phosphorylation |
| [44] |
| AZD8055 | mTOR | Inhibits mTORC1 and mTORC2/AKT activity, promotes proteasomal degradation | [45] | |
| XL388 | mTORC1 and mTORC2 | Induces apoptosis, suppresses autophagy | [46] | |
| NVP-BEZ235 (Dactolisib) | Dual PI3K/mTOR | Inhibits AKT activity, S6RP (S6) and 4E-BP1 phosphorylation, induces FKHRL1 nuclear translocation and cell cycle arrest | [47] | |
| PQR309 (Bimiralisib) | Dual PI3K/mTOR | Inhibits proliferation, and induces apoptosis and G1 cell cycle arrest | [48] | |
| PKI587 (Gedatolisib) | Dual PI3K/mTOR | Increases DNA damage | [49] | |
| JR-AB2-011 | mTORC-2 | Inhibits rictor-mTOR association, reduces migration and invasion | [50] | |
| RapaLink-1 | mTORC1 and mTORC2 | Reduces chemotherapeutic drug resistance | [51] |
| Drugs | Nanocarrier | Particle Size (nm) | Entrapment Efficiency (%) | Effects | References |
|---|---|---|---|---|---|
| Rapamycin | α,β-Poly(N-2-hydroxyethyl)-D,L-aspartamide (PHEA)-g-RhB | 100 | 82 | Efficient release and protection of Rapamycin from degradation | [85] |
| PI-103 | Supramolecular polysaccharide nanotheranostic (SPN) | 200 | - | Kinase inhibition and caspase-mediated apoptosis | [86] |
| Rapamycin | Biomimetic nanoparticle (Leukosome) | 108 | - | Decrease macrophage proliferation and proinflammatory cytokines | [87] |
| Rapamycin | Lipid nanocapsule (LNCs) | 110 | 69 | mTORC1 signaling inhibition | [80] |
| Rapamycin | P80-Par-PLGA-NPs or P80-CLD-PLGA-NPs | 110 | 69 | Anti-glioma activity | [88] |
| Rapamycin | NP-conjugated pericardial patche | 370 | 86 | Reduction of smooth muscle cell proliferation | [89] |
| Rapamycin | PLGA-LTTHYKL peptide | 122–130 | 88–91 | Phospho-S6 Inhibition | [90] |
| Sirolimus | Cholesterol-PEG-NH2 or Cholesterol-PEG-amine | 12–14 | 77–82 | Scleral permeation and retention | [91] |
| Sirolimus | D-α-tocopheryl polyethylene glycol succinate (TPGS) | 11 | 97 | Improve oral absorption | [92] |
| Rapamycin | O-octanoyl-chitosan-polyethylene glycol (OChiPEG) | 44 | 86 | Scleral permeation and retention | [93] |
| Rapamycin | Pluronic block copolymer | - | - | Increase solubility and oral administration, enhance absorption | [94] |
| Sirolimus | Polymeric nanoparticle (PNP) | 35–38 | - | Increase radiosensitivity | [95] |
| Rapamycin +Tacrolimus | Poly(ethylene glycol)-poly(pro-pylene sulfide) (PEG-PPS) | 39 | 41 | Allograft survival | [96] |
| Rapamycin | Poly(ethylene glycol)-block-poly(2-methyl-2-benzoxycar-bonyl-propylene carbonate) (PEG-b-PBC) | 66–76 | 15–88 | Toxicity reduction | [97] |
| Rapamycin | PLGA | 180 | 88 | Reduce neointimal hyperplasia | [98] |
| Rapamycin + Paclitaxel | Methoxyl-poly(ethylene glycol)-succinic acid | 56–94 | - | Reduce multi-drug resistance | [99] |
| Rapamycin + Piperine | PLGA | 150 | - | Improve oral bioavailability | [100] |
| Rapamycin + Cisplatin | PLGA | 12–75 | 93 | Alteration of tumor microenvironment | [101] |
| 17-AAG + Paclitaxel | PEG-PLA | 37–44 | - | Increase apoptosis | [102] |
| Therapy Category | Therapeutic Class | Therapeutic Agent | EV Source | EV Type | Packaging Method | Effects | References |
|---|---|---|---|---|---|---|---|
| Small molecules | mTOR inhibitor | Rapamycin | MSC | Small EV | Ultra sonication | Autoimmune response inhibition | [108] |
| Sirolimus | Fibroblast | Exosome | Electroporation and ultra sonication | Arterial restenosis inhibition | [109] | ||
| Rapamycin | Macrophage | Exosome | Extrusion (EB-AM) | Reduce proliferation and induce apoptosis | [107] | ||
| Rapamycin | 4T1-breast cancer | Exosome | Co-culture of 4T1-with Rapamycin | Increase M1 marker and decrease M2 marker expression | [106] | ||
| Chemotherapy | Doxorubicin | MSC | Exosome | Electroporation | Tumor growth inhibition | [110] | |
| Paclitaxel | PC-3 | Exosome | Co-culture | Induce cytotoxicity | [111] | ||
| Cisplantin | Macrophage | Exosome | Co-culture | Tumor growth inhibition | [112] | ||
| Curcumin | PANC-1 | Exosome | Co-culture | Induce apoptosis | [113] | ||
| TMZ | Glioma cell | Exosome | Co-culture | Tumor growth inhibition | [103] | ||
| Camptothecin | 4T1 | Apoptotic body | Co-culture | Tumor growth inhibition | [114] | ||
| Kinase inhibitor | TGFβRI | FBS | Exosome | Electroporation | Tumor growth inhibition | [115] | |
| Immune inhibitor | TLR7/8 | FBS | Exosome | Electroporation | Tumor growth inhibition | [115] | |
| Lapatinib | MCF10A | Exosome | Electroporation | T cell activation | [116] | ||
| CpG | EL4 | Apoptotic body | Co-culture | Prevent tumor metastasis and recurrence | [117] | ||
| cGAMP | Breast cancer cell | Apoptotic body | Active loading | STING activation and antigen representation | [118] | ||
| Antibodies | A33Ab | LIM125 | Exosome | Co-culture | Tumor targeting | [119] | |
| MHC, CD86, αCD3, αEGFR | DC | Exosome | Co-culture | Tumor growth inhibition | [120] | ||
| CD3, CD28 | HEK293T | Exosome | Transfection | T cell activation | [121] | ||
| RNA | miRNA | miR-138-5p | ADSCs | Exosome | Transduction | Tumor growth inhibition | [122] |
| miR-497 | HEK293T | Exosome | Transfection | Tumor growth inhibition | [123] | ||
| miR-199a | AMSC | Exosome | Transduction | Doxycycline sensitivity | [124] | ||
| miR-146b | MSC | Exosome | Electroporation | Tumor growth inhibition | [125] | ||
| miR-21 | HEK293T | Exosome | Electroporation | Tumor growth inhibition | [126] | ||
| siRNA | siS100A4 | Breast cancer cell | Exosome | Co-culture and Extrusion | Tumor growth inhibition | [127] | |
| siSTAT3 | RAW | Exosome | Ultra-sonication | Tumor growth inhibition | [128] | ||
| siCDK1 | Sk-hep1 | EV | Electroporation | Tumor growth inhibition | [129] | ||
| mRNA | PTEN | MEF and DC | Exosome | Nanaoporation | Tumor growth inhibition | [130] | |
| UPRT | HEK293 | Microvesicle | Co-culture | Tumor growth inhibition | [131] | ||
| Anti-sense | STAT6 | HEK293/M2 macrophage | Exosome | Co-incubation | Tumor growth inhibition | [132] | |
| Gene editing tool | CRISPR-Cas9 | CRISPR-PARP1 | SKOV3 | Exosome | Electroporation | Induce apoptosis | [133] |
| CRISPR-WNT10B | HEK293 | EV | Ultra-sonication | Tumor growth inhibition | [134] | ||
| Protein | Transferrin receptor binding peptide | MDA-MB-231 | Exosome | Co-incubation | Tumor growth inhibition | [126] | |
| Tlyp-1 | M1 macrophage | Exosome | Co-incubation | Tumor growth inhibition | [135] | ||
| CD63/EGFR | M1 macrophage | EV | Electroporation | Tumor growth inhibition | [136] | ||
| Combination | CPPO/Ce6/Dox-EMCH | THLG-HEK293 | EV | Electroporation | Induce drug sensitivity | [137] | |
| Dox/Cho-miR-159 | THP-15 | Exosome | Co-incubation | Tumor growth inhibition | [138] | ||
| 5-FU/miR21 | HEK293 | Exosome | Co-incubation | Tumor growth inhibition | [139] | ||
| CPT-SS-PR104A | HEK293 | Exosome | Co-incubation | Tumor growth inhibition | [114] | ||
| siGPX4/Fe3O4 | Tumor cell | Apoptotic body | Active loading | Tumor growth inhibition | [140] | ||
| Drugs | Registration No. | Stage | Disease Type | Target | Status |
|---|---|---|---|---|---|
| Afatinib Dasatinib Palbociclib Everolimus Olaparib | NCT05432518 | Early Phase I | Glioblastoma Recurrent disease Recurrent glioblastoma | mTOR and Tyrosine kinase | Not yet recruiting |
| AZD2014 | NCT02619864 | I | Glioblastoma multiforme | mTOR | Completed |
| AZD8055 | NCT01316809 | I | Glioblastoma multiform Anaplastic astrocytoma Anaplastic oligodendroglioma Malignant glioma Brain stem glioma | mTOR | Completed |
| XL765 (SAR245409) | NCT00704080 | I | Mixed gliomas Malignant gliomas Glioblastoma multiforme | Dual PI3K and mTOR | Completed |
| Everolimus Temozolomide | NCT00387400 | I | Brain and central nervous system Tumors | mTOR | Completed |
| XL765 (SAR245409) XL147 (SAR245408) | NCT01240460 | I | Glioma Glioblastoma Astrocytoma grade IV | Dual PI3K and mTOR | Completed |
| CC-115 | NCT01353625 | I | Glioblastoma multiforme | Dual pan-PI3K and mTOR | Completed |
| DEC-205/NY-ESO-1 fusion protein CDX-1401 Sirolimus | NCT01522820 | I | Glioblastoma Anaplastic astrocytoma | mTOR | Completed |
| GDC-0084 | NCT03696355 | I | Brain and central nervous system Tumors | Dual PI3K and mTOR | Active, Not recruiting |
| RMC-5552 | NCT05557292 | I | Glioblastoma Recurrent glioblastoma | mTOR | Not yet recruiting |
| MLN0128 | NCT02142803 | I | Adult glioblastoma | mTOR | Active, Not recruiting |
| Perifosine Temsirolimus | NCT02238496 | I | Brain tumor, Recurrent glioblastoma Anaplastic astrocytoma Anaplastic oligodendroglioma Mixed glioma | Dual Akt and mTOR | Active, Not recruiting |
| PQR309 | NCT02850744 | II | Glioblastoma multiforme | Dual pan-PI3K and mTOR | Terminated |
| Everolimus | NCT00515086 | II | Glioblastoma multiforme | mTOR | Terminated |
| CC-223 | NCT01177397 | I/II | Multiple myeloma Diffuse large B-Cell lymphoma Glioblastoma multiforme Hepatocellular carcinoma Non-small cell lung cancer Neuroendocrine tumors of non-pancreatic origin Hormone receptor-positive breast cancer | Dual PI3K and mTOR | Completed |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Singh, S.; Barik, D.; Lawrie, K.; Mohapatra, I.; Prasad, S.; Naqvi, A.R.; Singh, A.; Singh, G. Unveiling Novel Avenues in mTOR-Targeted Therapeutics: Advancements in Glioblastoma Treatment. Int. J. Mol. Sci. 2023, 24, 14960. https://doi.org/10.3390/ijms241914960
Singh S, Barik D, Lawrie K, Mohapatra I, Prasad S, Naqvi AR, Singh A, Singh G. Unveiling Novel Avenues in mTOR-Targeted Therapeutics: Advancements in Glioblastoma Treatment. International Journal of Molecular Sciences. 2023; 24(19):14960. https://doi.org/10.3390/ijms241914960
Chicago/Turabian StyleSingh, Shilpi, Debashis Barik, Karl Lawrie, Iteeshree Mohapatra, Sujata Prasad, Afsar R. Naqvi, Amar Singh, and Gatikrushna Singh. 2023. "Unveiling Novel Avenues in mTOR-Targeted Therapeutics: Advancements in Glioblastoma Treatment" International Journal of Molecular Sciences 24, no. 19: 14960. https://doi.org/10.3390/ijms241914960
APA StyleSingh, S., Barik, D., Lawrie, K., Mohapatra, I., Prasad, S., Naqvi, A. R., Singh, A., & Singh, G. (2023). Unveiling Novel Avenues in mTOR-Targeted Therapeutics: Advancements in Glioblastoma Treatment. International Journal of Molecular Sciences, 24(19), 14960. https://doi.org/10.3390/ijms241914960