
Three-Dimensional Gene Regulation Network in Glioblastoma Ferroptosis

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
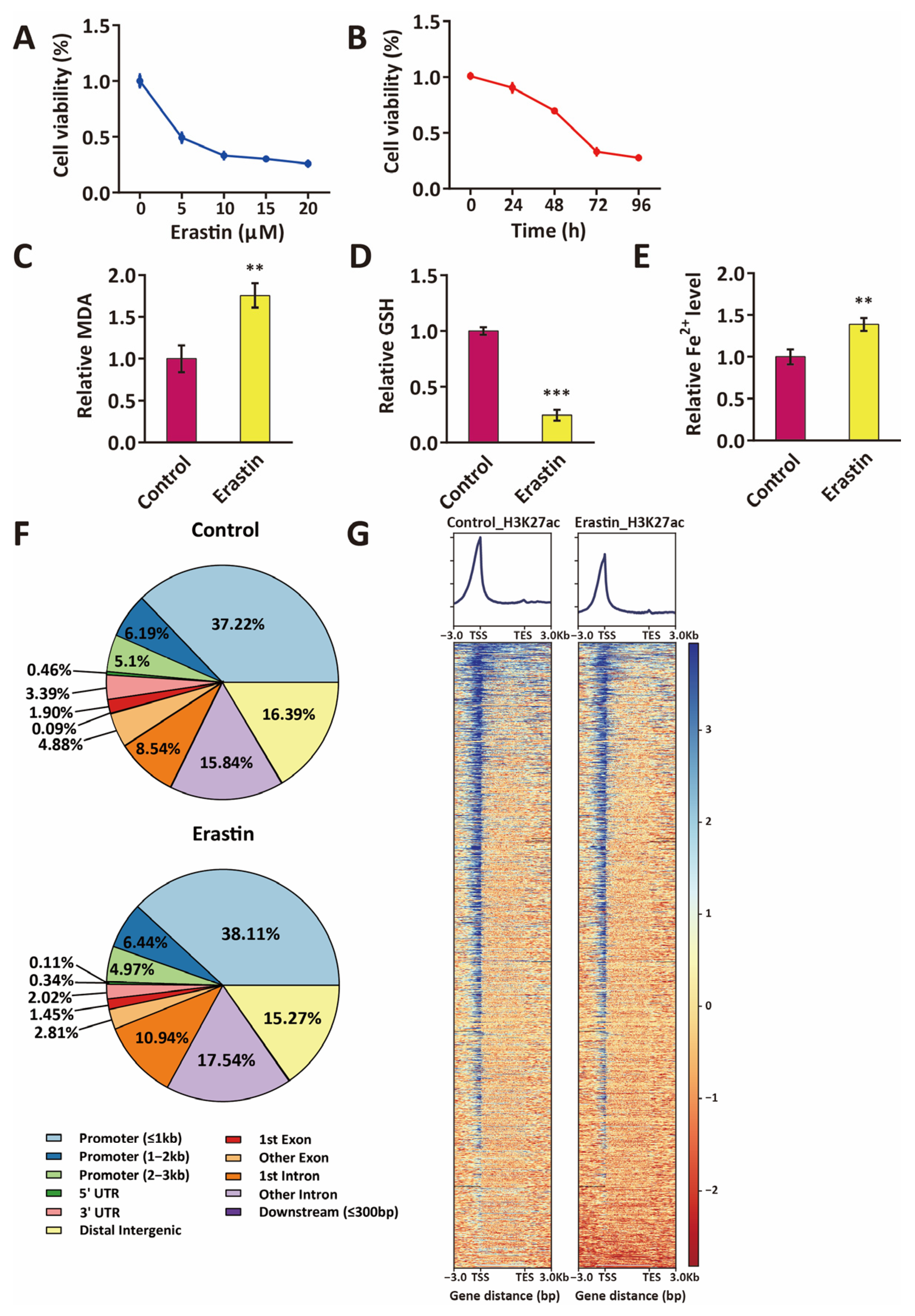
2.1. Distribution of Active Enhancers in Ferroptotic Glioblastoma Cells
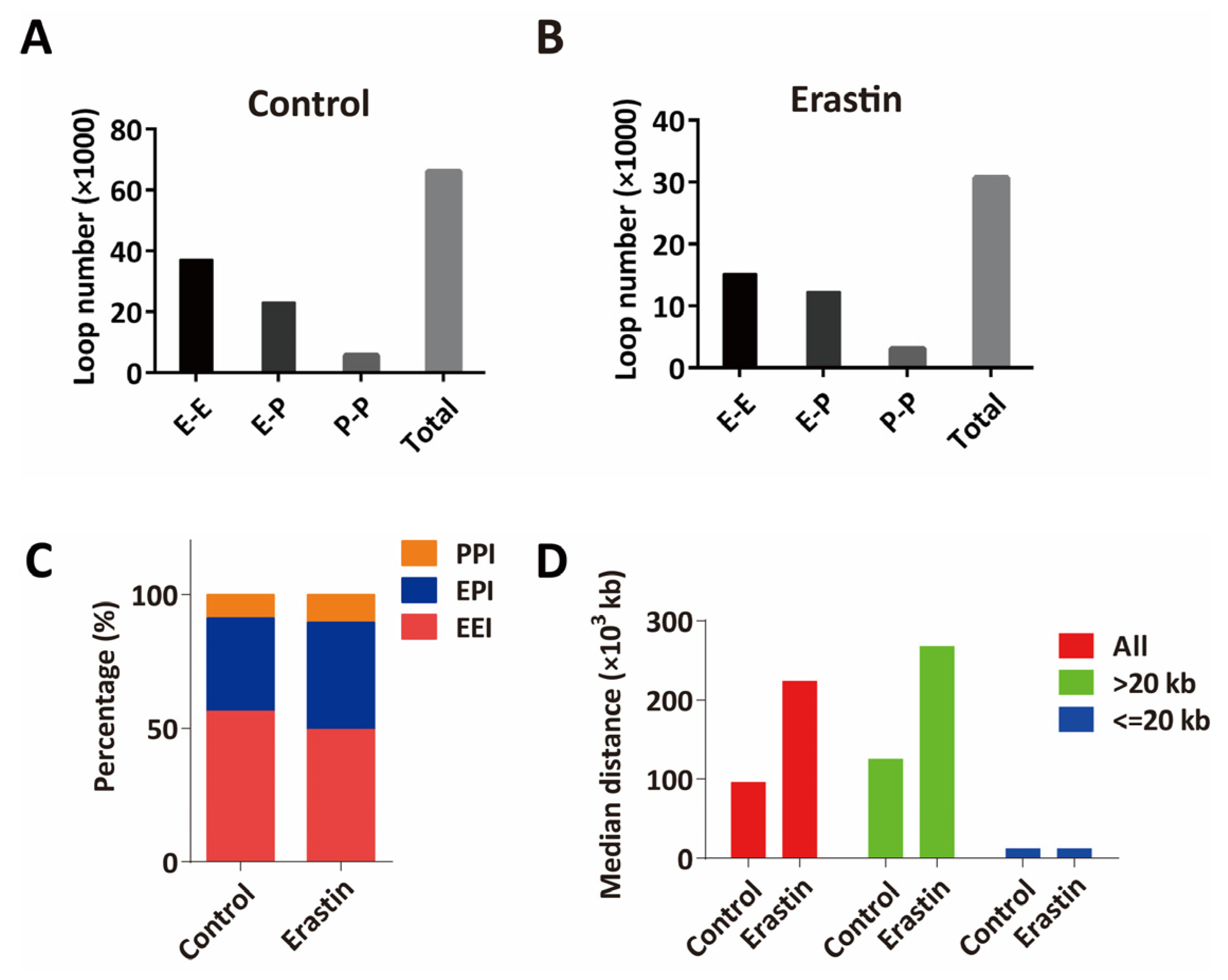
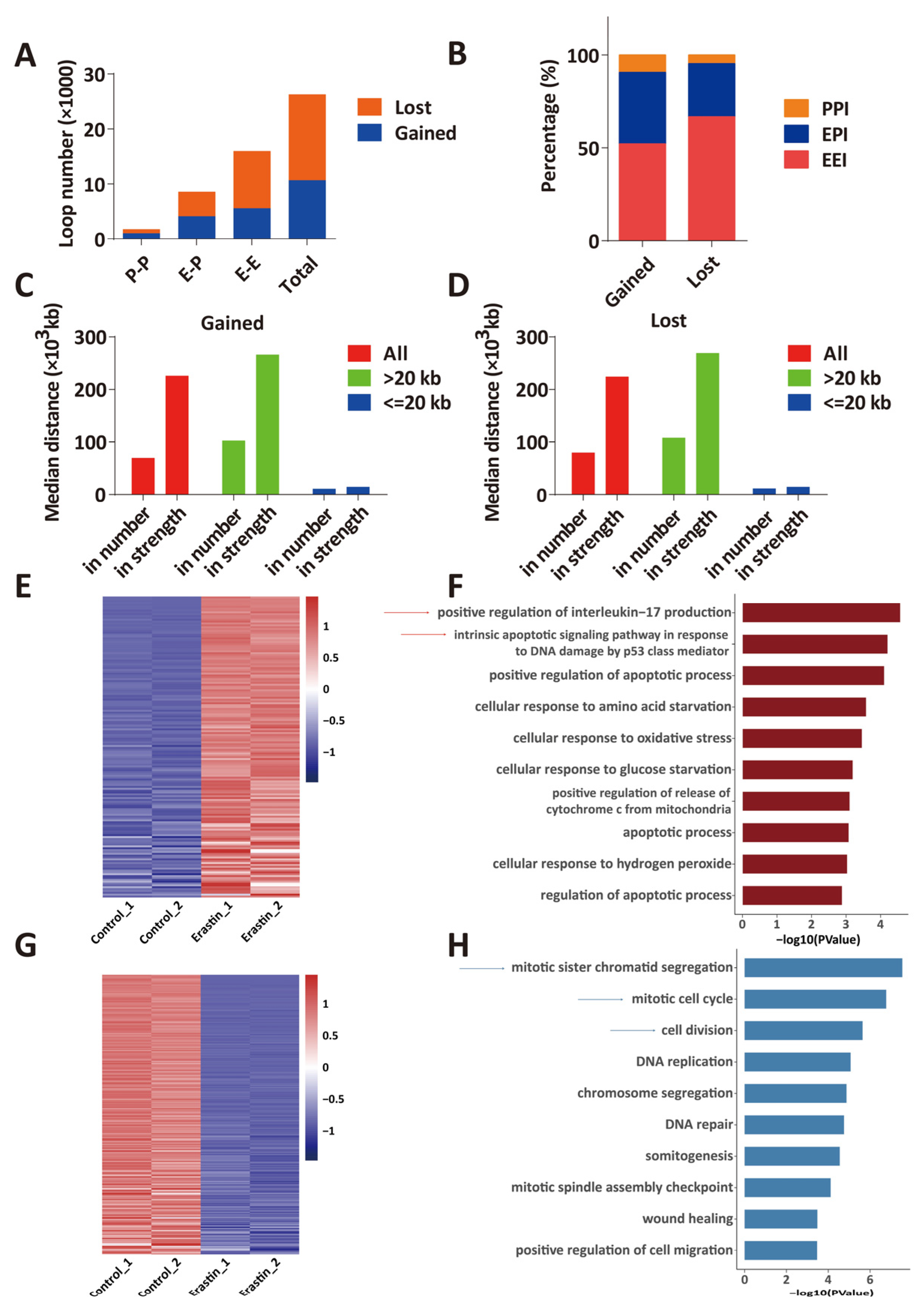
2.2. HiChIP Identifies the Change of 3D Chromatin Structure in Glioblastoma Cell Ferroptosis
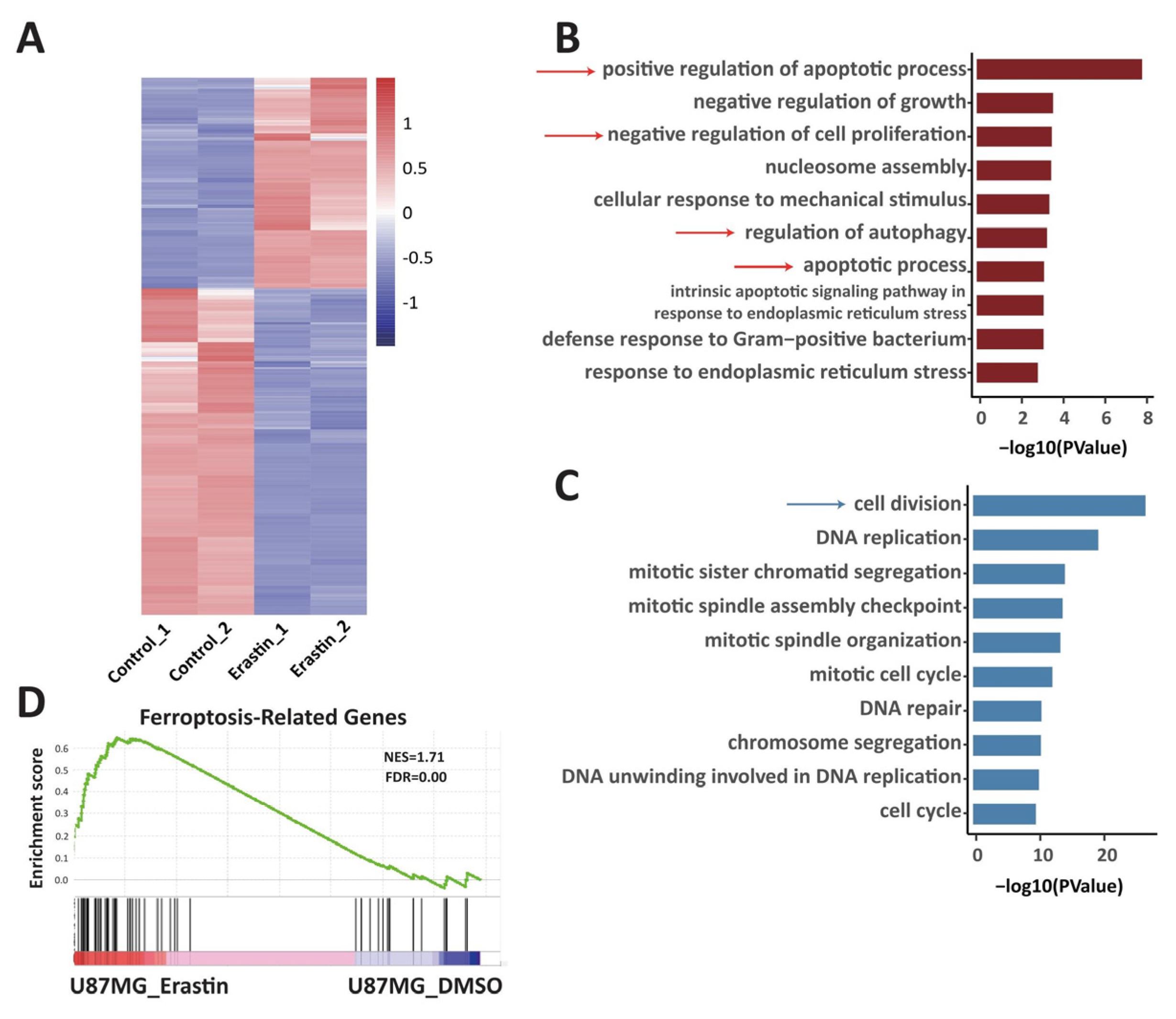
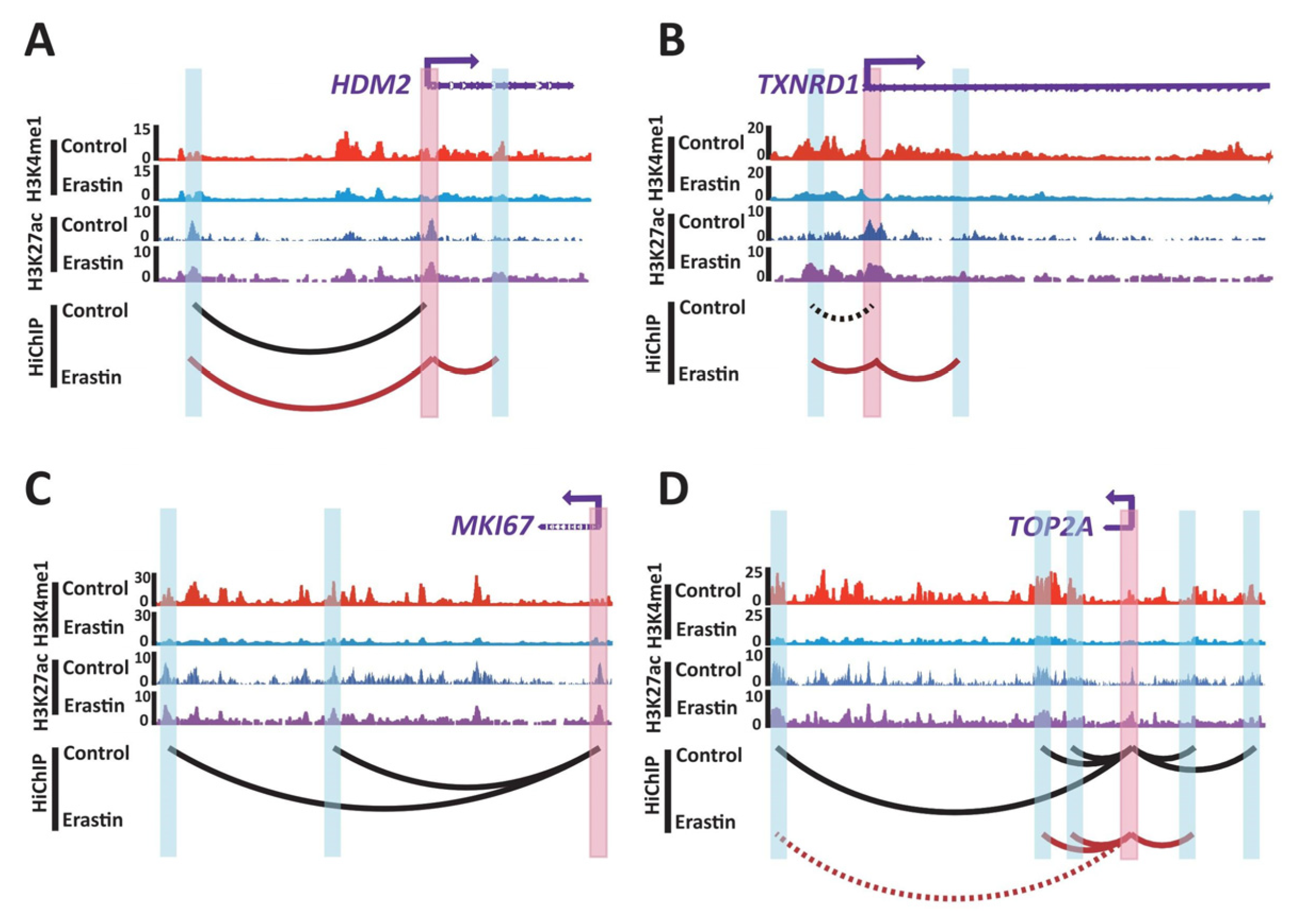
2.3. Transcriptome Change in Ferroptotic Glioblastoma Cells Is Associated with Changes of 3D Chromatin Structure
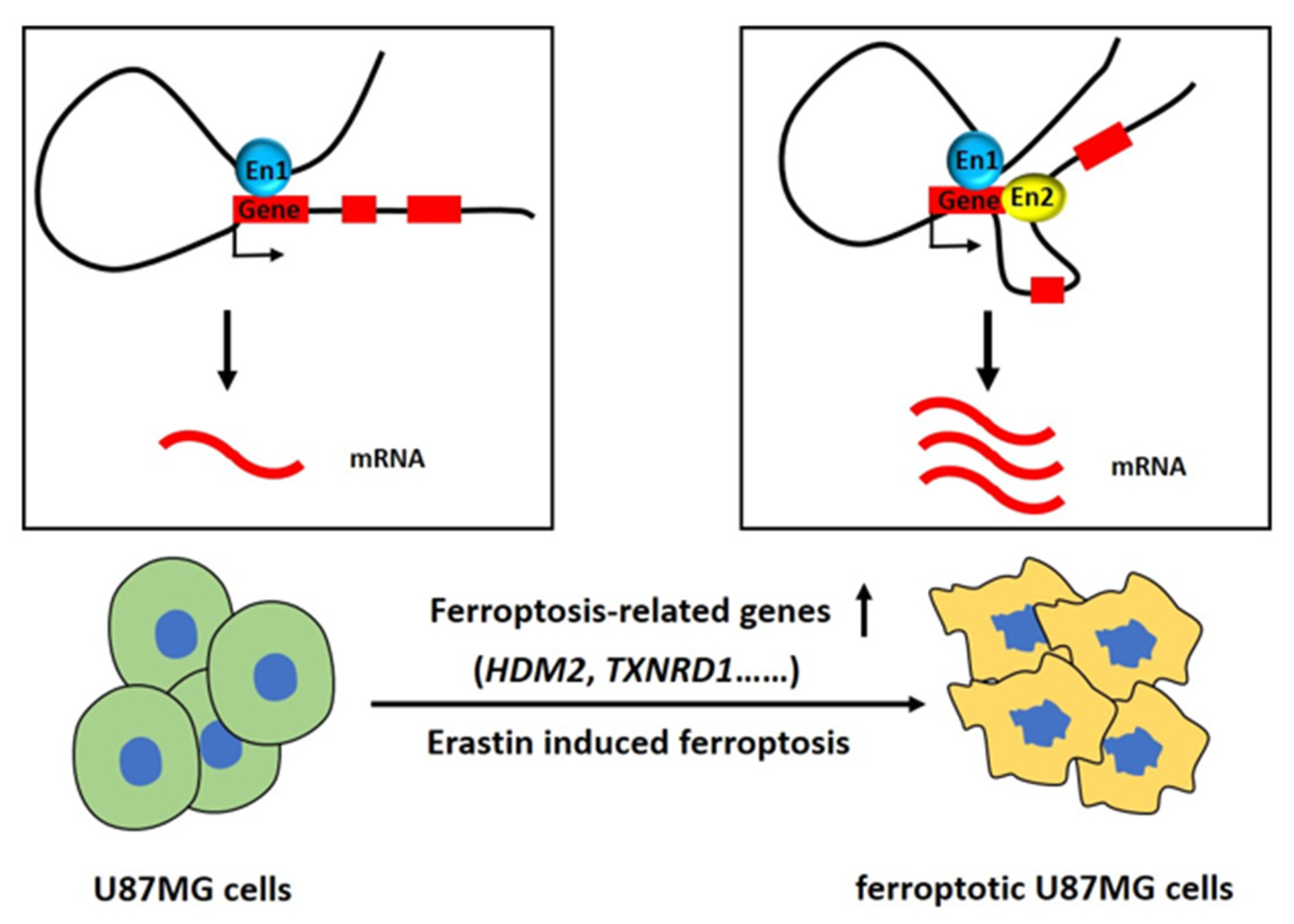
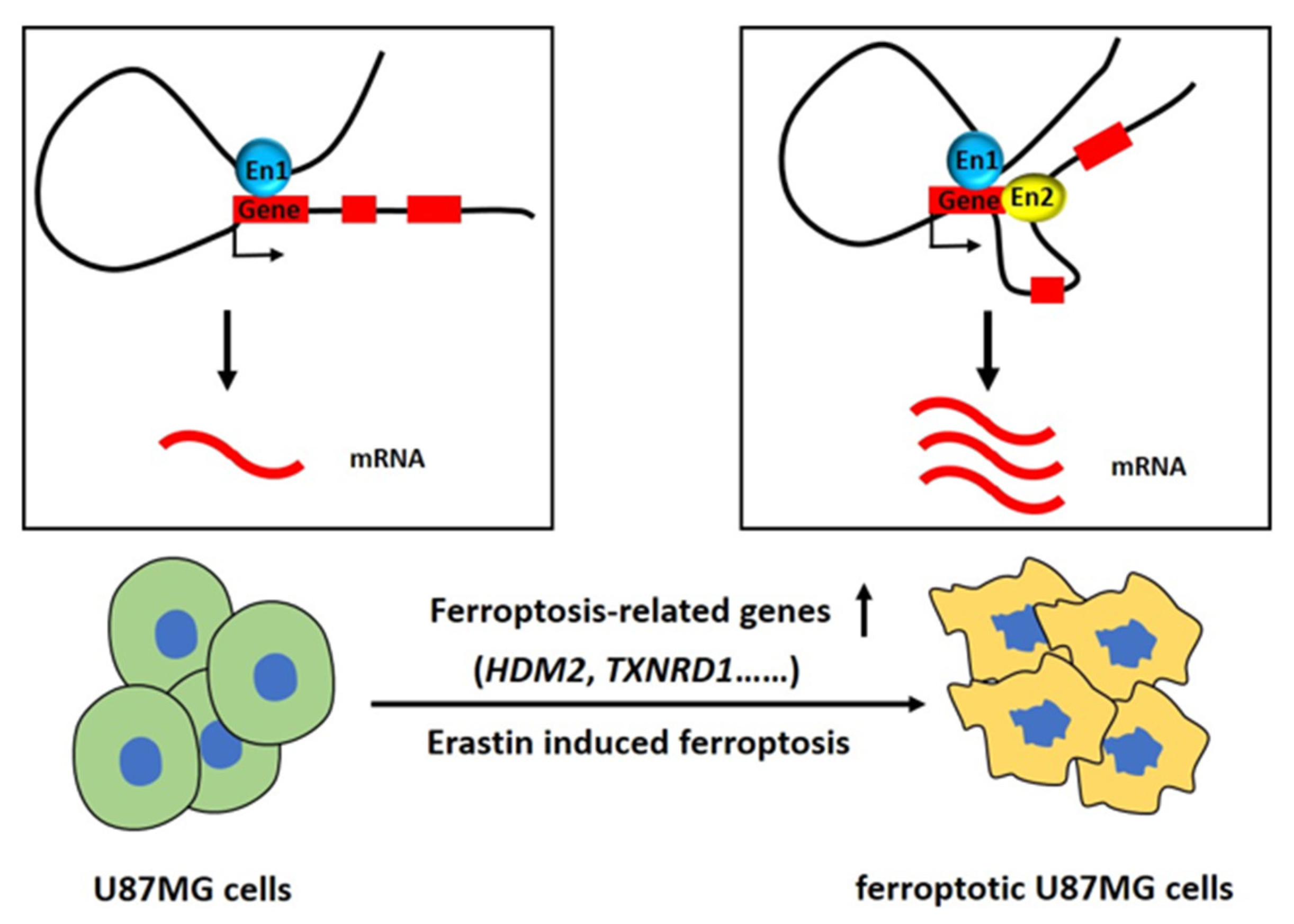
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. MTS Assay
4.3. Lipid Peroxidation Assay
4.4. Glutathione and Iron Assay
4.5. Western Blot
4.6. RNA-Seq
4.7. ChIP-Seq
4.8. HiChIP
4.9. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Weller, M.; Wick, W.; Aldape, K.; Brada, M.; Berger, M.; Pfister, S.M.; Nishikawa, R.; Rosenthal, M.; Wen, P.Y.; Stupp, R.; et al. Glioma. Nat. Rev. Dis. Primers 2015, 1, 15017. [Google Scholar] [CrossRef] [PubMed]
- Koneru, T.; McCord, E.; Pawar, S.; Tatiparti, K.; Sau, S.; Iyer, A.K. Transferrin: Biology and Use in Receptor-Targeted Nanotherapy of Gliomas. ACS Omega 2021, 6, 8727–8733. [Google Scholar] [CrossRef]
- Reifenberger, G.; Wirsching, H.G.; Knobbe-Thomsen, C.B.; Weller, M. Advances in the molecular genetics of gliomas—Implications for classification and therapy. Nat. Rev. Clin. Oncol. 2017, 14, 434–452. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Kang, R.; Kroemer, G.; Tang, D. Broadening horizons: The role of ferroptosis in cancer. Nat. Rev. Clin. Oncol. 2021, 18, 280–296. [Google Scholar] [CrossRef] [PubMed]
- Hassannia, B.; Vandenabeele, P.; Vanden Berghe, T. Targeting Ferroptosis to Iron Out Cancer. Cancer Cell 2019, 35, 830–849. [Google Scholar] [CrossRef]
- Mitre, A.O.; Florian, A.I.; Buruiana, A.; Boer, A.; Moldovan, I.; Soritau, O.; Florian, S.I.; Susman, S. Ferroptosis Involvement in Glioblastoma Treatment. Medicina 2022, 58, 319. [Google Scholar] [CrossRef]
- Belaghzal, H.; Dekker, J.; Gibcus, J.H. Hi-C 2.0: An optimized Hi-C procedure for high-resolution genome-wide mapping of chromosome conformation. Methods 2017, 123, 56–65. [Google Scholar] [CrossRef]
- Dekker, J.; Rippe, K.; Dekker, M.; Kleckner, N. Capturing chromosome conformation. Science 2002, 295, 1306–1311. [Google Scholar] [CrossRef]
- Mumbach, M.R.; Rubin, A.J.; Flynn, R.A.; Dai, C.; Khavari, P.A.; Greenleaf, W.J.; Chang, H.Y. HiChIP: Efficient and sensitive analysis of protein-directed genome architecture. Nat. Methods 2016, 13, 919–922. [Google Scholar] [CrossRef]
- Flavahan, W.A.; Drier, Y.; Liau, B.B.; Gillespie, S.M.; Venteicher, A.S.; Stemmer-Rachamimov, A.O.; Suvà, M.L.; Bernstein, B.E. Insulator dysfunction and oncogene activation in IDH mutant gliomas. Nature 2016, 529, 110–114. [Google Scholar] [CrossRef]
- Ahn, J.H.; Davis, E.S.; Daugird, T.A.; Zhao, S.; Quiroga, I.Y.; Uryu, H.; Li, J.; Storey, A.J.; Tsai, Y.H.; Keeley, D.P.; et al. Phase separation drives aberrant chromatin looping and cancer development. Nature 2021, 595, 591–595. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Lee, D.S.; Chandran, S.; Le, V.T.; Bump, R.; Yasis, J.; Dallarda, S.; Marcotte, S.; Clock, B.; Haghani, N.; et al. Structural variants drive context-dependent oncogene activation in cancer. Nature 2022, 612, 564–572. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Gu, Z.; Li, Z.; Yuan, Z.; Zhao, Y.; Zheng, X.; Bo, X.; Chen, H.; Wang, C. Dynamic Interplay between Structural Variations and 3D Genome Organization in Pancreatic Cancer. Adv. Sci. 2022, 9, e2200818. [Google Scholar] [CrossRef]
- Debruyne, D.N.; Dries, R.; Sengupta, S.; Seruggia, D.; Gao, Y.; Sharma, B.; Huang, H.; Moreau, L.; McLane, M.; Day, D.S.; et al. BORIS promotes chromatin regulatory interactions in treatment-resistant cancer cells. Nature 2019, 572, 676–680. [Google Scholar] [CrossRef] [PubMed]
- Deng, S.; Feng, Y.; Pauklin, S. 3D chromatin architecture and transcription regulation in cancer. J. Hematol. Oncol. 2022, 15, 49. [Google Scholar] [CrossRef] [PubMed]
- Costa, I.; Barbosa, D.J.; Benfeito, S.; Silva, V.; Chavarria, D.; Borges, F.; Remião, F.; Silva, R. Molecular mechanisms of ferroptosis and their involvement in brain diseases. Pharmacol. Ther. 2023, 244, 108373. [Google Scholar] [CrossRef]
- Creyghton, M.P.; Cheng, A.W.; Welstead, G.G.; Kooistra, T.; Carey, B.W.; Steine, E.J.; Hanna, J.; Lodato, M.A.; Frampton, G.M.; Sharp, P.A.; et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. USA 2010, 107, 21931–21936. [Google Scholar] [CrossRef]
- Bogdanovic, O.; Fernandez-Miñán, A.; Tena, J.J.; de la Calle-Mustienes, E.; Hidalgo, C.; van Kruysbergen, I.; van Heeringen, S.J.; Veenstra, G.J.; Gómez-Skarmeta, J.L. Dynamics of enhancer chromatin signatures mark the transition from pluripotency to cell specification during embryogenesis. Genome Res. 2012, 22, 2043–2053. [Google Scholar] [CrossRef]
- Chen, X.; Song, X.; Li, J.; Zhang, R.; Yu, C.; Zhou, Z.; Liu, J.; Liao, S.; Klionsky, D.J.; Kroemer, G.; et al. Identification of HPCAL1 as a specific autophagy receptor involved in ferroptosis. Autophagy 2023, 19, 54–74. [Google Scholar] [CrossRef]
- Li, J.; Liu, J.; Xu, Y.; Wu, R.; Chen, X.; Song, X.; Zeh, H.; Kang, R.; Klionsky, D.J.; Wang, X.; et al. Tumor heterogeneity in autophagy-dependent ferroptosis. Autophagy 2021, 17, 3361–3374. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wu, D.; Fu, Q.; Hao, S.; Gu, Y.; Zhao, W.; Chen, S.; Sheng, F.; Xu, Y.; Chen, Z.; et al. CHAC1 as a Novel Contributor of Ferroptosis in Retinal Pigment Epithelial Cells with Oxidative Damage. Int. J. Mol. Sci. 2023, 24, 1582. [Google Scholar] [CrossRef] [PubMed]
- Li, J.Y.; Ren, C.; Wang, L.X.; Yao, R.Q.; Dong, N.; Wu, Y.; Tian, Y.P.; Yao, Y.M. Sestrin2 protects dendrite cells against ferroptosis induced by sepsis. Cell Death Dis. 2021, 12, 834. [Google Scholar] [CrossRef] [PubMed]
- Dang, X.; Huan, X.; Du, X.; Chen, X.; Bi, M.; Yan, C.; Jiao, Q.; Jiang, H. Correlation of Ferroptosis and Other Types of Cell Death in Neurodegenerative Diseases. Neurosci. Bull. 2022, 38, 938–952. [Google Scholar] [CrossRef]
- Gao, M.; Monian, P.; Pan, Q.; Zhang, W.; Xiang, J.; Jiang, X. Ferroptosis is an autophagic cell death process. Cell Res. 2016, 26, 1021–1032. [Google Scholar] [CrossRef]
- Hou, W.; Xie, Y.; Song, X.; Sun, X.; Lotze, M.T.; Zeh, H.J., 3rd; Kang, R.; Tang, D. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 2016, 12, 1425–1428. [Google Scholar] [CrossRef]
- Zhuo, S.; Chen, Z.; Yang, Y.; Zhang, J.; Tang, J.; Yang, K. Clinical and Biological Significances of a Ferroptosis-Related Gene Signature in Glioma. Front. Oncol. 2020, 10, 590861. [Google Scholar] [CrossRef]
- Guo, B.; Zuo, Z.; Di, X.; Huang, Y.; Gong, G.; Xu, B.; Wang, L.; Zhang, X.; Liang, Z.; Hou, Y.; et al. Salidroside attenuates HALI via IL-17A-mediated ferroptosis of alveolar epithelial cells by regulating Act1-TRAF6-p38 MAPK pathway. Cell Commun. Signal. CCS 2022, 20, 183. [Google Scholar] [CrossRef]
- Panatta, E.; Zampieri, C.; Melino, G.; Amelio, I. Understanding p53 tumour suppressor network. Biol. Direct 2021, 16, 14. [Google Scholar] [CrossRef]
- Venkatesh, D.; O'Brien, N.A.; Zandkarimi, F.; Tong, D.R.; Stokes, M.E.; Dunn, D.E.; Kengmana, E.S.; Aron, A.T.; Klein, A.M.; Csuka, J.M.; et al. MDM2 and MDMX promote ferroptosis by PPARα-mediated lipid remodeling. Genes Dev. 2020, 34, 526–543. [Google Scholar] [CrossRef]
- Nguyen, P.; Awwad, R.T.; Smart, D.D.; Spitz, D.R.; Gius, D. Thioredoxin reductase as a novel molecular target for cancer therapy. Cancer Lett. 2006, 236, 164–174. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Wang, H.; Yang, X.; Wu, Q.; An, P.; Jin, X.; Liu, W.; Huang, X.; Li, Y.; Yan, S.; et al. Auranofin mitigates systemic iron overload and induces ferroptosis via distinct mechanisms. Signal Transduct. Target. Ther. 2020, 5, 138. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Wu, W.; Chen, Q.; Zheng, Z.; Jiang, X.; Xue, Y.; Lin, D. TXNRD1: A Key Regulator Involved in the Ferroptosis of CML Cells Induced by Cysteine Depletion In Vitro. Oxidative Med. Cell. Longev. 2021, 2021, 7674565. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhang, M.; Gan, H.; Wang, H.; Lee, J.H.; Fang, D.; Kitange, G.J.; He, L.; Hu, Z.; Parney, I.F.; et al. A novel enhancer regulates MGMT expression and promotes temozolomide resistance in glioblastoma. Nat. Commun. 2018, 9, 2949. [Google Scholar] [CrossRef] [PubMed]
- Qin, S.; Yuan, Y.; Huang, X.; Tan, Z.; Hu, X.; Liu, H.; Pu, Y.; Ding, Y.Q.; Su, Z.; He, C. Topoisomerase IIA in adult NSCs regulates SVZ neurogenesis by transcriptional activation of Usp37. Nucleic Acids Res. 2022, 50, 9319–9338. [Google Scholar] [CrossRef]
- Thakurela, S.; Garding, A.; Jung, J.; Schübeler, D.; Burger, L.; Tiwari, V.K. Gene regulation and priming by topoisomerase IIα in embryonic stem cells. Nat. Commun. 2013, 4, 2478. [Google Scholar] [CrossRef]
- Zhang, Y.W.; Wang, M.B.; Li, S.C. SuperTAD: Robust detection of hierarchical topologically associated domains with optimized structural information. Genome Biol. 2021, 22, 45. [Google Scholar] [CrossRef]
- Li, L.X.; Zhou, J.X.; Wang, X.; Zhang, H.; Harris, P.C.; Calvet, J.P.; Li, X. Cross-talk between CDK4/6 and SMYD2 regulates gene transcription, tubulin methylation, and ciliogenesis. Sci. Adv. 2020, 6, eabb3154. [Google Scholar] [CrossRef]
- Wang, H.; Yang, J.; Zhang, Y.; Qian, J.; Wang, J. Reconstruct high-resolution 3D genome structures for diverse cell-types using FLAMINGO. Nat. Commun. 2022, 13, 2645. [Google Scholar] [CrossRef]
- Qian, Y.; Zhang, L.; Cai, M.; Li, H.; Xu, H.; Yang, H.; Zhao, Z.; Rhie, S.K.; Farnham, P.J.; Shi, J.; et al. The prostate cancer risk variant rs55958994 regulates multiple gene expression through extreme long-range chromatin interaction to control tumor progression. Sci. Adv. 2019, 5, eaaw6710. [Google Scholar] [CrossRef]
- Feng, Y.; Cai, L.; Hong, W.; Zhang, C.; Tan, N.; Wang, M.; Wang, C.; Liu, F.; Wang, X.; Ma, J.; et al. Rewiring of 3D Chromatin Topology Orchestrates Transcriptional Reprogramming and the Development of Human Dilated Cardiomyopathy. Circulation 2022, 145, 1663–1683. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Huang, K.; Hu, G.; Babarinde, I.A.; Li, Y.; Dong, X.; Chen, Y.S.; Shang, L.; Guo, W.; Wang, J.; et al. An alternative CTCF isoform antagonizes canonical CTCF occupancy and changes chromatin architecture to promote apoptosis. Nat. Commun. 2019, 10, 1535. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.Y.; Erkek, S.; Tong, Y.; Yin, L.; Federation, A.J.; Zapatka, M.; Haldipur, P.; Kawauchi, D.; Risch, T.; Warnatz, H.J.; et al. Active medulloblastoma enhancers reveal subgroup-specific cellular origins. Nature 2016, 530, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Ing-Simmons, E.; Seitan, V.C.; Faure, A.J.; Flicek, P.; Carroll, T.; Dekker, J.; Fisher, A.G.; Lenhard, B.; Merkenschlager, M. Spatial enhancer clustering and regulation of enhancer-proximal genes by cohesin. Genome Res. 2015, 25, 504–513. [Google Scholar] [CrossRef]
- Bahr, C.; von Paleske, L.; Uslu, V.V.; Remeseiro, S.; Takayama, N.; Ng, S.W.; Murison, A.; Langenfeld, K.; Petretich, M.; Scognamiglio, R.; et al. A Myc enhancer cluster regulates normal and leukaemic haematopoietic stem cell hierarchies. Nature 2018, 553, 515–520. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Liu, Y.; Liu, S.; Zhu, X.; Wu, L.; Zhu, Y.; Zhao, D.; Xu, X.; Chemparathy, A.; Wang, H.; et al. Nested epistasis enhancer networks for robust genome regulation. Science 2022, 377, 1077–1085. [Google Scholar] [CrossRef]
- Miguel-Escalada, I.; Bonàs-Guarch, S.; Cebola, I.; Ponsa-Cobas, J.; Mendieta-Esteban, J.; Atla, G.; Javierre, B.M.; Rolando, D.M.Y.; Farabella, I.; Morgan, C.C.; et al. Human pancreatic islet three-dimensional chromatin architecture provides insights into the genetics of type 2 diabetes. Nat. Genet. 2019, 51, 1137–1148. [Google Scholar] [CrossRef]
- Lou, J.; Hao, Y.; Lin, K.; Lyu, Y.; Chen, M.; Wang, H.; Zou, D.; Jiang, X.; Wang, R.; Jin, D.; et al. Circular RNA CDR1as disrupts the p53/MDM2 complex to inhibit Gliomagenesis. Mol. Cancer 2020, 19, 138. [Google Scholar] [CrossRef]
- Levine, A.J. p53: 800 million years of evolution and 40 years of discovery. Nat. Rev. Cancer 2020, 20, 471–480. [Google Scholar] [CrossRef]
- Rayburn, E.R.; Ezell, S.J.; Zhang, R. Recent advances in validating MDM2 as a cancer target. Anti-Cancer Agents Med. Chem. 2009, 9, 882–903. [Google Scholar] [CrossRef]
- Rayburn, E.; Zhang, R.; He, J.; Wang, H. MDM2 and human malignancies: Expression, clinical pathology, prognostic markers, and implications for chemotherapy. Curr. Cancer Drug Targets 2005, 5, 27–41. [Google Scholar] [CrossRef] [PubMed]
- Karni-Schmidt, O.; Lokshin, M.; Prives, C. The Roles of MDM2 and MDMX in Cancer. Annu. Rev. Pathol. 2016, 11, 617–644. [Google Scholar] [CrossRef]
- Wade, M.; Li, Y.C.; Wahl, G.M. MDM2, MDMX and p53 in oncogenesis and cancer therapy. Nat. Rev. Cancer 2013, 13, 83–96. [Google Scholar] [CrossRef] [PubMed]
- Bohlman, S.; Manfredi, J.J. p53-independent effects of Mdm2. Sub-Cell. Biochem. 2014, 85, 235–246. [Google Scholar] [CrossRef]
- Nag, S.; Qin, J.; Srivenugopal, K.S.; Wang, M.; Zhang, R. The MDM2-p53 pathway revisited. J. Biomed. Res. 2013, 27, 254–271. [Google Scholar] [CrossRef]
- Chen, X.; Tai, L.; Gao, J.; Qian, J.; Zhang, M.; Li, B.; Xie, C.; Lu, L.; Lu, W.; Lu, W. A stapled peptide antagonist of MDM2 carried by polymeric micelles sensitizes glioblastoma to temozolomide treatment through p53 activation. J. Control. Release Off. J. Control. Release Soc. 2015, 218, 29–35. [Google Scholar] [CrossRef]
- Shi, C.; Rao, C.; Sun, C.; Yu, L.; Zhou, X.; Hua, D.; Wang, R.; Luo, W.; Jiang, Z.; Zhou, J.; et al. miR-29s function as tumor suppressors in gliomas by targeting TRAF4 and predict patient prognosis. Cell Death Dis. 2018, 9, 1078. [Google Scholar] [CrossRef]
- Jiang, Y.; Wang, Z.; Ying, C.; Hu, J.; Zeng, T.; Gao, L. FMR1/circCHAF1A/miR-211-5p/HOXC8 feedback loop regulates proliferation and tumorigenesis via MDM2-dependent p53 signaling in GSCs. Oncogene 2021, 40, 4094–4110. [Google Scholar] [CrossRef]
- Cheff, D.M.; Huang, C.; Scholzen, K.C.; Gencheva, R.; Ronzetti, M.H.; Cheng, Q.; Hall, M.D.; Arnér, E.S.J. The ferroptosis inducing compounds RSL3 and ML162 are not direct inhibitors of GPX4 but of TXNRD1. Redox Biol. 2023, 62, 102703. [Google Scholar] [CrossRef]
- Cadenas, C.; Franckenstein, D.; Schmidt, M.; Gehrmann, M.; Hermes, M.; Geppert, B.; Schormann, W.; Maccoux, L.J.; Schug, M.; Schumann, A.; et al. Role of thioredoxin reductase 1 and thioredoxin interacting protein in prognosis of breast cancer. Breast Cancer Res. BCR 2010, 12, R44. [Google Scholar] [CrossRef]
- Lincoln, D.T.; Ali Emadi, E.M.; Tonissen, K.F.; Clarke, F.M. The thioredoxin-thioredoxin reductase system: Over-expression in human cancer. Anticancer Res. 2003, 23, 2425–2433. [Google Scholar] [PubMed]
- Bhatia, M.; McGrath, K.L.; Di Trapani, G.; Charoentong, P.; Shah, F.; King, M.M.; Clarke, F.M.; Tonissen, K.F. The thioredoxin system in breast cancer cell invasion and migration. Redox Biol. 2016, 8, 68–78. [Google Scholar] [CrossRef]
- Leone, A.; Roca, M.S.; Ciardiello, C.; Costantini, S.; Budillon, A. Oxidative Stress Gene Expression Profile Correlates with Cancer Patient Poor Prognosis: Identification of Crucial Pathways Might Select Novel Therapeutic Approaches. Oxidative Med. Cell. Longev. 2017, 2017, 2597581. [Google Scholar] [CrossRef]
- Guo, W.; Wu, Z.; Chen, J.; Guo, S.; You, W.; Wang, S.; Ma, J.; Wang, H.; Wang, X.; Wang, H.; et al. Nanoparticle delivery of miR-21-3p sensitizes melanoma to anti-PD-1 immunotherapy by promoting ferroptosis. J. Immunother. Cancer 2022, 10, e004381. [Google Scholar] [CrossRef] [PubMed]
- Youssef, L.A.; Rebbaa, A.; Pampou, S.; Weisberg, S.P.; Stockwell, B.R.; Hod, E.A.; Spitalnik, S.L. Increased erythrophagocytosis induces ferroptosis in red pulp macrophages in a mouse model of transfusion. Blood 2018, 131, 2581–2593. [Google Scholar] [CrossRef] [PubMed]
- Koh, E.K.; Ryu, B.K.; Jeong, D.Y.; Bang, I.S.; Nam, M.H.; Chae, K.S. A 60-Hz sinusoidal magnetic field induces apoptosis of prostate cancer cells through reactive oxygen species. Int. J. Radiat. Biol. 2008, 84, 945–955. [Google Scholar] [CrossRef]
- Guachalla, L.M.; Rudolph, K.L. ROS induced DNA damage and checkpoint responses: Influences on aging? Cell Cycle 2010, 9, 4058–4060. [Google Scholar] [CrossRef]
- Ishikawa, K.; Takenaga, K.; Akimoto, M.; Koshikawa, N.; Yamaguchi, A.; Imanishi, H.; Nakada, K.; Honma, Y.; Hayashi, J. ROS-generating mitochondrial DNA mutations can regulate tumor cell metastasis. Science 2008, 320, 661–664. [Google Scholar] [CrossRef]
- He, L.; Lai, H.; Chen, T. Dual-function nanosystem for synergetic cancer chemo-/radiotherapy through ROS-mediated signaling pathways. Biomaterials 2015, 51, 30–42. [Google Scholar] [CrossRef]
- Chen, L.; Chou, C.L.; Knepper, M.A. Targeted Single-Cell RNA-seq Identifies Minority Cell Types of Kidney Distal Nephron. J. Am. Soc. Nephrol. 2021, 32, 886–896. [Google Scholar] [CrossRef]
- Sohlberg, E.; Pfefferle, A.; Heggernes Ask, E.; Tschan-Plessl, A.; Jacobs, B.; Netskar, H.; Lorenz, S.; Kanaya, M.; Kosugi-Kanaya, M.; Meinke, S.; et al. Perturbed NK-cell homeostasis associated with disease severity in chronic neutropenia. Blood 2022, 139, 704–716. [Google Scholar] [CrossRef] [PubMed]
- Lévy, P.; Vidaud, D.; Leroy, K.; Laurendeau, I.; Wechsler, J.; Bolasco, G.; Parfait, B.; Wolkenstein, P.; Vidaud, M.; Bièche, I. Molecular profiling of malignant peripheral nerve sheath tumors associated with neurofibromatosis type 1, based on large-scale real-time RT-PCR. Mol. Cancer 2004, 3, 20. [Google Scholar] [CrossRef] [PubMed]
- Pabla, S.; Conroy, J.M.; Nesline, M.K.; Glenn, S.T.; Papanicolau-Sengos, A.; Burgher, B.; Hagen, J.; Giamo, V.; Andreas, J.; Lenzo, F.L.; et al. Proliferative potential and resistance to immune checkpoint blockade in lung cancer patients. J. Immunother. Cancer 2019, 7, 27. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Gong, K.; Yang, Y.; Wu, Q.; Wang, Q.; Shen, Y.; Xie, L.; Gong, Y.; Liu, H.; Liu, J. MKI67 as a potential diagnostic biomarker in pulmonary hypertension. Front. Pediatr. 2022, 10, 1016889. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.Y.; Cao, W.W.; Li, L.; Li, S.P.; Liu, T.; Wan, H.Y.; Liu, M.; Li, X.; Tang, H. MicroRNA-519d targets MKi67 and suppresses cell growth in the hepatocellular carcinoma cell line QGY-7703. Cancer Lett. 2011, 307, 182–190. [Google Scholar] [CrossRef]
- Lei, G.; Zhuang, L.; Gan, B. Targeting ferroptosis as a vulnerability in cancer. Nat. Rev. Cancer 2022, 22, 381–396. [Google Scholar] [CrossRef]
- Jiang, L.; Kon, N.; Li, T.; Wang, S.J.; Su, T.; Hibshoosh, H.; Baer, R.; Gu, W. Ferroptosis as a p53-mediated activity during tumour suppression. Nature 2015, 520, 57–62. [Google Scholar] [CrossRef]
- Yu, Q. Restoring p53-mediated apoptosis in cancer cells: New opportunities for cancer therapy. Drug Resist. Updates Rev. Comment. Antimicrob. Anticancer Chemother. 2006, 9, 19–25. [Google Scholar] [CrossRef]
- Su, W.; Huang, B.; Zhang, Q.; Han, W.; An, L.; Guan, Y.; Ji, J.; Yu, D. Exploring Potential Biomarkers, Ferroptosis Mechanisms, and Therapeutic Targets Associated with Cutaneous Squamous Cell Carcinoma via Integrated Transcriptomic Analysis. J. Healthc. Eng. 2022, 2022, 3524022. [Google Scholar] [CrossRef]
- Li, C.; Cui, X.; Li, Y.; Guo, D.; He, S. Identification of ferroptosis and drug resistance related hub genes to predict the prognosis in Hepatocellular Carcinoma. Sci. Rep. 2023, 13, 8681. [Google Scholar] [CrossRef]
- Yuan, F.; Sun, Q.; Zhang, S.; Ye, L.; Xu, Y.; Deng, G.; Xu, Z.; Zhang, S.; Liu, B.; Chen, Q. The dual role of p62 in ferroptosis of glioblastoma according to p53 status. Cell Biosci. 2022, 12, 20. [Google Scholar] [CrossRef] [PubMed]
- de Souza, I.; Monteiro, L.K.S.; Guedes, C.B.; Silva, M.M.; Andrade-Tomaz, M.; Contieri, B.; Latancia, M.T.; Mendes, D.; Porchia, B.; Lazarini, M.; et al. High levels of NRF2 sensitize temozolomide-resistant glioblastoma cells to ferroptosis via ABCC1/MRP1 upregulation. Cell Death Dis. 2022, 13, 591. [Google Scholar] [CrossRef] [PubMed]
- Song, Q.; Peng, S.; Sun, Z.; Heng, X.; Zhu, X. Temozolomide Drives Ferroptosis via a DMT1-Dependent Pathway in Glioblastoma Cells. Yonsei Med. J. 2021, 62, 843–849. [Google Scholar] [CrossRef] [PubMed]
- Fan, R.; Chen, C.; Mu, M.; Chuan, D.; Liu, H.; Hou, H.; Huang, J.; Tong, A.; Guo, G.; Xu, J. Engineering MMP-2 Activated Nanoparticles Carrying B7-H3 Bispecific Antibodies for Ferroptosis-Enhanced Glioblastoma Immunotherapy. ACS Nano 2023, 17, 9126–9139. [Google Scholar] [CrossRef]
- Zhang, J.; Han, L.; Wu, H.; Zhong, Y.; Shangguan, P.; Liu, Y.; He, M.; Sun, H.; Song, C.; Wang, X.; et al. A Brain-Targeting NIR-II Ferroptosis System: Effective Visualization and Oncotherapy for Orthotopic Glioblastoma. Adv. Sci. 2023, 10, e2206333. [Google Scholar] [CrossRef]
- Lang, X.; Green, M.D.; Wang, W.; Yu, J.; Choi, J.E.; Jiang, L.; Liao, P.; Zhou, J.; Zhang, Q.; Dow, A.; et al. Radiotherapy and Immunotherapy Promote Tumoral Lipid Oxidation and Ferroptosis via Synergistic Repression of SLC7A11. Cancer Discov. 2019, 9, 1673–1685. [Google Scholar] [CrossRef]
- Lei, G.; Zhang, Y.; Koppula, P.; Liu, X.; Zhang, J.; Lin, S.H.; Ajani, J.A.; Xiao, Q.; Liao, Z.; Wang, H.; et al. The role of ferroptosis in ionizing radiation-induced cell death and tumor suppression. Cell Res. 2020, 30, 146–162. [Google Scholar] [CrossRef]
- Ke, K.; Li, L.; Lu, C.; Zhu, Q.; Wang, Y.; Mou, Y.; Wang, H.; Jin, W. The crosstalk effect between ferrous and other ions metabolism in ferroptosis for therapy of cancer. Front. Oncol. 2022, 12, 916082. [Google Scholar] [CrossRef]
- Kim, Y.J.; Hyun, J. Mechanosensitive ion channels in apoptosis and ferroptosis: Focusing on the role of Piezo1. BMB Rep. 2023, 56, 145–152. [Google Scholar] [CrossRef]
- Zhao, Y.; Li, Y.; Zhang, R.; Wang, F.; Wang, T.; Jiao, Y. The Role of Erastin in Ferroptosis and Its Prospects in Cancer Therapy. OncoTargets Ther. 2020, 13, 5429–5441. [Google Scholar] [CrossRef]
- Brandalise, F.; Ratto, D.; Leone, R.; Olivero, F.; Roda, E.; Locatelli, C.A.; Grazia Bottone, M.; Rossi, P. Deeper and Deeper on the Role of BK and Kir4.1 Channels in Glioblastoma Invasiveness: A Novel Summative Mechanism? Front. Neurosci. 2020, 14, 595664. [Google Scholar] [CrossRef] [PubMed]
- Kantidze, O.L.; Gurova, K.V.; Studitsky, V.M.; Razin, S.V. The 3D Genome as a Target for Anticancer Therapy. Trends Mol. Med. 2020, 26, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, E.; Iwanaga, M. Comparison of bacteria in the tympanic cavity and the mastoid antrum in chronic otitis media. Am. J. Otolaryngol. 1986, 7, 298–301. [Google Scholar] [CrossRef]
- Morgan, S.L.; Mariano, N.C.; Bermudez, A.; Arruda, N.L.; Wu, F.; Luo, Y.; Shankar, G.; Jia, L.; Chen, H.; Hu, J.F.; et al. Manipulation of nuclear architecture through CRISPR-mediated chromosomal looping. Nat. Commun. 2017, 8, 15993. [Google Scholar] [CrossRef] [PubMed]
- Deng, W.; Rupon, J.W.; Krivega, I.; Breda, L.; Motta, I.; Jahn, K.S.; Reik, A.; Gregory, P.D.; Rivella, S.; Dean, A.; et al. Reactivation of developmentally silenced globin genes by forced chromatin looping. Cell 2014, 158, 849–860. [Google Scholar] [CrossRef] [PubMed]
- Deng, W.; Lee, J.; Wang, H.; Miller, J.; Reik, A.; Gregory, P.D.; Dean, A.; Blobel, G.A. Controlling long-range genomic interactions at a native locus by targeted tethering of a looping factor. Cell 2012, 149, 1233–1244. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Cai, M.; Liu, M.; Su, G.; An, D.; Moon, B.; Lyu, G.; Si, Y.; Chen, L.; Lu, W. lncRNA 5430416N02Rik Promotes the Proliferation of Mouse Embryonic Stem Cells by Activating Mid1 Expression through 3D Chromatin Architecture. Stem Cell Rep. 2020, 14, 493–505. [Google Scholar] [CrossRef]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq--a Python framework to work with high-throughput sequencing data. Bioinform. 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Mootha, V.K.; Lindgren, C.M.; Eriksson, K.F.; Subramanian, A.; Sihag, S.; Lehar, J.; Puigserver, P.; Carlsson, E.; Ridderstråle, M.; Laurila, E.; et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003, 34, 267–273. [Google Scholar] [CrossRef]
- Bi, J.; Wang, W.; Zhang, M.; Zhang, B.; Liu, M.; Su, G.; Chen, F.; Chen, B.; Shi, T.; Zheng, Y.; et al. KLF4 inhibits early neural differentiation of ESCs by coordinating specific 3D chromatin structure. Nucleic Acids Res. 2022, 50, 12235–12250. [Google Scholar] [CrossRef]
- Grytten, I.; Rand, K.D.; Nederbragt, A.J.; Storvik, G.O.; Glad, I.K.; Sandve, G.K. Graph Peak Caller: Calling ChIP-seq peaks on graph-based reference genomes. PLoS Comput. Biol. 2019, 15, e1006731. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, T.; Meyer, C.A.; Eeckhoute, J.; Johnson, D.S.; Bernstein, B.E.; Nusbaum, C.; Myers, R.M.; Brown, M.; Li, W.; et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008, 9, R137. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinform. 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Servant, N.; Varoquaux, N.; Lajoie, B.R.; Viara, E.; Chen, C.J.; Vert, J.P.; Heard, E.; Dekker, J.; Barillot, E. HiC-Pro: An optimized and flexible pipeline for Hi-C data processing. Genome Biol. 2015, 16, 259. [Google Scholar] [CrossRef]
- Lareau, C.A.; Aryee, M.J. hichipper: A preprocessing pipeline for calling DNA loops from HiChIP data. Nat. Methods 2018, 15, 155–156. [Google Scholar] [CrossRef]
- Robinson, J.T.; Turner, D.; Durand, N.C.; Thorvaldsdóttir, H.; Mesirov, J.P.; Aiden, E.L. Juicebox.js Provides a Cloud-Based Visualization System for Hi-C Data. Cell Syst. 2018, 6, 256–258.e251. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, M.; Wang, W.; Zhang, H.; Bi, J.; Zhang, B.; Shi, T.; Su, G.; Zheng, Y.; Fan, S.; Huang, X.; et al. Three-Dimensional Gene Regulation Network in Glioblastoma Ferroptosis. Int. J. Mol. Sci. 2023, 24, 14945. https://doi.org/10.3390/ijms241914945
Liu M, Wang W, Zhang H, Bi J, Zhang B, Shi T, Su G, Zheng Y, Fan S, Huang X, et al. Three-Dimensional Gene Regulation Network in Glioblastoma Ferroptosis. International Journal of Molecular Sciences. 2023; 24(19):14945. https://doi.org/10.3390/ijms241914945
Chicago/Turabian StyleLiu, Man, Wenbin Wang, Han Zhang, Jinfang Bi, Baoying Zhang, Tengfei Shi, Guangsong Su, Yaoqiang Zheng, Sibo Fan, Xiaofeng Huang, and et al. 2023. "Three-Dimensional Gene Regulation Network in Glioblastoma Ferroptosis" International Journal of Molecular Sciences 24, no. 19: 14945. https://doi.org/10.3390/ijms241914945
APA StyleLiu, M., Wang, W., Zhang, H., Bi, J., Zhang, B., Shi, T., Su, G., Zheng, Y., Fan, S., Huang, X., Chen, B., Song, Y., Zhao, Z., Shi, J., Li, P., Lu, W., & Zhang, L. (2023). Three-Dimensional Gene Regulation Network in Glioblastoma Ferroptosis. International Journal of Molecular Sciences, 24(19), 14945. https://doi.org/10.3390/ijms241914945