
Proteomics Investigation of the Impact of the Enterococcus faecalis Secretome on MCF-7 Tumor Cells

 , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Results
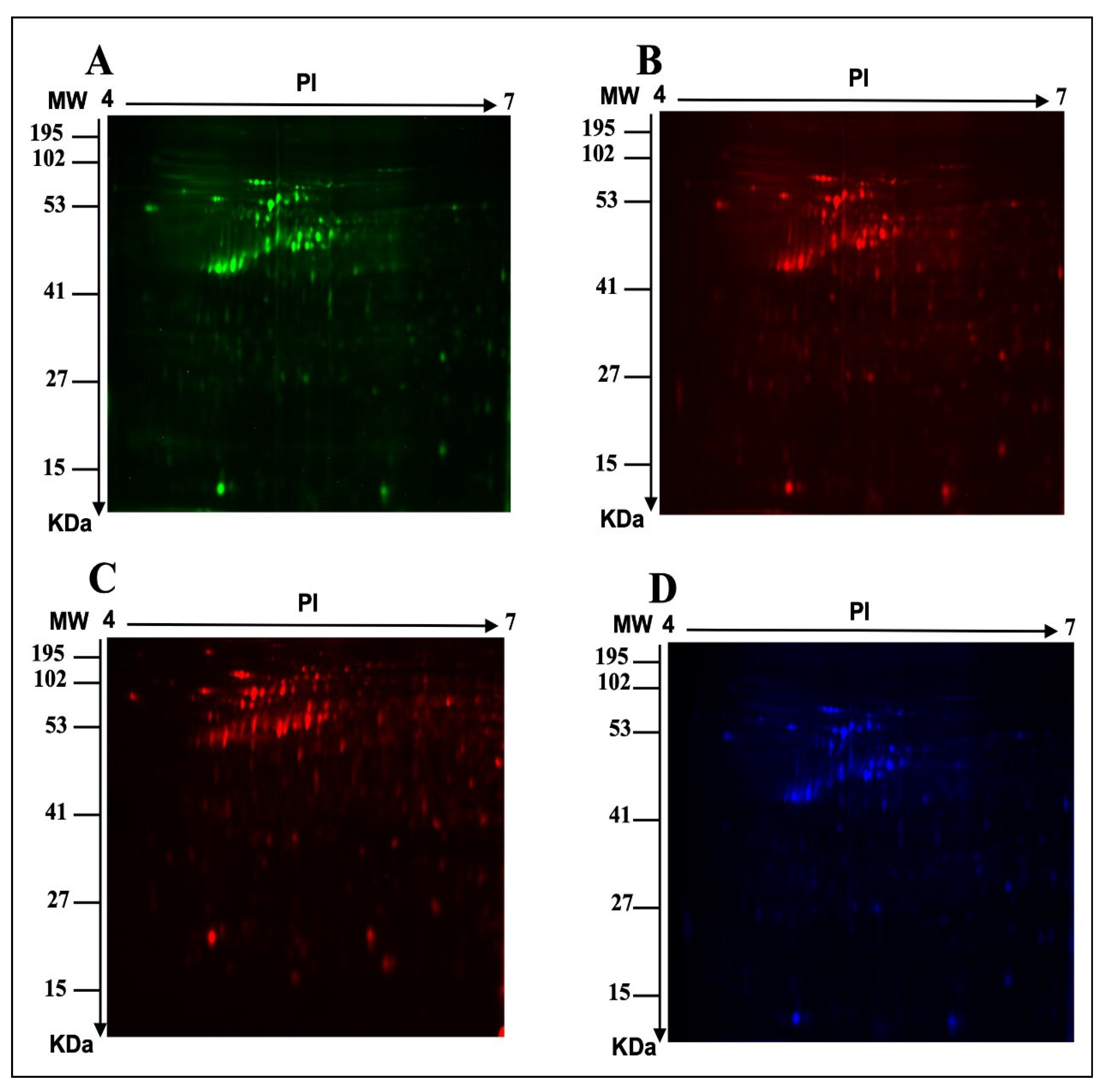
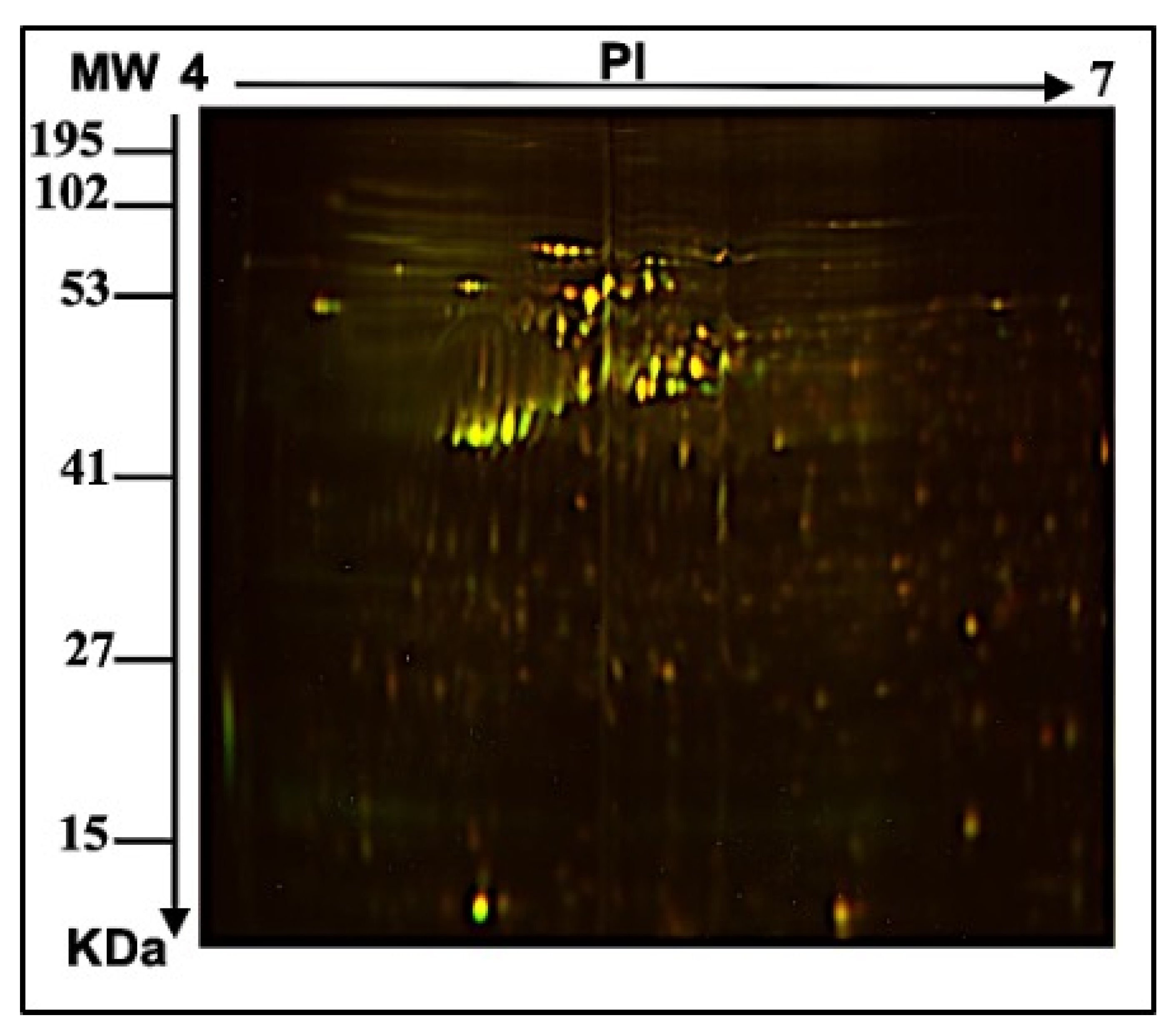
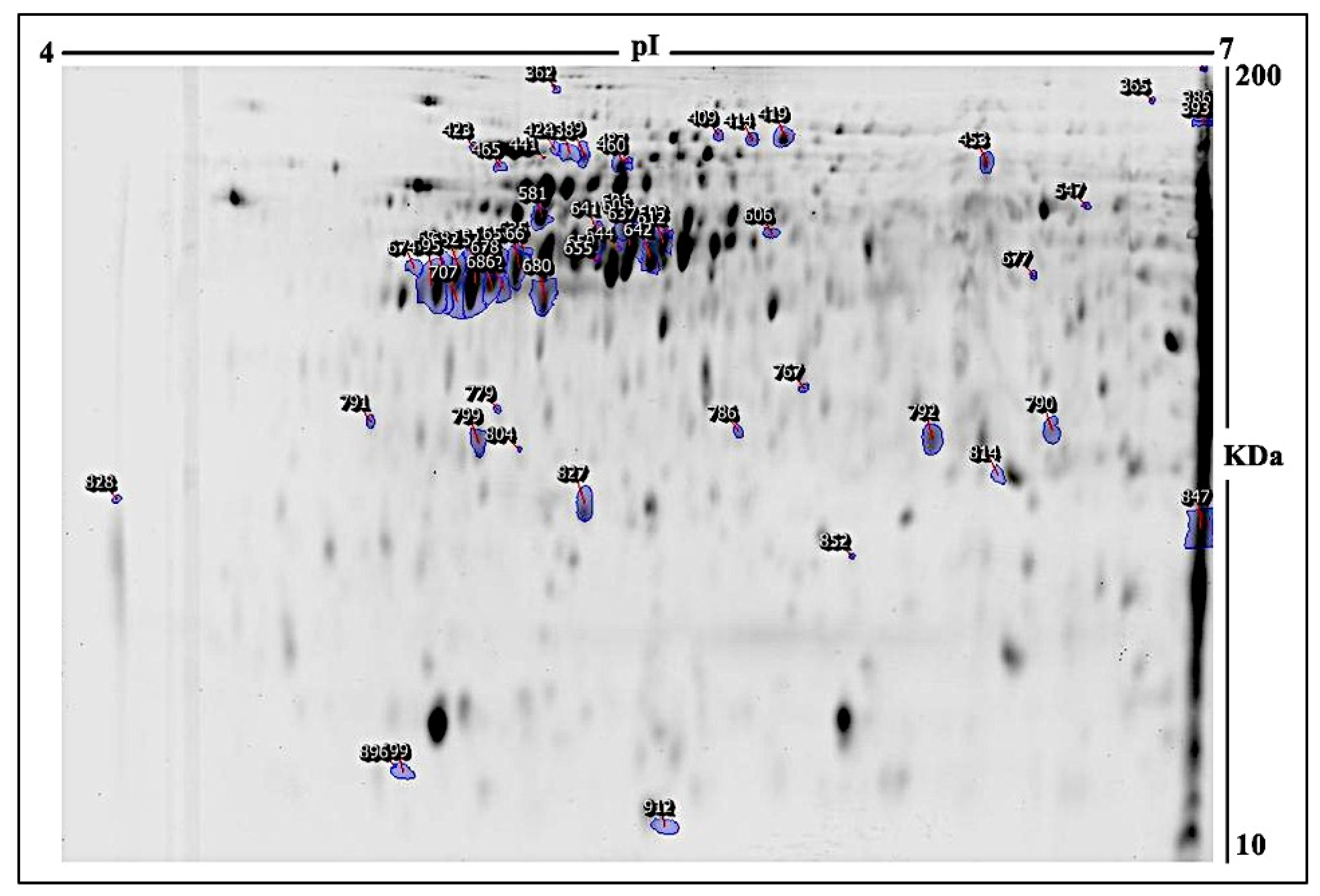
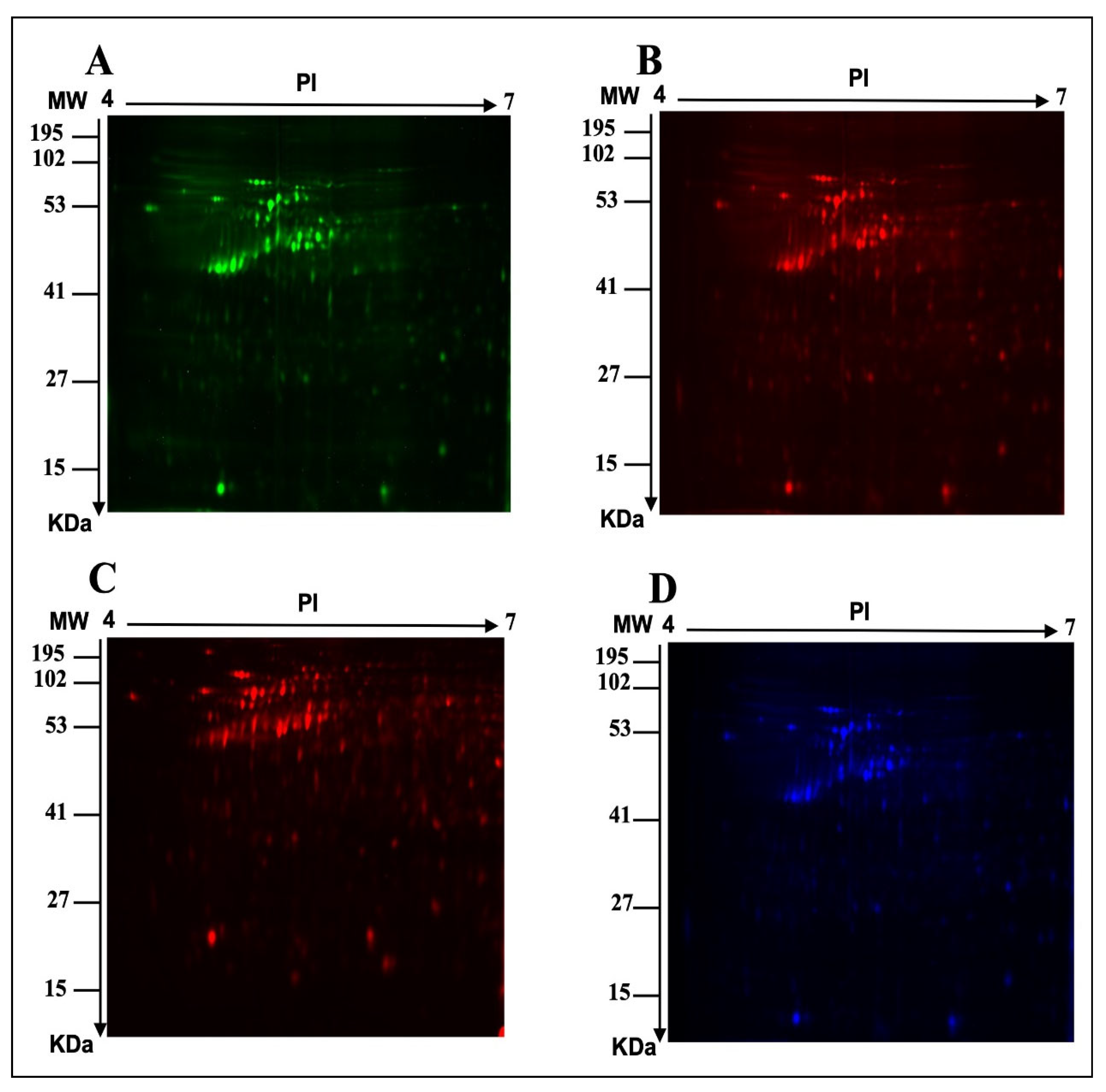
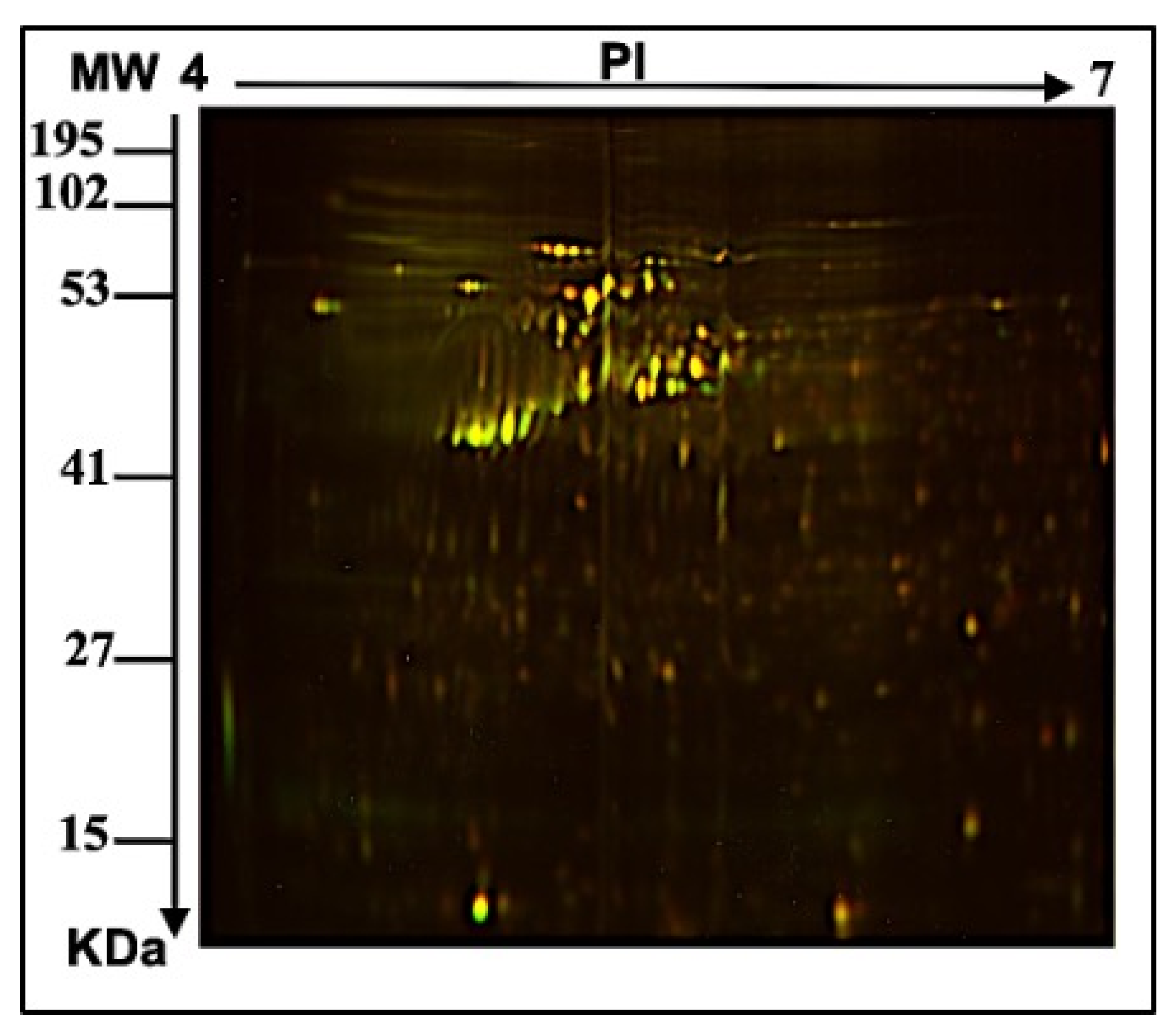
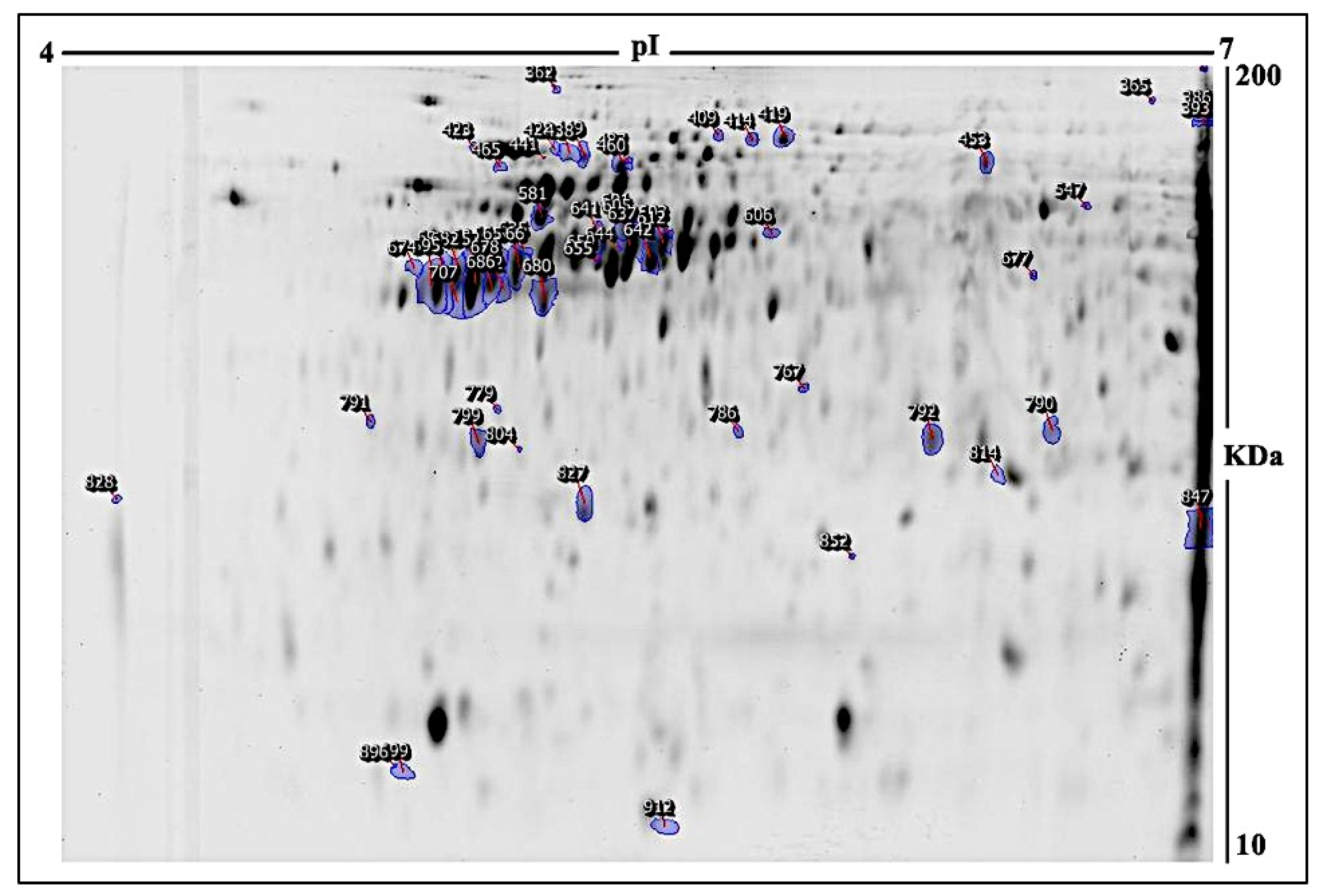
2.1. E. faecalis Supernatant Effects the Expression of MCF-7 Cell Line Proteins
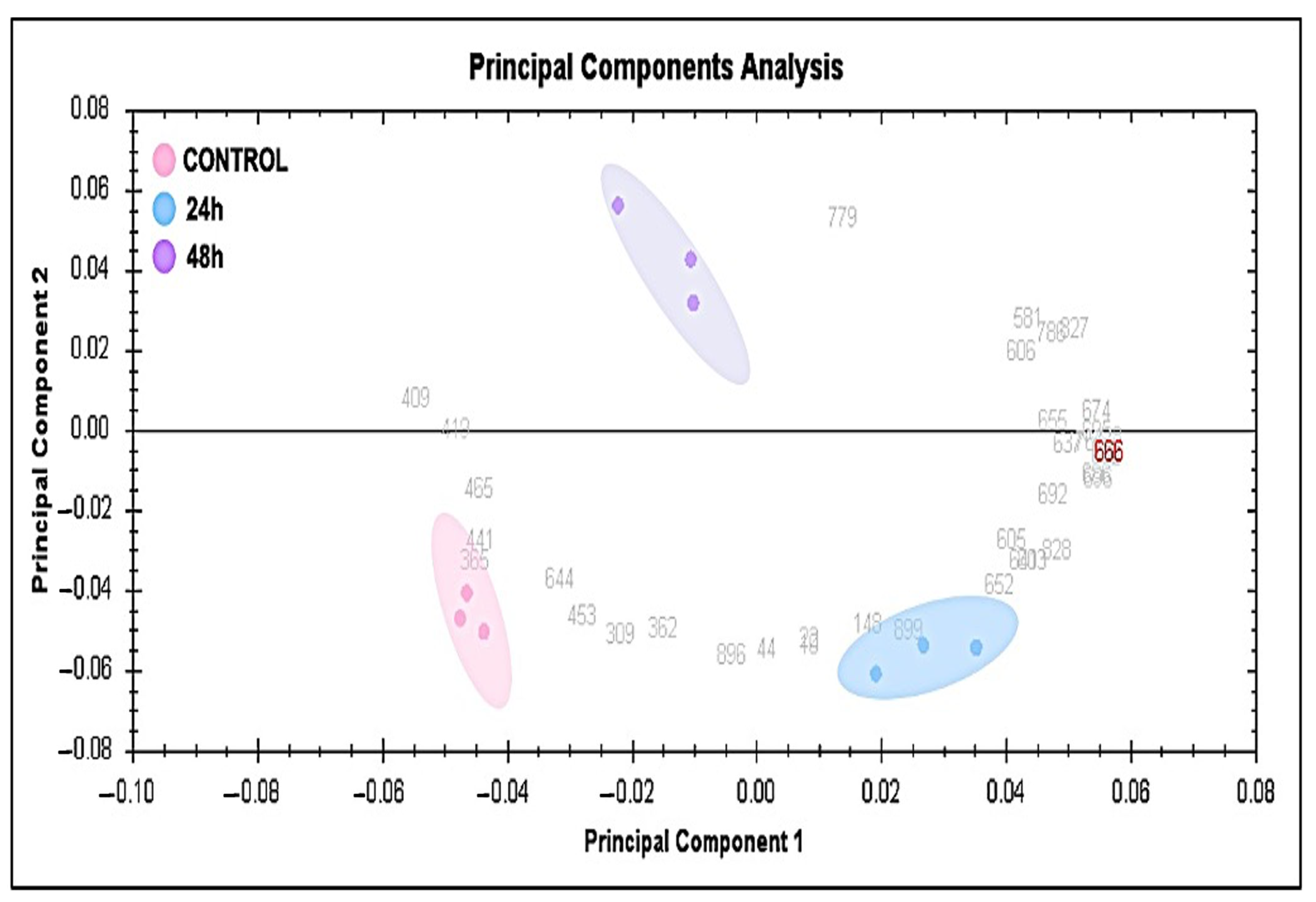
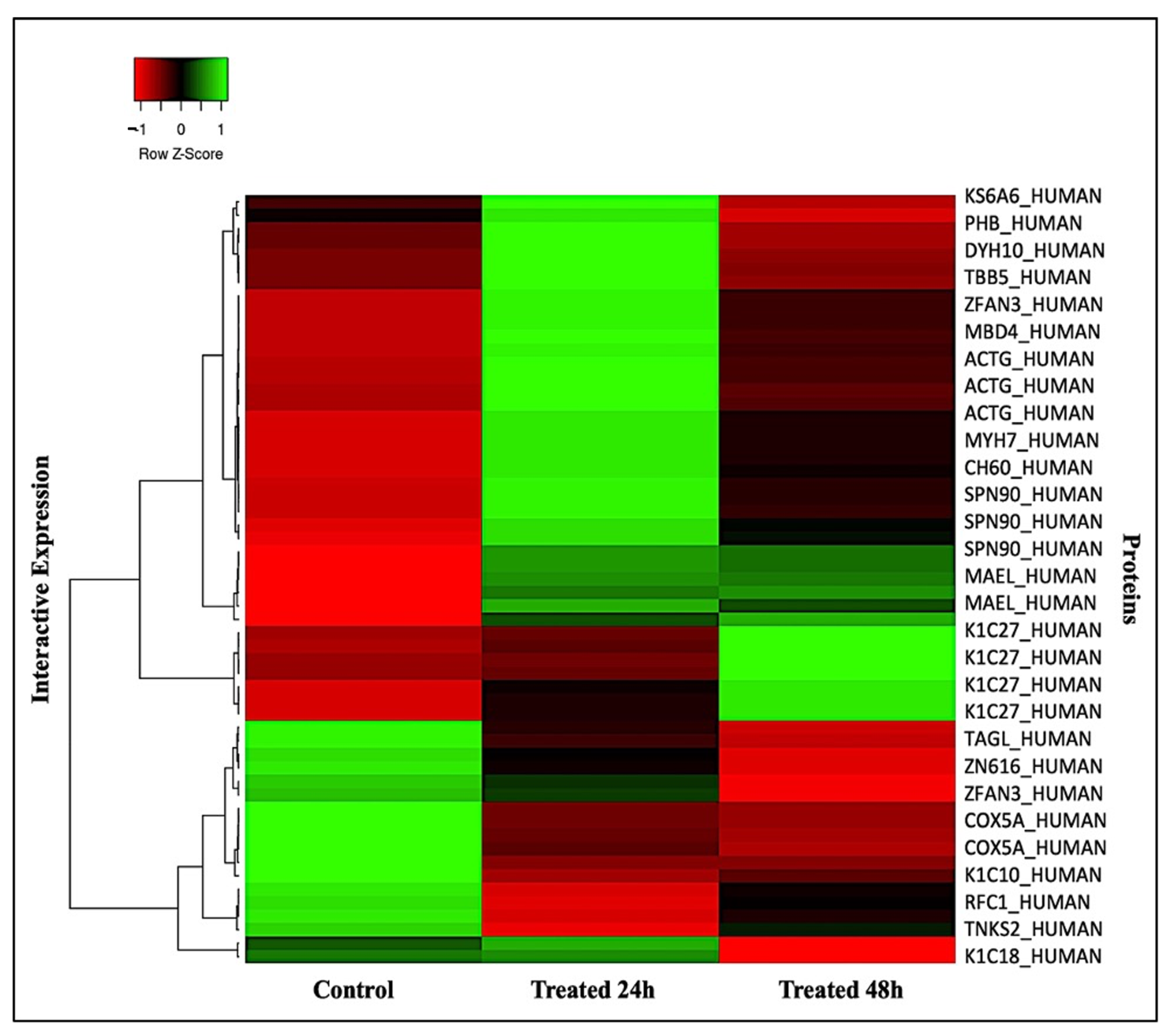
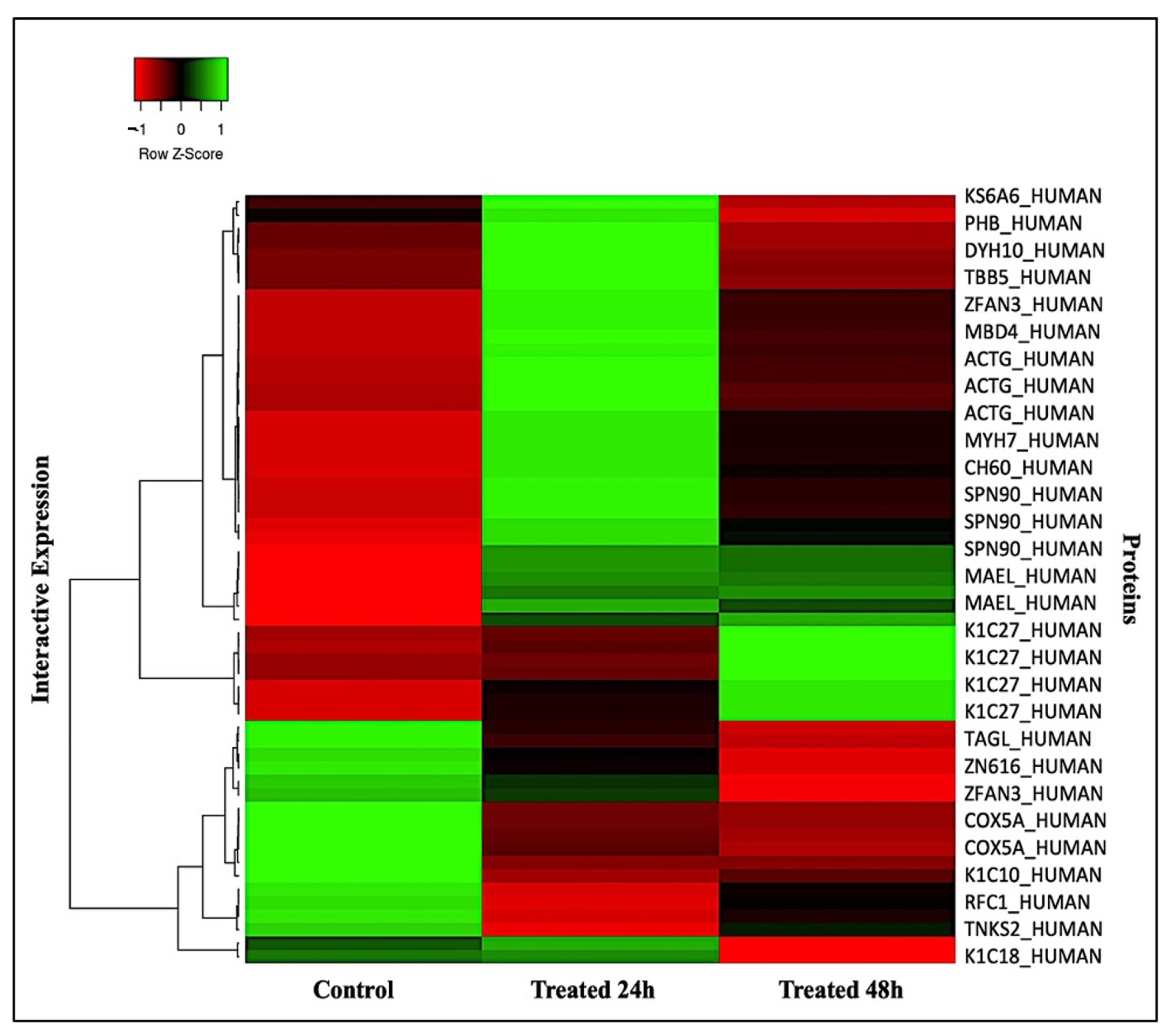
2.2. Principal Component, Cluster Analysis, and Heatmap of Significant Proteins
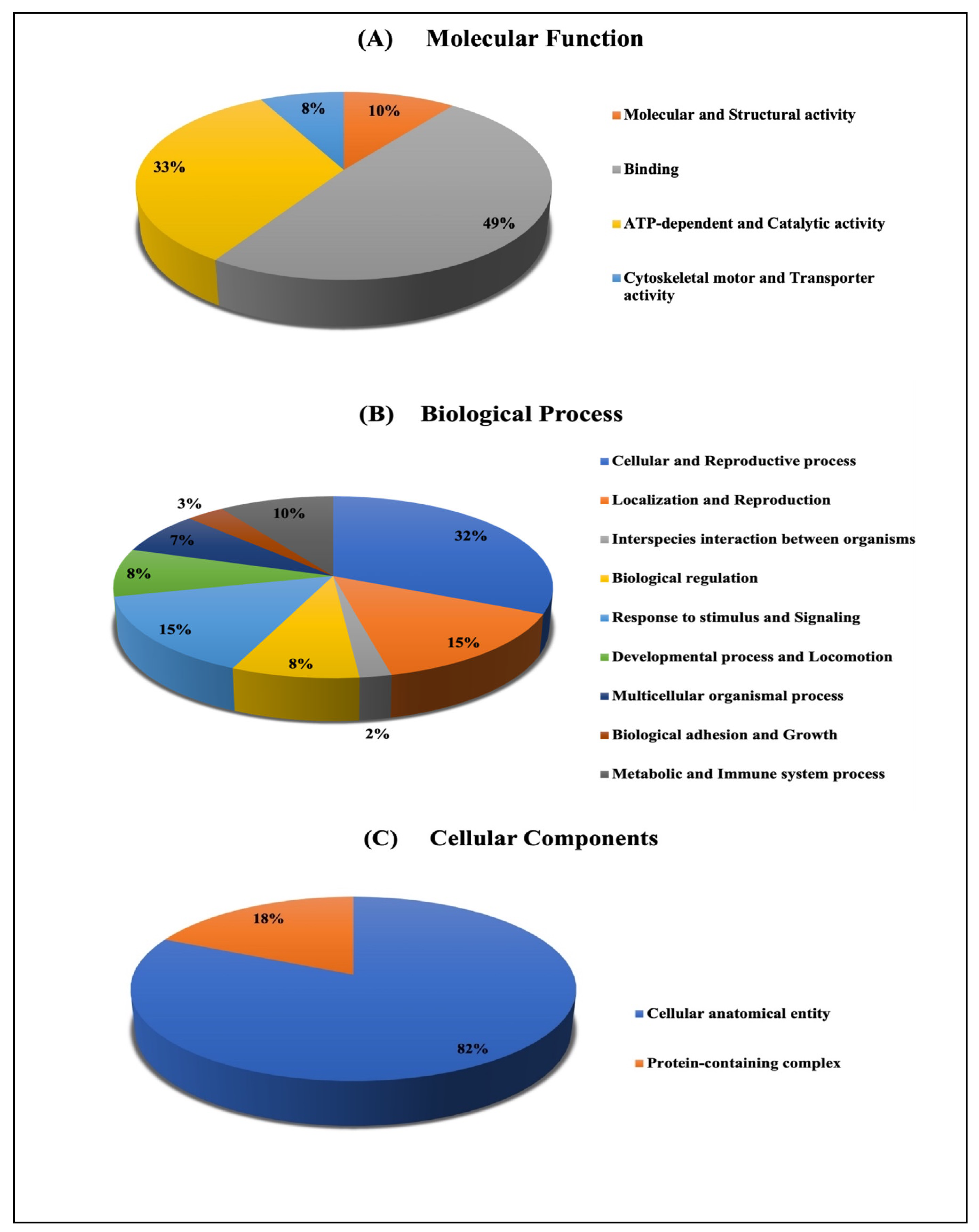
2.3. Bioinformatics Analysis
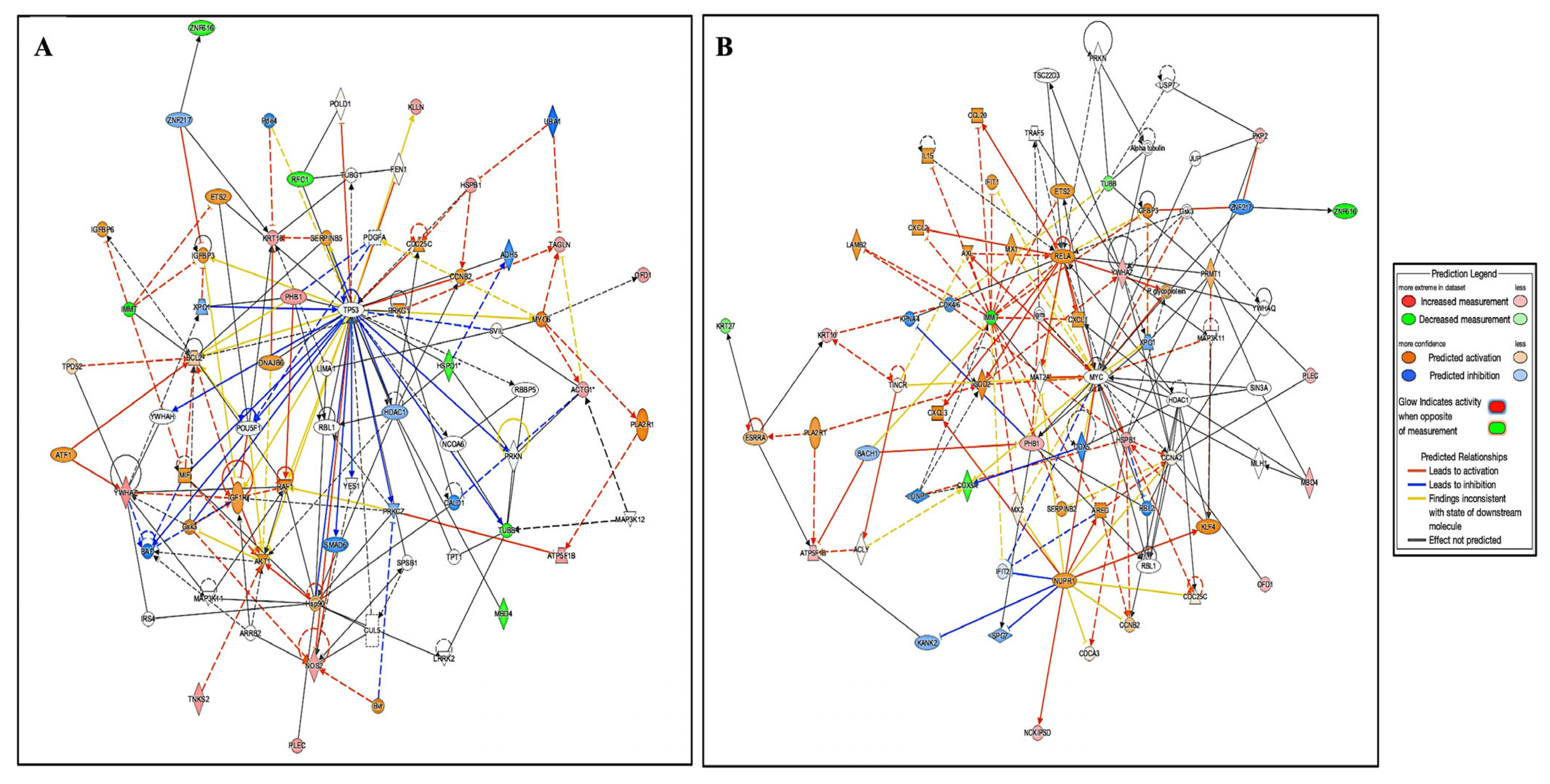
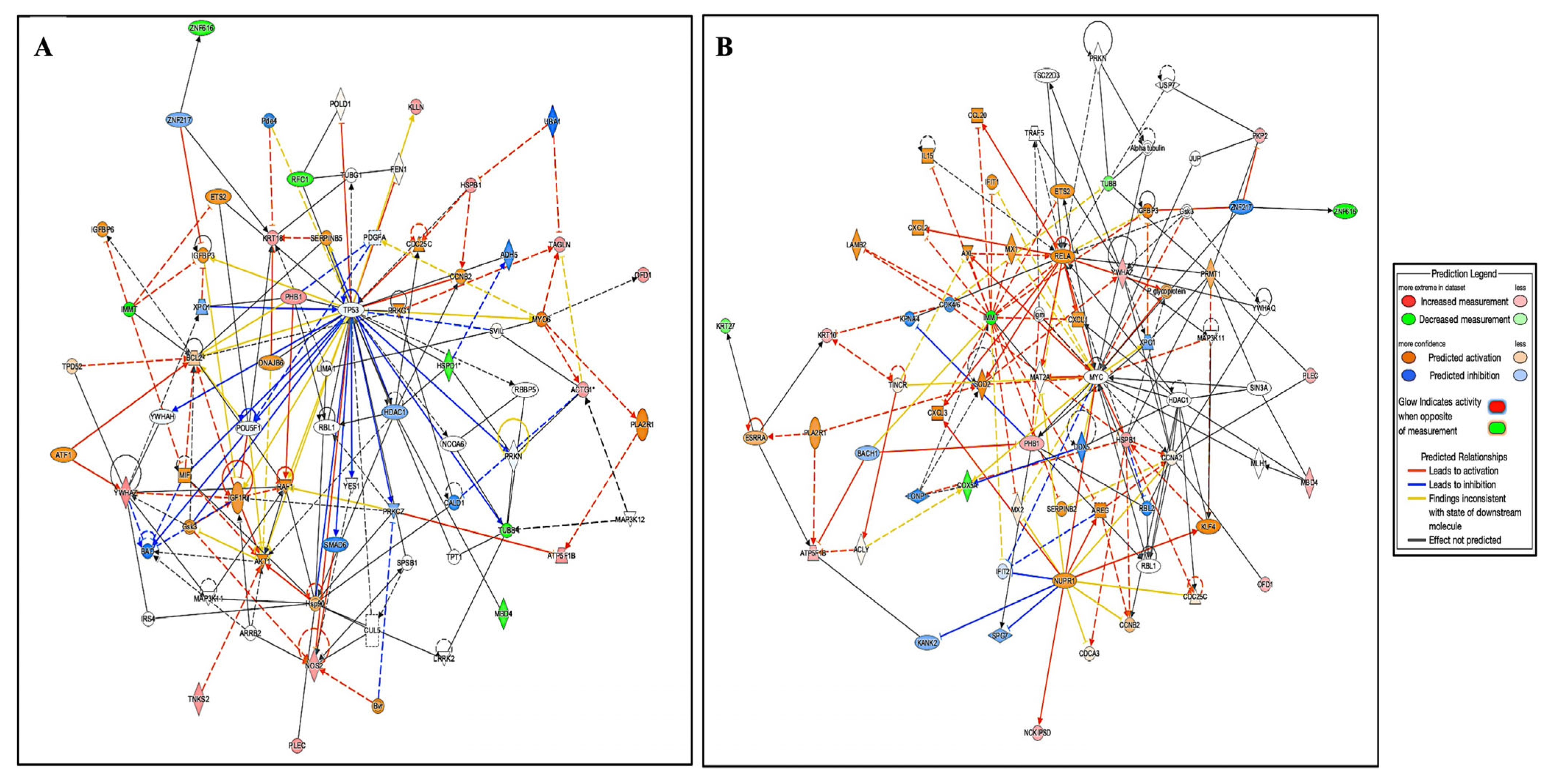
2.3.1. Interactions of Identified Proteins and Network Connectivity Mapping Using Ingenuity Pathway Analysis (IPA)
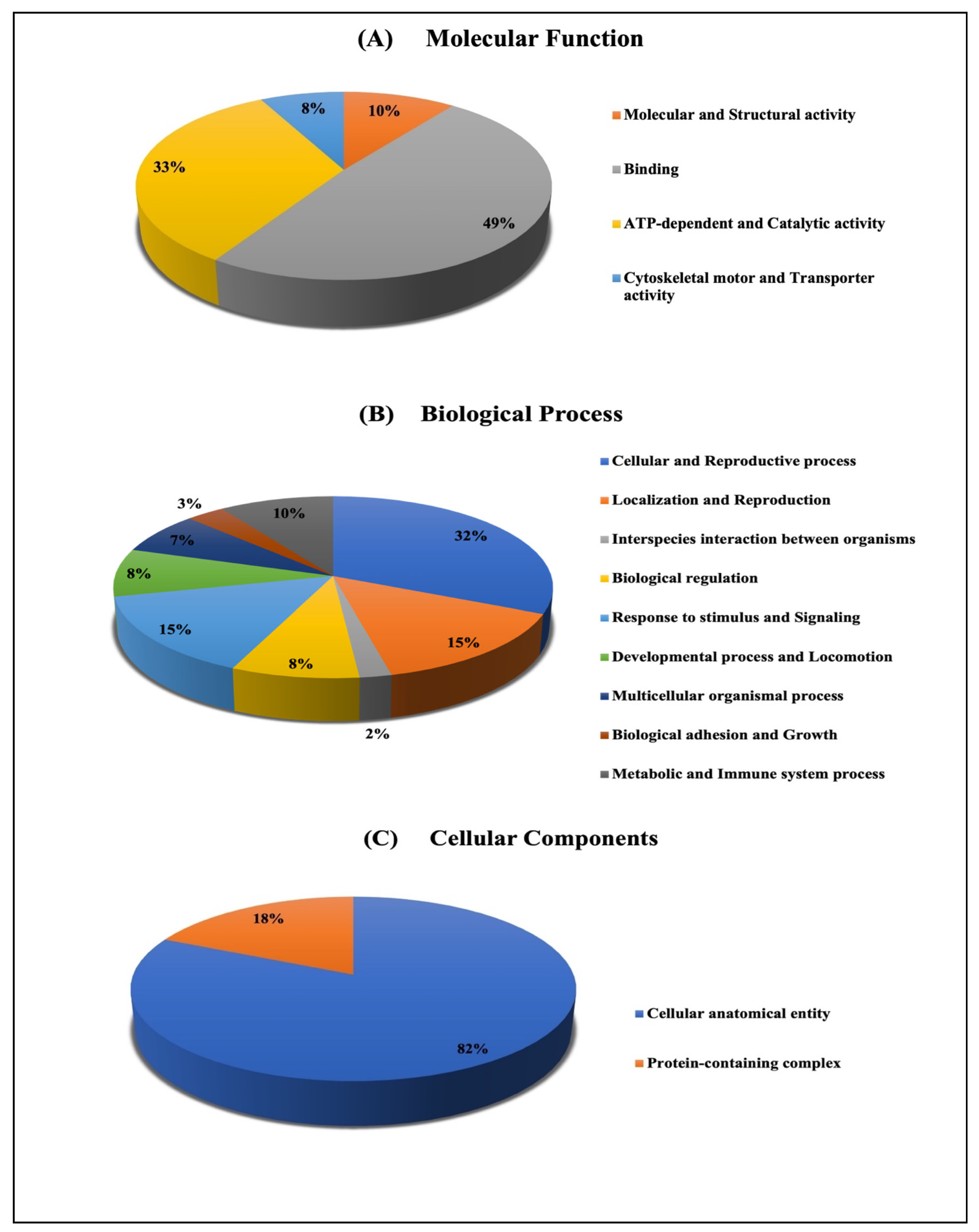
2.3.2. Classification of Key Proteins Based on Function
3. Materials and Methods
3.1. Cell and Bacterial Culture
3.1.1. Cells and Cell Culture
3.1.2. Bacterial Culture and Preparation
3.1.3. Indirect Bacterial Effect Using SF-CM Media on MCF-7 Cells
3.2. Proteomic Analysis
3.2.1. Protein Extraction
3.2.2. Protein Labeling with Cyanine Dyes
3.2.3. Two-Dimensional (2D) Electrophoresis, Image Scanning, and Preparative Gel
3.2.4. Statistical Analysis
3.2.5. Protein Identification via Matrix-Assisted Laser Desorption/Ionization–Time-of-Flight Mass Spectrometry (MALDI-TOF-MS)
3.2.6. Principal Component Analysis, Cluster Analysis, and Heatmap
3.2.7. Bioinformatics Analysis
4. Discussion
4.1. Significant Upregulated and Downregulated Proteins in the Samples Treated for 24 and 48 h
4.2. Functions and Interactions of Identified Proteins
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pleasance, E. Pan-cancer analysis of whole genomes. Nature 2020, 578, 82–93. [Google Scholar]
- Yang, S.-S.; Ma, S.; Dou, H.; Liu, F.; Zhang, S.-Y.; Jiang, C.; Xiao, M.; Huang, Y.-X. Breast cancer-derived exosomes regulate cell invasion and metastasis in breast cancer via miR-146a to activate cancer associated fibroblasts in tumor microenvironment. Exp. Cell Res. 2020, 391, 111983. [Google Scholar] [CrossRef] [PubMed]
- Chopra, S. Eleri Lloyd Davies. Breast Cancer. Med. 2020, 48, 113–118. [Google Scholar]
- Rontogianni, S.; Synadaki, E.; Li, B.; Liefaard, M.C.; Lips, E.H.; Wesseling, J.; Wu, W.; Altelaar, M. Proteomic profiling of extracellular vesicles allows for human breast cancer subtyping. Commun. Biol. 2019, 2, 325. [Google Scholar] [CrossRef] [PubMed]
- Veyssière, H.; Bidet, Y.; Penault-Llorca, F.; Radosevic-Robin, N.; Durando, X. Circulating proteins as predictive and prognostic biomarkers in breast cancer. Clin. Proteom. 2022, 19, 25. [Google Scholar] [CrossRef] [PubMed]
- Al-Ansari, M.M.; AlMalki, R.H.; Dahabiyeh, L.A.; Abdel Rahman, A.M. Metabolomics-microbiome crosstalk in the breast cancer microenvironment. Metabolites 2021, 11, 758. [Google Scholar] [CrossRef]
- Wang, B.; Xi, C.; Liu, M.; Sun, H.; Liu, S.; Song, L.; Kang, H. Breast fibroblasts in both cancer and normal tissues induce phenotypic transformation of breast cancer stem cells: A preliminary study. PeerJ 2018, 6, e4805. [Google Scholar] [CrossRef]
- Erdogan, B.; Webb, D.J. Cancer-associated fibroblasts modulate growth factor signaling and extracellular matrix remodeling to regulate tumor metastasis. Biochem. Soc. Trans. 2017, 45, 229–236. [Google Scholar] [CrossRef]
- Eiro, N.; González, L.; Martínez-Ordoñez, A.; Fernandez-Garcia, B.; González, L.O.; Cid, S.; Dominguez, F.; Perez-Fernandez, R.; Vizoso, F.J. Cancer-associated fibroblasts affect breast cancer cell gene expression, invasion and angiogenesis. Cell. Oncol. 2018, 41, 369–378. [Google Scholar] [CrossRef]
- Thompson, K.J.; Ingle, J.N.; Tang, X.; Chia, N.; Jeraldo, P.R.; Walther-Antonio, M.R.; Kandimalla, K.K.; Johnson, S.; Yao, J.Z.; Harrington, S.C. A comprehensive analysis of breast cancer microbiota and host gene expression. PLoS ONE 2017, 12, e0188873. [Google Scholar] [CrossRef]
- Fernández, M.F.; Reina-Pérez, I.; Astorga, J.M.; Rodríguez-Carrillo, A.; Plaza-Díaz, J.; Fontana, L. Breast cancer and its relationship with the microbiota. Int. J. Environ. Res. Public Health 2018, 15, 1747. [Google Scholar] [CrossRef] [PubMed]
- Urbaniak, C.; Gloor, G.B.; Brackstone, M.; Scott, L.; Tangney, M.; Reid, G. The microbiota of breast tissue and its association with breast cancer. Appl. Environ. Microbiol. 2016, 82, 5039–5048. [Google Scholar] [CrossRef] [PubMed]
- Hieken, T.J.; Chen, J.; Hoskin, T.L.; Walther-Antonio, M.; Johnson, S.; Ramaker, S.; Xiao, J.; Radisky, D.C.; Knutson, K.L.; Kalari, K.R. The microbiome of aseptically collected human breast tissue in benign and malignant disease. Sci. Rep. 2016, 6, 30751. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Tian, T.; Wei, Z.; Shih, N.; Feldman, M.D.; Peck, K.N.; DeMichele, A.M.; Alwine, J.C.; Robertson, E.S. Distinct microbial signatures associated with different breast cancer types. Front. Microbiol. 2018, 9, 951. [Google Scholar] [CrossRef] [PubMed]
- Bøhle, L.A.; Riaz, T.; Egge-Jacobsen, W.; Skaugen, M.; Busk, Ø.L.; Eijsink, V.G.; Mathiesen, G. Identification of surface proteins in Enterococcus faecalis V583. BMC Genom. 2011, 12, 135. [Google Scholar] [CrossRef]
- Nami, Y.; Abdullah, N.; Haghshenas, B.; Radiah, D.; Rosli, R.; Yari Khosroushahi, A. A newly isolated probiotic Enterococcus faecalis strain from vagina microbiota enhances apoptosis of human cancer cells. J. Appl. Microbiol. 2014, 117, 498–508. [Google Scholar] [CrossRef]
- Alfadda, A.A.; Masood, A.; Al-Naami, M.Y.; Chaurand, P.; Benabdelkamel, H. A proteomics based approach reveals differential regulation of visceral adipose tissue proteins between metabolically healthy and unhealthy obese patients. Mol. Cells 2017, 40, 685. [Google Scholar]
- Zhou, J.; Wang, W.; Xie, Y.; Zhao, Y.; Chen, X.; Xu, W.; Wang, Y.; Guan, Z. Proteomics-based identification and analysis of proteins associated with Helicobacter pylori in gastric cancer. PLoS ONE 2016, 11, e0146521. [Google Scholar] [CrossRef]
- Risha, Y.; Minic, Z.; Ghobadloo, S.M.; Berezovski, M.V. The proteomic analysis of breast cell line exosomes reveals disease patterns and potential biomarkers. Sci. Rep. 2020, 10, 13572. [Google Scholar] [CrossRef]
- Benabdelkamel, H.; Rafiullah, M.; Masood, A.; Alsaif, A.; Musambil, M.; Alfadda, A.A. Proteomic profiling of thyroid tissue in patients with obesity and benign diffuse goiter. Front. Endocrinol. 2022, 13, 923465. [Google Scholar] [CrossRef]
- Kuczynski, J.; Lauber, C.L.; Walters, W.A.; Parfrey, L.W.; Clemente, J.C.; Gevers, D.; Knight, R. Experimental and analytical tools for studying the human microbiome. Nat. Rev. Genet. 2012, 13, 47–58. [Google Scholar] [CrossRef]
- AlMalki, R.H.; Sebaa, R.; Al-Ansari, M.M.; Al-Alwan, M.; Alwehaibi, M.A.; Rahman, A.M.A.E. E. coli Secretome Metabolically Modulates MDA-MB-231 Breast Cancer Cells’ Energy Metabolism. Int. J. Mol. Sci. 2023, 24, 4219. [Google Scholar] [CrossRef]
- Al-Ansari, M.M.; Aboussekhra, A. miR-146b-5p mediates p16-dependent repression of IL-6 and suppresses paracrine procarcinogenic effects of breast stromal fibroblasts. Oncotarget 2015, 6, 30006. [Google Scholar] [CrossRef]
- Alghanem, B.; Ali, R.; Nehdi, A.; Al Zahrani, H.; Altolayyan, A.; Shaibah, H.; Baz, O.; Alhallaj, A.; Moresco, J.J.; Diedrich, J.K. Proteomics Profiling of KAIMRC1 in Comparison to MDA-MB231 and MCF-7. Int. J. Mol. Sci. 2020, 21, 4328. [Google Scholar] [CrossRef]
- Rabiei, P.; Mohabatkar, H.; Behbahani, M. Studying the effects of several heat-inactivated bacteria on colon and breast cancer cells. Mol. Biol. Res. Commun. 2019, 8, 91. [Google Scholar]
- Bermudez-Brito, M.; Munoz-Quezada, S.; Gomez-Llorente, C.; Matencio, E.; Bernal, M.J.; Romero, F.; Gil, A. Cell-free culture supernatant of Bifidobacterium breve CNCM I-4035 decreases pro-inflammatory cytokines in human dendritic cells challenged with Salmonella typhi through TLR activation. PLoS ONE 2013, 8, e59370. [Google Scholar] [CrossRef] [PubMed]
- Kouitcheu Mabeku, L.B.; Ngue, S.; Bonsou Nguemo, I.; Leundji, H. Potential of selected lactic acid bacteria from Theobroma cacao fermented fruit juice and cell-free supernatants from cultures as inhibitors of Helicobacter pylori and as good probiotic. BMC Res. Notes 2020, 13, 64. [Google Scholar] [CrossRef] [PubMed]
- Chiba, A.; Bawaneh, A.; Velazquez, C.; Clear, K.Y.; Wilson, A.S.; Howard-McNatt, M.; Levine, E.A.; Levi-Polyachenko, N.; Yates-Alston, S.A.; Diggle, S.P. Neoadjuvant chemotherapy shifts breast tumor microbiota populations to regulate drug responsiveness and the development of metastasis. Mol. Cancer Res. 2020, 18, 130–139. [Google Scholar] [CrossRef] [PubMed]
- Al-Ansari, M.M.; Al-Saif, M.; Arafah, M.; Eldali, A.M.; Tulbah, A.; Al-Tweigeri, T.; Semlali, A.; Khabar, K.S.; Aboussekhra, A. Clinical and functional significance of tumor/stromal ATR expression in breast cancer patients. Breast Cancer Res. 2020, 22, 49. [Google Scholar] [CrossRef] [PubMed]
- Benabdelkamel, H.; Masood, A.; Almidani, G.M.; Alsadhan, A.A.; Bassas, A.F.; Duncan, M.W.; Alfadda, A.A. Mature adipocyte proteome reveals differentially altered protein abundances between lean, overweight and morbidly obese human subjects. Mol. Cell. Endocrinol. 2015, 401, 142–154. [Google Scholar] [CrossRef]
- Shevchenko, A.; Wilm, M.; Vorm, O.; Mann, M. Mass spectrometric sequencing of proteins from silver-stained polyacrylamide gels. Anal. Chem. 1996, 68, 850–858. [Google Scholar] [CrossRef] [PubMed]
- Masood, A.; Benabdelkamel, H.; Ekhzaimy, A.A.; Alfadda, A.A. Plasma-based proteomics profiling of patients with hyperthyroidism after antithyroid treatment. Molecules 2020, 25, 2831. [Google Scholar] [CrossRef]
- Benabdelkamel, H.; Masood, A.; Okla, M.; Al-Naami, M.Y.; Alfadda, A.A. A proteomics-based approach reveals differential regulation of urine proteins between metabolically healthy and unhealthy obese patients. Int. J. Mol. Sci. 2019, 20, 4905. [Google Scholar] [CrossRef] [PubMed]
- Hassan, Z.; Mustafa, S.; Rahim, R.A.; Isa, N.M. Anti-breast cancer effects of live, heat-killed and cytoplasmic fractions of Enterococcus faecalis and Staphylococcus hominis isolated from human breast milk. Vitr. Cell. Dev. Biol.-Anim. 2016, 52, 337–348. [Google Scholar] [CrossRef]
- Hudelist, G.; Singer, C.F.; Pischinger, K.I.; Kaserer, K.; Manavi, M.; Kubista, E.; Czerwenka, K.F. Proteomic analysis in human breast cancer: Identification of a characteristic protein expression profile of malignant breast epithelium. Proteomics 2006, 6, 1989–2002. [Google Scholar] [CrossRef] [PubMed]
- Ng, E.K.; Shin, V.Y.; Leung, C.P.; Chan, V.W.; Law, F.B.; Siu, M.T.; Lang, B.H.; Ma, E.S.; Kwong, A. Elevation of methylated DNA in KILLIN/PTEN in the plasma of patients with thyroid and/or breast cancer. OncoTargets Ther. 2014, 7, 2085–2092. [Google Scholar]
- Akkour, K.; Alanazi, I.O.; Alfadda, A.A.; Alhalal, H.; Masood, A.; Musambil, M.; Rahman, A.M.A.; Alwehaibi, M.A.; Arafah, M.; Bassi, A. Tissue-Based Proteomic Profiling in Patients with Hyperplasia and Endometrial Cancer. Cells 2022, 11, 2119. [Google Scholar] [CrossRef]
- Shields, J.M.; Rogers-Graham, K.; Der, C.J. Loss of transgelin in breast and colon tumors and in RIE-1 cells by Ras deregulation of gene expression through Raf-independent pathways. J. Biol. Chem. 2002, 277, 9790–9799. [Google Scholar] [CrossRef]
- Eisemann, T.; Pascal, J.M. Poly (ADP-ribose) polymerase enzymes and the maintenance of genome integrity. Cell. Mol. Life Sci. 2020, 77, 19–33. [Google Scholar] [CrossRef]
- Liao, W.; Zhao, R.; Lu, L.; Zhang, R.; Zou, J.; Xu, T.; Wu, C.; Tang, J.; Deng, Y.; Lu, X. Overexpression of a novel osteopetrosis-related gene CCDC154 suppresses cell proliferation by inducing G2/M arrest. Cell Cycle 2012, 11, 3270–3279. [Google Scholar] [CrossRef]
- Liu, R.; Liu, T.; Wei, W.; Guo, K.; Yang, N.; Tian, S.; Zhu, J.; Liu, Y.; Zhou, W.; Yang, H. Novel interacting proteins identified by tandem affinity purification coupled to nano LC–MS/MS interact with ribosomal S6 protein kinase 4 (RSK4) and its variant protein (RSK4m). Int. J. Biol. Macromol. 2017, 96, 421–428. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.-C. DDX3X multifunctionally modulates tumor progression and serves as a prognostic indicator to predict cancer outcomes. Int. J. Mol. Sci. 2019, 21, 281. [Google Scholar] [CrossRef] [PubMed]
- Galber, C.; Acosta, M.J.; Minervini, G.; Giorgio, V. The role of mitochondrial ATP synthase in cancer. Biol. Chem. 2020, 401, 1199–1214. [Google Scholar] [CrossRef]
- Wang, T.; Ma, F.; Qian, H.-L. Defueling the cancer: ATP synthase as an emerging target in cancer therapy. Mol. Ther.-Oncolytics 2021, 23, 82–95. [Google Scholar] [CrossRef] [PubMed]
- Speransky, S.; Serafini, P.; Caroli, J.; Bicciato, S.; Lippman, M.; Bishopric, N. A novel RNA aptamer identifies plasma membrane ATP synthase beta subunit as an early marker and therapeutic target in aggressive cancer. Breast Cancer Res. Treat. 2019, 176, 271–289. [Google Scholar] [CrossRef] [PubMed]
- Nayak, A.P.; Kapur, A.; Barroilhet, L.; Patankar, M.S. Oxidative phosphorylation: A target for novel therapeutic strategies against ovarian cancer. Cancers 2018, 10, 337. [Google Scholar] [CrossRef]
- Huang, Y.-J.; Jan, Y.-H.; Chang, Y.-C.; Tsai, H.-F.; Wu, A.T.; Chen, C.-L.; Hsiao, M. ATP synthase subunit epsilon overexpression promotes metastasis by modulating AMPK signaling to induce epithelial-to-mesenchymal transition and is a poor prognostic marker in colorectal cancer patients. J. Clin. Med. 2019, 8, 1070. [Google Scholar] [CrossRef]
- Qi, B.; Han, M. Microbial siderophore enterobactin promotes mitochondrial iron uptake and development of the host via interaction with ATP synthase. Cell 2018, 175, 571–582.e11. [Google Scholar] [CrossRef]
- Lee, K.Y.; Bignone, P.A.; Ganesan, T.S. p90 Ribosomal S6 kinases-eclectic members of the human kinome. Signal Transduct. 2007, 7, 225–239. [Google Scholar] [CrossRef]
- Yun, C.W.; Kim, H.J.; Lim, J.H.; Lee, S.H. Heat shock proteins: Agents of cancer development and therapeutic targets in anti-cancer therapy. Cells 2019, 9, 60. [Google Scholar] [CrossRef]
- Qu, Y.; Hao, C.; Xu, J.; Cheng, Z.; Wang, W.; Liu, H. ILK promotes cell proliferation in breast cancer cells by activating the PI3K/Akt pathway. Mol. Med. Rep. 2017, 16, 5036–5042. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| # | Spot No a | Protein Name | MASCOT ID | Ratio 24/C b | Exp c | Ratio 48/C b | Exp c | p Value d |
|---|---|---|---|---|---|---|---|---|
| 1 | 309 | Zinc finger protein 616 | ZN616_HUMAN | −1.5 | DOWN | −2.7 | DOWN | 0.003 |
| 2 | 641 | Nitric oxide synthase, inducible | NOS2_HUMAN | 1.5 | UP | −1 | NS | 0.003 |
| 3 | 652 | Transgelin | TAGL_HUMAN | 1.5 | UP | −1.1 | NS | 0.004 |
| 4 | 644 | Tektin-2 | TEKT2_HUMAN | −1.5 | DOWN | −1.9 | DOWN | 0.004 |
| 5 | 827 | Egl nine homolog 1 | EGLN1_HUMAN | 1.2 | NS | 1.2 | NS | 0.01 |
| 6 | 682 | Dynein axonemal heavy chain 10 | DYH10_HUMAN | 1.7 | UP | 1.2 | NS | 0.01 |
| 7 | 828 | Twinfilin-2 | TWF2_HUMAN | 3.2 | UP | −1.1 | NS | 0.01 |
| 8 | 899 | Spectrin beta chain, non-erythrocytic 5 | SPTN5_HUMAN | 1.2 | NS | −1.2 | NS | 0.01 |
| 9 | 655 | Keratin, type I cytoskeletal 18 | K1C18_HUMAN | 1.5 | UP | 1.2 | NS | 0.01 |
| 10 | 896 | Cytochrome c oxidase subunit 5A, mitochondrial | COX5A_HUMAN | −1 | NS | −1.9 | DOWN | 0.01 |
| 11 | 365 | MICOS complex subunit MIC60 | MIC60_HUMAN | −2.1 | DOWN | −2.4 | DOWN | 0.01 |
| 12 | 428 | Coiled-coil domain-containing protein 154 | CC154_HUMAN | 2 | UP | 1.4 | NS | 0.01 |
| 13 | 606 | Plectin | PLEC1_HUMAN | 1.5 | UP | 1.5 | UP | 0.02 |
| 14 | 686 | Zinc finger protein 600 | ZN600_HUMAN | 1.8 | UP | 1.2 | NS | 0.02 |
| 15 | 692 | Putative uncharacterized protein encoded by LINC02694 | CO053_HUMAN | 1.6 | UP | 1.2 | NS | 0.02 |
| 16 | 674 | Centriole and centriolar satellite protein OFD1 | OFD1_HUMAN | 1.5 | UP | 1.5 | UP | 0.02 |
| 17 | 696 | Protein maelstrom homolog | MAEL_HUMAN | 1.6 | UP | 1.1 | NS | 0.02 |
| 18 | 786 | Prohibitin 1 | PHB_HUMAN | 1.6 | UP | 1.6 | UP | 0.02 |
| 19 | 603 | Microtubule-associated protein 6 | MAP6_HUMAN | 1.5 | UP | −1.1 | NS | 0.02 |
| 20 | 767 | Cartilage matrix protein | MATN1_HUMAN | 1.5 | UP | 1.2 | NS | 0.02 |
| 21 | 581 | ATP synthase subunit beta, mitochondrial | ATPB_HUMAN | 1.5 | UP | 1.5 | UP | 0.02 |
| 22 | 779 | Keratin, type I cytoskeletal 10 | K1C10_HUMAN | 1.1 | NS | 1.5 | UP | 0.03 |
| 23 | 465 | Tubulin beta chain | TBB5_HUMAN | −1.5 | DOWN | −1.5 | DOWN | 0.03 |
| 24 | 605 | Poly [ADP-ribose] polymerase tankyrase-2 | TNKS2_HUMAN | 1.6 | UP | −1 | DOWN | 0.03 |
| 25 | 409 | Replication factor C subunit 1 | RFC1_HUMAN | −1.5 | DOWN | −1 | NS | 0.03 |
| 26 | 419 | 60 kDa heat shock protein, mitochondrial | CH60_HUMAN | −1.5 | DOWN | −1.2 | NS | 0.03 |
| 27 | 453 | Ankyrin repeat domain-containing protein 6 | ANKR6_HUMAN | −1.5 | DOWN | −1.9 | DOWN | 0.03 |
| 28 | 695 | AN1-type zinc finger protein 3 | ZFAN3_HUMAN | 1.7 | UP | 1.2 | NS | 0.04 |
| 29 | 362 | AN1-type zinc finger protein 3 | ZFAN3_HUMAN | −1.1 | NS | −1.7 | DOWN | 0.04 |
| 30 | 637 | Actin, cytoplasmic 2 | ACTG_HUMAN | 1.5 | UP | 1.2 | NS | 0.04 |
| 31 | 791 | 14-3-3 protein zeta/delta | 1433Z_HUMAN | 1.8 | UP | 1.6 | UP | 0.04 |
| 32 | 678 | N-acyl-aromatic-L-amino acid amidohydrolase (carboxylate-forming) | ACY3_HUMAN | 1.7 | UP | 1.1 | NS | 0.04 |
| 33 | 386 | Dynein axonemal heavy chain 9 | DYH9_HUMAN | −1.5 | DOWN | −1.7 | DOWN | 0.04 |
| 34 | 677 | Forkhead-associated domain-containing protein 1 | FHAD1_HUMAN | 1.2 | NS | 2.3 | UP | 0.05 |
| 35 | 847 | Keratin, type I cytoskeletal 27 | K1C27_HUMAN | −1.2 | NS | −1.5 | DOWN | 0.05 |
| 36 | 680 | Plakophilin-2 | PKP2_HUMAN | −1 | NS | 1.5 | UP | 0.05 |
| 37 | 601 | Methyl-CpG-binding domain protein 4 | MBD4_HUMAN | −1.5 | DOWN | 1.5 | UP | 0.05 |
| 38 | 665 | Ribosomal protein S6 kinase alpha-6 | KS6A6_HUMAN | 1.5 | UP | 1.1 | NS | 0.05 |
| 39 | 642 | Glycerol-3-phosphate dehydrogenase, mitochondrial | GPDM_HUMAN | 1.5 | UP | −1.1 | NS | 0.05 |
| 40 | 814 | Heat shock protein beta-1 | HSPB1_HUMAN | 1.5 | UP | 1.8 | UP | 0.05 |
| 41 | 612 | Actin, cytoplasmic 2 | ACTG_HUMAN | 1.5 | UP | 1.2 | NS | 0.05 |
| 42 | 691 | Killin | KILIN_HUMAN | 1.5 | UP | 1.1 | NS | 0.05 |
| 43 | 799 | NCK-interacting protein with SH3 domain | SPN90_HUMAN | 1.1 | NS | 1.5 | UP | 0.05 |
| 44 | 449 | Protein tyrosine phosphatase domain-containing protein 1 | PTPC1_HUMAN | 1.5 | UP | 1.1 | NS | 0.05 |
| 45 | 707 | Retinol dehydrogenase 13 | RDH13_HUMAN | 1.8 | UP | 1.2 | NS | 0.05 |
| 46 | 414 | Pericentrin | PCNT_HUMAN | −1.5 | DOWN | −1.2 | NS | 0.05 |
| 47 | 334 | Sciellin | SCEL_HUMAN | −1.2 | NS | −2.3 | DOWN | 0.05 |
| 48 | 423 | Protein disulfide-isomerase | PDIA1_HUMAN | 2.3 | UP | 4 | UP | 0.05 |
| 49 | 460 | Myosin-7 | MYH7_HUMAN | −1.6 | DOWN | −1.8 | DOWN | 0.05 |
| 50 | 804 | 28S ribosomal protein S18c, mitochondrial | RT18C_HUMAN | 2 | UP | 1.5 | UP | 0.05 |
| 51 | 393 | ATP synthase subunit alpha, mitochondrial | ATPA_HUMAN | −1.6 | DOWN | −1.5 | DOWN | 0.05 |
| 52 | 438 | AN1-type zinc finger protein 3 | ZFAN3_HUMAN | 1.5 | UP | 1.2 | NS | 0.05 |
| 53 | 912 | ATP-dependent RNA helicase DDX3X | DDX3X_HUMAN | 1.1 | NS | −1.5 | DOWN | 0.05 |
| 54 | 547 | 60 kDa heat shock protein, mitochondrial | CH60_HUMAN | 1.1 | NS | 1.5 | UP | 0.05 |
| 55 | 790 | Probable ATP-dependent RNA helicase DDX60 | DDX60_HUMAN | 1.5 | UP | 1.6 | UP | 0.05 |
| 56 | 792 | Myosin-13 | MYH13_HUMAN | 1.5 | UP | 1.8 | UP | 0.05 |
| 57 | 487 | 60 kDa heat shock protein, mitochondrial | CH60_HUMAN | −1.3 | DOWN | −1.3 | DOWN | 0.05 |
| 58 | 635 | Myosin-4 | MYH4_HUMAN | 1.5 | UP | 1.2 | NS | 0.05 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alwehaibi, M.A.; Al-Ansari, M.M.; Alfadda, A.A.; Al-Malki, R.; Masood, A.; Abdel Rahman, A.M.; Benabdelkamel, H. Proteomics Investigation of the Impact of the Enterococcus faecalis Secretome on MCF-7 Tumor Cells. Int. J. Mol. Sci. 2023, 24, 14937. https://doi.org/10.3390/ijms241914937
Alwehaibi MA, Al-Ansari MM, Alfadda AA, Al-Malki R, Masood A, Abdel Rahman AM, Benabdelkamel H. Proteomics Investigation of the Impact of the Enterococcus faecalis Secretome on MCF-7 Tumor Cells. International Journal of Molecular Sciences. 2023; 24(19):14937. https://doi.org/10.3390/ijms241914937
Chicago/Turabian StyleAlwehaibi, Moudi A., Mysoon M. Al-Ansari, Assim A. Alfadda, Reem Al-Malki, Afshan Masood, Anas M. Abdel Rahman, and Hicham Benabdelkamel. 2023. "Proteomics Investigation of the Impact of the Enterococcus faecalis Secretome on MCF-7 Tumor Cells" International Journal of Molecular Sciences 24, no. 19: 14937. https://doi.org/10.3390/ijms241914937
APA StyleAlwehaibi, M. A., Al-Ansari, M. M., Alfadda, A. A., Al-Malki, R., Masood, A., Abdel Rahman, A. M., & Benabdelkamel, H. (2023). Proteomics Investigation of the Impact of the Enterococcus faecalis Secretome on MCF-7 Tumor Cells. International Journal of Molecular Sciences, 24(19), 14937. https://doi.org/10.3390/ijms241914937