
Spinal Muscular Atrophy: An Evolving Scenario through New Perspectives in Diagnosis and Advances in Therapies
 , ,
, , 

Abstract
:1. Introduction
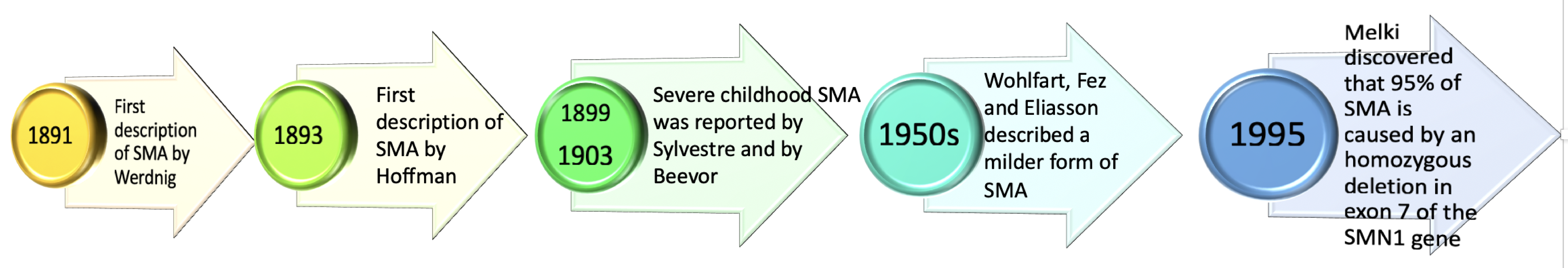
2. History of Spinal Muscular Atrophy
3. Clinical Features of SMA
4. SMA Diagnosis with Molecular Genetic Testing
5. Psychological Adjustment of Individuals with SMA
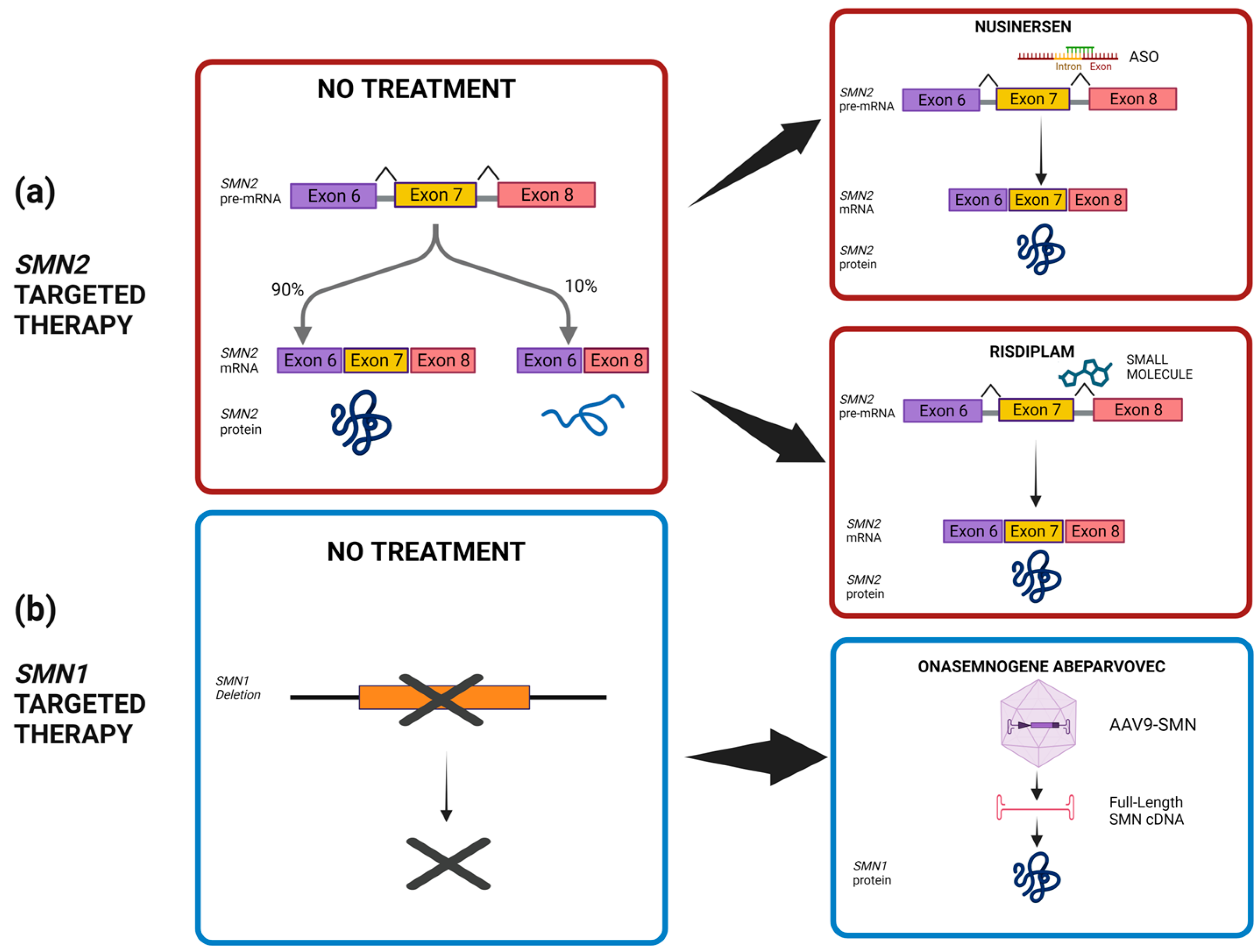
6. SMA Treatments and Therapies
6.1. FDA-Approved SMN-Based Therapies for SMA
6.2. Neuroprotective Drugs
6.3. Neuromuscular Junction Drugs
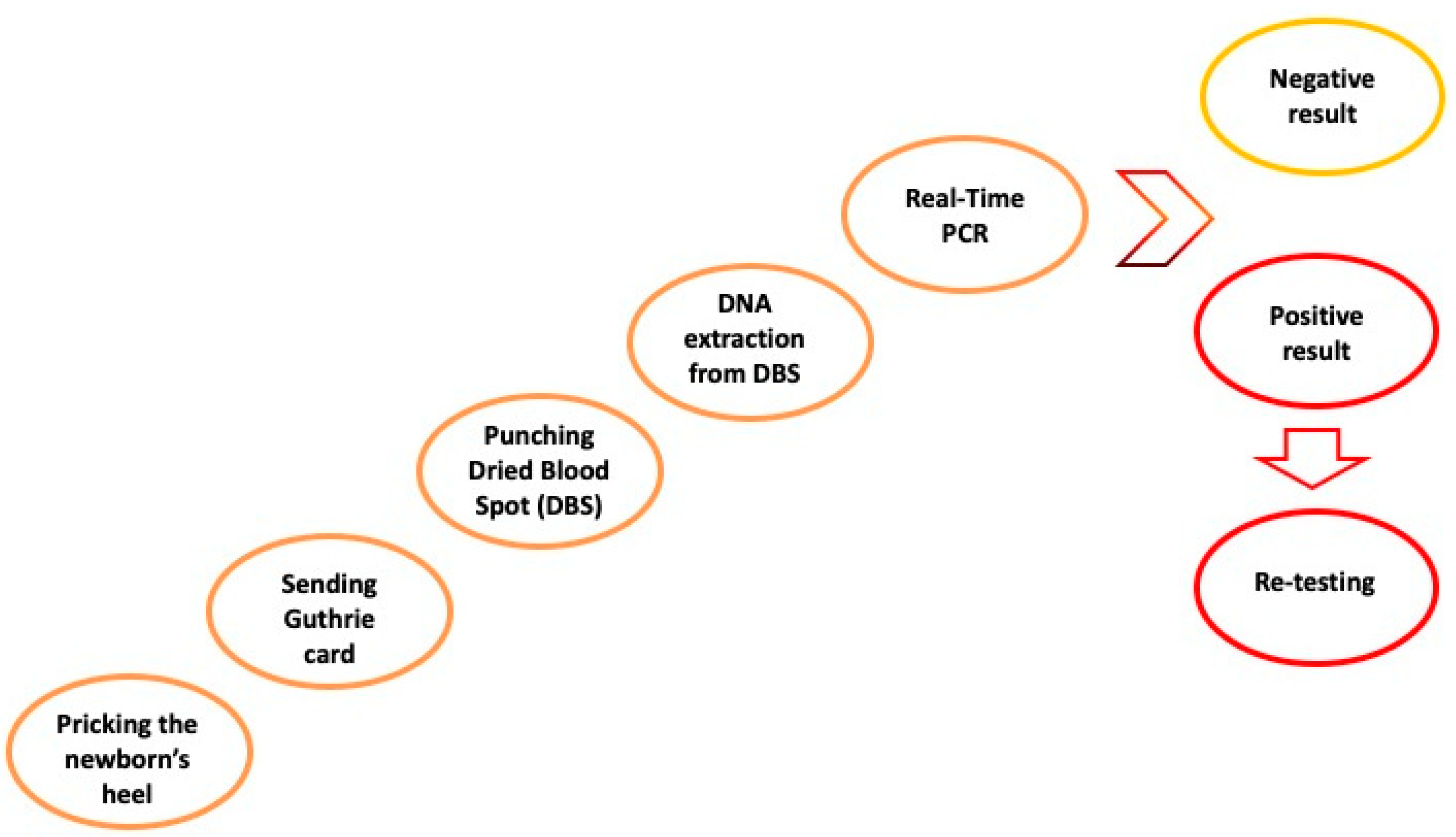
7. Newborn Screening for SMA
7.1. New Perspectives on SMA through Newborn Screening
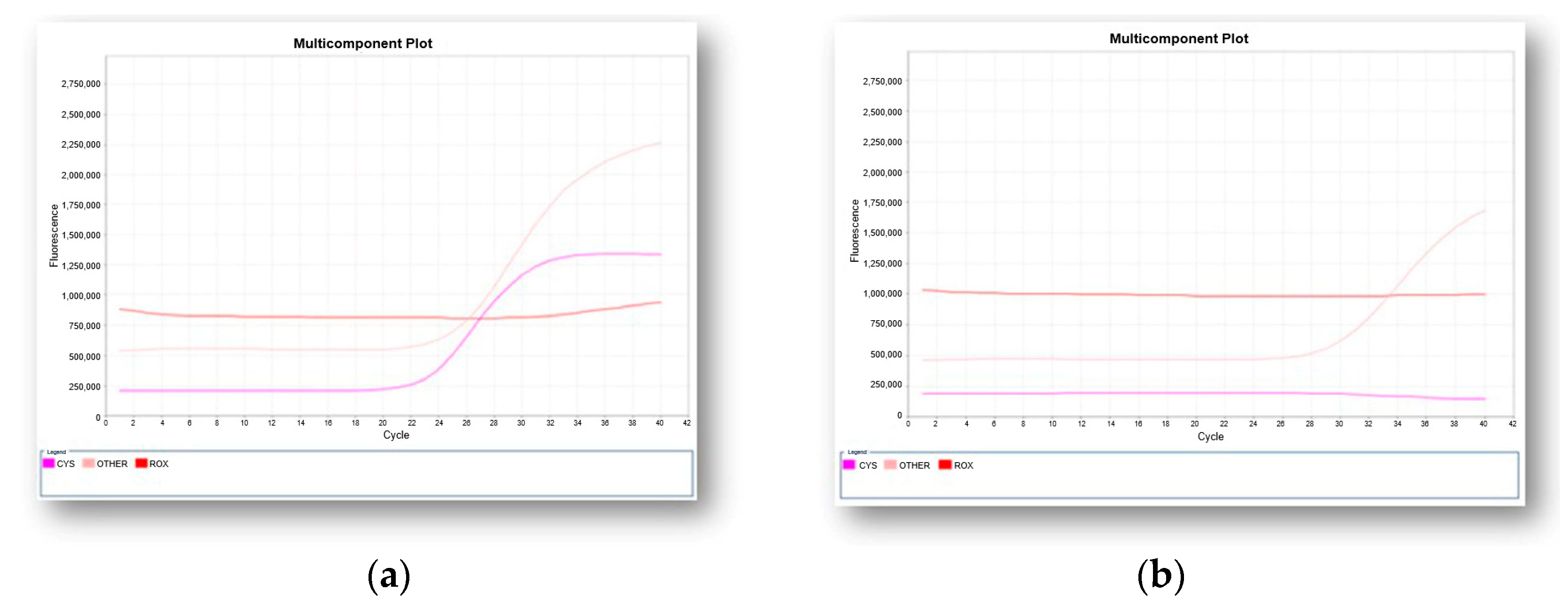
7.2. Molecular Analysis as Newborn Blood Screening Test for SMA
8. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Scarciolla, O.; Stuppia, L.; De Angelis, M.V.; Murru, S.; Palka, C.; Giuliani, R.; Pace, M.; Di Muzio, A.; Torrente, I.; Morella, A.; et al. Spinal muscular atrophy genotyping by gene dosage using multiple ligation-dependent probe amplification. Neurogenetics 2006, 7, 269–276. [Google Scholar] [CrossRef] [PubMed]
- Glascock, J.; Sampson, J.; Haidet-Phillips, A.; Connolly, A.; Darras, B.; Day, J.; Finkel, R.; Howell, R.R.; Klinger, K.; Kuntz, N.; et al. Treatment Algorithm for Infants Diagnosed with Spinal Muscular Atrophy through Newborn Screening. J. Neuromuscul. Dis. 2018, 5, 145–158. [Google Scholar] [CrossRef] [PubMed]
- Butchbach, M.E.R. Genomic Variability in the Survival Motor Neuron Genes (SMN1 and SMN2): Implications for Spinal Muscular Atrophy Phenotype and Therapeutics Development. Int. J. Mol. Sci. 2021, 22, 7896. [Google Scholar] [CrossRef]
- Kolb, S.J.; Kissel, J.T. Spinal muscular atrophy: A timely review. Arch. Neurol. 2011, 68, 979–984. [Google Scholar] [CrossRef] [PubMed]
- Kolb, S.J.; Kissel, J.T. Spinal Muscular Atrophy. Neurol. Clin. 2015, 33, 831–846. [Google Scholar] [CrossRef]
- Brzustowicz, L.M.; Lehner, T.; Castilla, L.H.; Penchaszadeh, G.K.; Wilhelmsen, K.C.; Daniels, R.; Davies, K.E.; Leppert, M.; Ziter, F.; Wood, D. Genetic mapping of chronic childhood-onset spinal muscular atrophy to chromosome 5q11.2-13.3. Nature 1990, 344, 540–541. [Google Scholar] [CrossRef]
- Groen, E.J.N.; Talbot, K.; Gillingwater, T.H. Advances in therapy for spinal muscular atrophy: Promises and challenges. Nat. Rev. Neurol. 2018, 14, 214–224. [Google Scholar] [CrossRef]
- Phan, H.C.; Taylor, J.L.; Hannon, H.; Howell, R. Newborn screening for spinal muscular atrophy: Anticipating an imminent need. Semin. Perinatol. 2015, 39, 217–229. [Google Scholar] [CrossRef]
- Ogino, S.; Wilson, R.B. Genetic testing and risk assessment for spinal muscular atrophy (SMA). Hum. Genet. 2002, 111, 477–500. [Google Scholar] [CrossRef]
- Butterfield, R.J. Spinal Muscular Atrophy Treatments, Newborn Screening, and the Creation of a Neurogenetics Urgency. Semin. Pediatr. Neurol. 2021, 38, 100899. [Google Scholar] [CrossRef]
- Iannaccone, S.T. Spinal muscular atrophy. Semin. Neurol. 1998, 18, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Nadeau, A.; D’Anjou, G.; Debray, G.; Robitaille, Y.; Simard, L.R.; Vanasse, M. A newborn with spinal muscular atrophy type 0 presenting with a clinicopathological picture suggestive of myotubular myopathy. J. Child. Neurol. 2007, 22, 1301–1304. [Google Scholar] [CrossRef] [PubMed]
- Munsat, T.L.; Davies, K.E. International SMA consortium meeting. (26–28 June 1992, Bonn, Germany). Neuromuscul. Disord. 1992, 2, 423–428. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, S.; Bürglen, L.; Reboullet, S.; Clermont, O.; Burlet, P.; Viollet, L.; Benichou, B.; Cruaud, C.; Millasseau, P.; Zeviani, M.; et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell 1995, 80, 155–165. [Google Scholar] [CrossRef]
- Mailman, M.D.; Heinz, J.W.; Papp, A.C.; Snyder, P.J.; Sedra, M.S.; Wirth, B.; Burghes, A.H.M.; Prior, T.W. Molecular analysis of spinal muscular atrophy and modification of the phenotype by SMN2. Genet. Med. 2002, 4, 20–26. [Google Scholar] [CrossRef]
- Prior, T.W.; Krainer, A.R.; Hua, Y.; Swoboda, K.J.; Snyder, P.C.; Bridgeman, S.J.; Burghes, A.H.M.; Kissel, J.T. A positive modifier of spinal muscular atrophy in the SMN2 gene. Am. J. Hum. Genet. 2009, 85, 408–413. [Google Scholar] [CrossRef]
- Monani, U.R.; Sendtner, M.; Coovert, D.D.; Parsons, D.W.; Andreassi, C.; Le, T.T.; Jablonka, S.; Schrank, B.; Rossoll, W.; Prior, T.W.; et al. The human centromeric survival motor neuron gene (SMN2) rescues embryonic lethality in Smn(-/-) mice and results in a mouse with spinal muscular atrophy. Hum. Mol. Genet. 2000, 9, 333–339. [Google Scholar] [CrossRef]
- Rouzier, C.; Chaussenot, A.; Paquis-Flucklinger, V. Molecular diagnosis and genetic counseling for spinal muscular atrophy (SMA). Arch. Pediatr. 2020, 27, 7S9–7S14. [Google Scholar] [CrossRef]
- Ojala, K.S.; Reedich, E.J.; DiDonato, C.J.; Meriney, S.D. In Search of a Cure: The Development of Therapeutics to Alter the Progression of Spinal Muscular Atrophy. Brain Sci. 2021, 11. [Google Scholar] [CrossRef]
- Prior, T.W.; Leach, M.E.; Finanger, E. Spinal Muscular Atrophy. In GeneReviews; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Polido, G.J.; Miranda, M.M.V.D.; Carvas, N.; Mendonça, R.D.H.; Caromano, F.A.; Reed, U.C.; Zanoteli, E.; Voos, M.C. Cognitive performance of children with spinal muscular atrophy: A systematic review. Dement. Neuropsychol. 2019, 13, 436–443. [Google Scholar] [CrossRef]
- Darras, B.T.; Farrar, M.A.; Mercuri, E.; Finkel, R.S.; Foster, R.; Hughes, S.G.; Bhan, I.; Farwell, W.; Gheuens, S. An Integrated Safety Analysis of Infants and Children with Symptomatic Spinal Muscular Atrophy (SMA) Treated with Nusinersen in Seven Clinical Trials. CNS Drugs 2019, 33, 919–932. [Google Scholar] [CrossRef] [PubMed]
- Prior, T.W.; Nagan, N.; Sugarman, E.A.; Batish, S.D.; Braastad, C. Technical standards and guidelines for spinal muscular atrophy testing. Genet. Med. 2011, 13, 686–694. [Google Scholar] [CrossRef] [PubMed]
- Babić, M.; Banović, M.; Berečić, I.; Banić, T.; Babić Leko, M.; Ulamec, M.; Junaković, A.; Kopić, J.; Sertić, J.; Barišić, N.; et al. Molecular Biomarkers for the Diagnosis, Prognosis, and Pharmacodynamics of Spinal Muscular Atrophy. J. Clin. Med. 2023, 12. [Google Scholar] [CrossRef]
- Burr, P.; Reddivari, A.K.R. Spinal Muscle Atrophy. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- D’Amico, A.; Mercuri, E.; Tiziano, F.D.; Bertini, E. Spinal muscular atrophy. Orphanet J. Rare Dis. 2011, 6, 71. [Google Scholar] [CrossRef] [PubMed]
- Bartels, B.; Montes, J.; van der Pol, W.L.; de Groot, J.F. Physical exercise training for type 3 spinal muscular atrophy. Cochrane Database Syst. Rev. 2019, 3, CD012120. [Google Scholar] [CrossRef] [PubMed]
- Strunk, A.; Abbes, A.; Stuitje, A.; Hettinga, C.; Sepers, E.; Snetselaar, R.; Schouten, J.; Asselman, F.-L.; Cuppen, I.; Lemmink, H.; et al. Validation of a Fast, Robust, Inexpensive, Two-Tiered Neonatal Screening Test algorithm on Dried Blood Spots for Spinal Muscular Atrophy. Int. J. Neonatal Screen. 2019, 5, 21. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Liu, L.; Peter, I.; Zhu, J.; Scott, S.A.; Zhao, G.; Eversley, C.; Kornreich, R.; Desnick, R.J.; Edelmann, L. An Ashkenazi Jewish SMN1 haplotype specific to duplication alleles improves pan-ethnic carrier screening for spinal muscular atrophy. Genet. Med. 2014, 16, 149–156. [Google Scholar] [CrossRef]
- Mercuri, E.; Finkel, R.S.; Muntoni, F.; Wirth, B.; Montes, J.; Main, M.; Mazzone, E.S.; Vitale, M.; Snyder, B.; Quijano-Roy, S.; et al. Diagnosis and management of spinal muscular atrophy: Part 1: Recommendations for diagnosis, rehabilitation, orthopedic and nutritional care. Neuromuscul. Disord. 2018, 28, 103–115. [Google Scholar] [CrossRef]
- Vezain, M.; Saugier-Veber, P.; Goina, E.; Touraine, R.; Manel, V.; Toutain, A.; Fehrenbach, S.; Frébourg, T.; Pagani, F.; Tosi, M.; et al. A rare SMN2 variant in a previously unrecognized composite splicing regulatory element induces exon 7 inclusion and reduces the clinical severity of spinal muscular atrophy. Hum. Mutat. 2010, 31, E1110–E1125. [Google Scholar] [CrossRef]
- Arkblad, E.L.; Darin, N.; Berg, K.; Kimber, E.; Brandberg, G.; Lindberg, C.; Holmberg, E.; Tulinius, M.; Nordling, M. Multiplex ligation-dependent probe amplification improves diagnostics in spinal muscular atrophy. Neuromuscul. Disord. 2006, 16, 830–838. [Google Scholar] [CrossRef]
- Passon, N.; Dubsky de Wittenau, G.; Jurman, I.; Radovic, S.; Bregant, E.; Molinis, C.; Damante, G.; Lonigro, I.R. Quick MLPA test for quantification of SMN1 and SMN2 copy numbers. Mol. Cell. Probes 2010, 24, 310–314. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M.J.; Asselman, F.-L.; Kruitwagen-van Reenen, E.T.; Verhoef, M.; Wadman, R.I.; Visser-Meily, J.M.A.; van der Pol, W.L.; Schröder, C.D. Psychological well-being in adults with spinal muscular atrophy: The contribution of participation and psychological needs. Disabil. Rehabil. 2020, 42, 2262–2270. [Google Scholar] [CrossRef] [PubMed]
- Sari, D.M.; Wijaya, L.C.G.; Sitorus, W.D.R.; Dewi, M.M. Psychological burden in spinal muscular atrophy patients and their families: A systematic review. Egypt. J. Neurol. Psychiatry Neurosurg. 2022, 58, 140. [Google Scholar] [CrossRef]
- Wan, H.W.Y.; Carey, K.A.; D’Silva, A.; Kasparian, N.A.; Farrar, M.A. “Getting ready for the adult world”: How adults with spinal muscular atrophy perceive and experience healthcare, transition and well-being. Orphanet J. Rare Dis. 2019, 14, 74. [Google Scholar] [CrossRef] [PubMed]
- Kruitwagen-Van Reenen, E.T.; Wadman, R.I.; Visser-Meily, J.M.; van den Berg, L.H.; Schröder, C.; van der Pol, W.L. Correlates of health related quality of life in adult patients with spinal muscular atrophy. Muscle Nerve 2016, 54, 850–855. [Google Scholar] [CrossRef]
- Iannaccone, S.T.; Hynan, L.S.; Morton, A.; Buchanan, R.; Limbers, C.A.; Varni, J.W. The PedsQLTM in pediatric patients with Spinal Muscular Atrophy: Feasibility, reliability, and validity of the Pediatric Quality of Life InventoryTM Generic Core Scales and Neuromuscular Module. Neuromuscul. Disord. 2009, 19, 805–812. [Google Scholar] [CrossRef]
- Ditchman, N.; Sung, C.; Easton, A.B.; Johnson, K.S.; Batchos, E. Symptom severity and life satisfaction in brain injury: The mediating role of disability acceptance and social self-efficacy. NeuroRehabilitation 2017, 40, 531–543. [Google Scholar] [CrossRef]
- Park, E.-Y. Rasch Analysis of the Disability Acceptance Scale for Individuals With Cerebral Palsy. Front. Neurol. 2019, 10. [Google Scholar] [CrossRef]
- Steger, M.F.; Oishi, S.; Kashdan, T.B. Meaning in Life across the Life Span: Levels and Correlates of Meaning in Life from Emerging Adulthood to Older Adulthood. J. Posit. Psychol. 2009, 4, 43–52. [Google Scholar] [CrossRef]
- Szcześniak, M.; Świątek, A.H.; Cieślak, M.; Świdurska, D. Disease Acceptance and Eudemonic Well-Being Among Adults With Physical Disabilities: The Mediator Effect of Meaning in Life. Front. Psychol. 2020, 11, 525560. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, Y.; Deng, X.; Deng, C.; Pan, Y.; Hu, A. The Correlation Between Quality of Life and Acceptability of Disability in Patients With Facial Burn Scars. Front. Bioeng. Biotechnol. 2019, 7. [Google Scholar] [CrossRef] [PubMed]
- Wilson, S.; Washington, L.A.; Engel, J.M.; Ciol, M.A.; Jensen, M.P. Perceived social support, psychological adjustment, and functional ability in youths with physical disabilities. Rehabil. Psychol. 2006, 51, 322–330. [Google Scholar] [CrossRef]
- Lamb, C.; Peden, A. Understanding the Experience of Living with Spinal Muscular Atrophy. J. Neurosci. Nurs. 2008, 40, 250–256. [Google Scholar] [CrossRef]
- Ho, H.-M.; Tseng, Y.-H.; Hsin, Y.-M.; Chou, F.-H.; Lin, W.-T. Living with illness and self-transcendence: The lived experience of patients with spinal muscular atrophy. J. Adv. Nurs. 2016, 72, 2695–2705. [Google Scholar] [CrossRef]
- Post, M.W.M.; van der Zee, C.H.; Hennink, J.; Schafrat, C.G.; Visser-Meily, J.M.A.; van Berlekom, S.B. Validity of the utrecht scale for evaluation of rehabilitation-participation. Disabil. Rehabil. 2012, 34, 478–485. [Google Scholar] [CrossRef] [PubMed]
- Brandt, M.; Johannsen, L.; Inhestern, L.; Bergelt, C. Parents as informal caregivers of children and adolescents with spinal muscular atrophy: A systematic review of quantitative and qualitative data on the psychosocial situation, caregiver burden, and family needs. Orphanet J. Rare Dis. 2022, 17, 274. [Google Scholar] [CrossRef] [PubMed]
- Bramanti, S.M.; Trumello, C.; Lombardi, L.; Babore, A. COVID-19 and chronic disease patients: Perceived stress, worry, and emotional regulation strategies. Rehabil. Psychol. 2021, 66, 380–385. [Google Scholar] [CrossRef] [PubMed]
- Kruitwagen-van Reenen, E.T.; van der Pol, L.; Schröder, C.; Wadman, R.I.; van den Berg, L.H.; Visser-Meily, J.M.A.; Post, M.W.M. Social participation of adult patients with spinal muscular atrophy: Frequency, restrictions, satisfaction, and correlates. Muscle Nerve 2018, 58, 805–811. [Google Scholar] [CrossRef]
- Boardman, F.K.; Young, P.J.; Griffiths, F.E. Impairment Experiences, Identity and Attitudes Towards Genetic Screening: The Views of People with Spinal Muscular Atrophy. J. Genet. Couns. 2018, 27, 69–84. [Google Scholar] [CrossRef]
- Günther, R.; Wurster, C.D.; Cordts, I.; Koch, J.C.; Kamm, C.; Petzold, D.; Aust, E.; Deschauer, M.; Lingor, P.; Ludolph, A.C.; et al. Patient-Reported Prevalence of Non-motor Symptoms Is Low in Adult Patients Suffering From 5q Spinal Muscular Atrophy. Front. Neurol. 2019, 10. [Google Scholar] [CrossRef]
- Yao, M.; Xia, Y.; Feng, Y.; Ma, Y.; Hong, Y.; Zhang, Y.; Chen, J.; Yuan, C.; Mao, S. Anxiety and depression in school-age patients with spinal muscular atrophy: A cross-sectional study. Orphanet J. Rare Dis. 2021, 16, 385. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.H.; Finkel, R.S.; Bertini, E.S.; Schroth, M.; Simonds, A.; Wong, B.; Aloysius, A.; Morrison, L.; Main, M.; Crawford, T.O.; et al. Consensus statement for standard of care in spinal muscular atrophy. J. Child. Neurol. 2007, 22, 1027–1049. [Google Scholar] [CrossRef] [PubMed]
- Bladen, C.L.; Thompson, R.; Jackson, J.M.; Garland, C.; Wegel, C.; Ambrosini, A.; Pisano, P.; Walter, M.C.; Schreiber, O.; Lusakowska, A.; et al. Mapping the differences in care for 5,000 spinal muscular atrophy patients, a survey of 24 national registries in North America, Australasia and Europe. J. Neurol. 2014, 261, 152–163. [Google Scholar] [CrossRef] [PubMed]
- Finkel, R.S.; Chiriboga, C.A.; Vajsar, J.; Day, J.W.; Montes, J.; De Vivo, D.C.; Yamashita, M.; Rigo, F.; Hung, G.; Schneider, E.; et al. Treatment of infantile-onset spinal muscular atrophy with nusinersen: A phase 2, open-label, dose-escalation study. Lancet 2016, 388, 3017–3026. [Google Scholar] [CrossRef]
- Day, J.W.; Howell, K.; Place, A.; Long, K.; Rossello, J.; Kertesz, N.; Nomikos, G. Advances and limitations for the treatment of spinal muscular atrophy. BMC Pediatr. 2022, 22, 632. [Google Scholar] [CrossRef]
- Ramos-Platt, L.; Elman, L.; Shieh, P.B. Experience and Perspectives in the US on the Evolving Treatment Landscape in Spinal Muscular Atrophy. Int. J. Gen. Med. 2022, 15, 7341–7353. [Google Scholar] [CrossRef]
- Nishio, H.; Niba, E.T.E.; Saito, T.; Okamoto, K.; Takeshima, Y.; Awano, H. Spinal Muscular Atrophy: The Past, Present, and Future of Diagnosis and Treatment. Int. J. Mol. Sci. 2023, 24, 11939. [Google Scholar] [CrossRef]
- Ottesen, E.W. ISS-N1 makes the First FDA-approved Drug for Spinal Muscular Atrophy. Transl. Neurosci. 2017, 8, 1–6. [Google Scholar] [CrossRef]
- Qiu, J.; Wu, L.; Qu, R.; Jiang, T.; Bai, J.; Sheng, L.; Feng, P.; Sun, J. History of development of the life-saving drug “Nusinersen” in spinal muscular atrophy. Front. Cell. Neurosci. 2022, 16, 942976. [Google Scholar] [CrossRef]
- Vázquez-Costa, J.F.; Povedano, M.; Nascimiento-Osorio, A.E.; Moreno Escribano, A.; Kapetanovic Garcia, S.; Dominguez, R.; Exposito, J.M.; González, L.; Marco, C.; Medina Castillo, J.; et al. Nusinersen in adult patients with 5q spinal muscular atrophy: A multicenter observational cohorts’ study. Eur. J. Neurol. 2022, 29, 3337–3346. [Google Scholar] [CrossRef]
- Park, J.-M.; Min, Y.-S.; Park, D.; Park, J.-S. Effect of Nusinersen in a late onset spinal muscular atrophy patient for 14 months: A case report. Medicine 2021, 100, e24236. [Google Scholar] [CrossRef] [PubMed]
- Belančić, A.; Strbad, T.; Kučan Štiglić, M.; Vitezić, D. Effectiveness of Nusinersen in Type 1, 2 and 3 Spinal Muscular Atrophy: Croatian Real-World Data. J. Clin. Med. 2023, 12, 2839. [Google Scholar] [CrossRef] [PubMed]
- Dunaway Young, S.; Montes, J.; Glanzman, A.M.; Gee, R.; Day, J.W.; Finkel, R.S.; Darras, B.T.; De Vivo, D.C.; Gambino, G.; Foster, R.; et al. Nusinersen Treatment of Children with Later-Onset Spinal Muscular Atrophy and Scoliosis Is Associated with Improvements or Stabilization of Motor Function. J. Clin. Med. 2023, 12, 4901. [Google Scholar] [CrossRef]
- Crawford, T.O.; Swoboda, K.J.; De Vivo, D.C.; Bertini, E.; Hwu, W.-L.; Finkel, R.S.; Kirschner, J.; Kuntz, N.L.; Nazario, A.N.; Parsons, J.A.; et al. Continued benefit of nusinersen initiated in the presymptomatic stage of spinal muscular atrophy: 5-year update of the NURTURE study. Muscle Nerve 2023, 68, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Schorling, D.C.; Pechmann, A.; Kirschner, J. Advances in Treatment of Spinal Muscular Atrophy—New Phenotypes, New Challenges, New Implications for Care. J. Neuromuscul. Dis. 2020, 7, 1–13. [Google Scholar] [CrossRef]
- Zhong, Z.-J.; Zheng, P.-M.; Dou, H.-H.; Wang, J.-G. Adverse events in the treatment of spinal muscular atrophy in children and adolescents with nusinersen: A systematic review and meta-analysis. Front. Pediatr. 2023, 11, 1152318. [Google Scholar] [CrossRef] [PubMed]
- Stolte, B.; Nonnemacher, M.; Kizina, K.; Bolz, S.; Totzeck, A.; Thimm, A.; Wagner, B.; Deuschl, C.; Kleinschnitz, C.; Hagenacker, T. Nusinersen treatment in adult patients with spinal muscular atrophy: A safety analysis of laboratory parameters. J. Neurol. 2021, 268, 4667–4679. [Google Scholar] [CrossRef]
- Wu, H.; Wahane, A.; Alhamadani, F.; Zhang, K.; Parikh, R.; Lee, S.; McCabe, E.M.; Rasmussen, T.P.; Bahal, R.; Zhong, X.-B.; et al. Nephrotoxicity of marketed antisense oligonucleotide drugs. Curr. Opin. Toxicol. 2022, 32, 100373. [Google Scholar] [CrossRef]
- Albrechtsen, S.S.; Born, A.P.; Boesen, M.S. Nusinersen treatment of spinal muscular atrophy—A systematic review. Dan. Med. J. 2020, 67, A02200100. [Google Scholar]
- Gidaro, T.; Servais, L. Nusinersen treatment of spinal muscular atrophy: Current knowledge and existing gaps. Dev. Med. Child. Neurol. 2019, 61, 19–24. [Google Scholar] [CrossRef]
- Mahajan, R. Onasemnogene Abeparvovec for Spinal Muscular Atrophy: The Costlier Drug Ever. Int. J. Appl. Basic Med. Res. 2019, 9, 127–128. [Google Scholar] [CrossRef] [PubMed]
- Kotulska, K.; Fattal-Valevski, A.; Haberlova, J. Recombinant Adeno-Associated Virus Serotype 9 Gene Therapy in Spinal Muscular Atrophy. Front. Neurol. 2021, 12, 726468. [Google Scholar] [CrossRef] [PubMed]
- Kichula, E.A.; Proud, C.M.; Farrar, M.A.; Kwon, J.M.; Saito, K.; Desguerre, I.; McMillan, H.J. Expert recommendations and clinical considerations in the use of onasemnogene abeparvovec gene therapy for spinal muscular atrophy. Muscle Nerve 2021, 64, 413–427. [Google Scholar] [CrossRef] [PubMed]
- Strauss, K.A.; Farrar, M.A.; Muntoni, F.; Saito, K.; Mendell, J.R.; Servais, L.; McMillan, H.J.; Finkel, R.S.; Swoboda, K.J.; Kwon, J.M.; et al. Onasemnogene abeparvovec for presymptomatic infants with three copies of SMN2 at risk for spinal muscular atrophy: The Phase III SPR1NT trial. Nat. Med. 2022, 28, 1390–1397. [Google Scholar] [CrossRef]
- Jędrzejowska, M. Advances in Newborn Screening and Presymptomatic Diagnosis of Spinal Muscular Atrophy. Degener. Neurol. Neuromuscul. Dis. 2020, 10, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Panagiotou, P.; Kanaka-Gantenbein, C.; Kaditis, A.G. Changes in Ventilatory Support Requirements of Spinal Muscular Atrophy (SMA) Patients Post Gene-Based Therapies. Children 2022, 9, 1207. [Google Scholar] [CrossRef] [PubMed]
- Mendell, J.R.; Connolly, A.M.; Lehman, K.J.; Griffin, D.A.; Khan, S.Z.; Dharia, S.D.; Quintana-Gallardo, L.; Rodino-Klapac, L.R. Testing preexisting antibodies prior to AAV gene transfer therapy: Rationale, lessons and future considerations. Mol. Ther. Methods Clin. Dev. 2022, 25, 74–83. [Google Scholar] [CrossRef] [PubMed]
- Gorovits, B.; Azadeh, M.; Buchlis, G.; Harrison, T.; Havert, M.; Jawa, V.; Long, B.; McNally, J.; Milton, M.; Nelson, R.; et al. Evaluation of the Humoral Response to Adeno-Associated Virus-Based Gene Therapy Modalities Using Total Antibody Assays. AAPS J. 2021, 23, 108. [Google Scholar] [CrossRef]
- Chand, D.H.; Zaidman, C.; Arya, K.; Millner, R.; Farrar, M.A.; Mackie, F.E.; Goedeker, N.L.; Dharnidharka, V.R.; Dandamudi, R.; Reyna, S.P. Thrombotic Microangiopathy Following Onasemnogene Abeparvovec for Spinal Muscular Atrophy: A Case Series. J. Pediatr. 2021, 231, 265–268. [Google Scholar] [CrossRef]
- Guillou, J.; de Pellegars, A.; Porcheret, F.; Frémeaux-Bacchi, V.; Allain-Launay, E.; Debord, C.; Denis, M.; Péréon, Y.; Barnérias, C.; Desguerre, I.; et al. Fatal thrombotic microangiopathy case following adeno-associated viral SMN gene therapy. Blood Adv. 2022, 6, 4266–4270. [Google Scholar] [CrossRef]
- Dhillon, S. Risdiplam: First Approval. Drugs 2020, 80, 1853–1858. [Google Scholar] [CrossRef] [PubMed]
- Kakazu, J.; Walker, N.L.; Babin, K.C.; Trettin, K.A.; Lee, C.; Sutker, P.B.; Kaye, A.M.; Kaye, A.D. Risdiplam for the Use of Spinal Muscular Atrophy. Orthop. Rev. 2021, 13, 25579. [Google Scholar] [CrossRef] [PubMed]
- Menduti, G.; Rasà, D.M.; Stanga, S.; Boido, M. Drug Screening and Drug Repositioning as Promising Therapeutic Approaches for Spinal Muscular Atrophy Treatment. Front. Pharmacol. 2020, 11, 592234. [Google Scholar] [CrossRef] [PubMed]
- Ribero, V.A.; Daigl, M.; Martí, Y.; Gorni, K.; Evans, R.; Scott, D.A.; Mahajan, A.; Abrams, K.R.; Hawkins, N. How does risdiplam compare with other treatments for Types 1-3 spinal muscular atrophy: A systematic literature review and indirect treatment comparison. J. Comp. Eff. Res. 2022, 11, 347–370. [Google Scholar] [CrossRef]
- Bowerman, M.; Becker, C.G.; Yáñez-Muñoz, R.J.; Ning, K.; Wood, M.J.A.; Gillingwater, T.H.; Talbot, K.; UK SMA Research Consortium. Therapeutic strategies for spinal muscular atrophy: SMN and beyond. Dis. Model. Mech. 2017, 10, 943–954. [Google Scholar] [CrossRef]
- Deng, S.; Lee, B.H.; Ciafaloni, E. Parent Perceptions in Choosing Treatment for Infants With Spinal Muscular Atrophy Diagnosed Through Newborn Screening. J. Child. Neurol. 2022, 37, 43–49. [Google Scholar] [CrossRef]
- Abiusi, E.; Vaisfeld, A.; Fiori, S.; Novelli, A.; Spartano, S.; Faggiano, M.V.; Giovanniello, T.; Angeloni, A.; Vento, G.; Santoloci, R.; et al. Experience of a 2-year spinal muscular atrophy NBS pilot study in Italy: Towards specific guidelines and standard operating procedures for the molecular diagnosis. J. Med. Genet. 2023, 60, 697–705. [Google Scholar] [CrossRef]
- Farrar, M.A.; Park, S.B.; Vucic, S.; Carey, K.A.; Turner, B.J.; Gillingwater, T.H.; Swoboda, K.J.; Kiernan, M.C. Emerging therapies and challenges in spinal muscular atrophy. Ann. Neurol. 2017, 81, 355–368. [Google Scholar] [CrossRef]
- Swoboda, K.J.; Prior, T.W.; Scott, C.B.; McNaught, T.P.; Wride, M.C.; Reyna, S.P.; Bromberg, M.B. Natural history of denervation in SMA: Relation to age, SMN2 copy number, and function. Ann. Neurol. 2005, 57, 704–712. [Google Scholar] [CrossRef]
- De Vivo, D.C.; Bertini, E.; Swoboda, K.J.; Hwu, W.-L.; Crawford, T.O.; Finkel, R.S.; Kirschner, J.; Kuntz, N.L.; Parsons, J.A.; Ryan, M.M.; et al. Nusinersen initiated in infants during the presymptomatic stage of spinal muscular atrophy: Interim efficacy and safety results from the Phase 2 NURTURE study. Neuromuscul. Disord. 2019, 29, 842–856. [Google Scholar] [CrossRef]
- McMillan, H.J.; Kernohan, K.D.; Yeh, E.; Amburgey, K.; Boyd, J.; Campbell, C.; Dowling, J.J.; Gonorazky, H.; Marcadier, J.; Tarnopolsky, M.A.; et al. Newborn Screening for Spinal Muscular Atrophy: Ontario Testing and Follow-up Recommendations. Can. J. Neurol. Sci. 2021, 48, 504–511. [Google Scholar] [CrossRef] [PubMed]
- Kernohan, K.D.; McMillan, H.J.; Yeh, E.; Lacaria, M.; Kowalski, M.; Campbell, C.; Dowling, J.J.; Gonorazky, H.; Marcadier, J.; Tarnopolsky, M.A.; et al. Ontario Newborn Screening for Spinal Muscular Atrophy: The First Year. Can. J. Neurol. Sci. 2022, 49, 821–823. [Google Scholar] [CrossRef] [PubMed]
- Andermann, A.; Blancquaert, I.; Beauchamp, S.; Déry, V. Revisiting Wilson and Jungner in the genomic age: A review of screening criteria over the past 40 years. Bull. World Health Organ. 2008, 86, 317–319. [Google Scholar] [CrossRef]
- Prior, T.W.; Nagan, N. Spinal Muscular Atrophy: Overview of Molecular Diagnostic Approaches. Curr. Protoc. Hum. Genet. 2016, 88, 9.27.1–9.27.13. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.M.; Jungner, Y.G. Principles and practice of mass screening for disease. Bol. Oficina Sanit. Panam. 1968, 65, 281–393. [Google Scholar]
- Dangouloff, T.; Vrščaj, E.; Servais, L.; Osredkar, D.; SMA NBS World Study Group. Newborn screening programs for spinal muscular atrophy worldwide: Where we stand and where to go. Neuromuscul. Disord. 2021, 31, 574–582. [Google Scholar] [CrossRef]
- Kariyawasam, D.S.T.; Russell, J.S.; Wiley, V.; Alexander, I.E.; Farrar, M.A. The implementation of newborn screening for spinal muscular atrophy: The Australian experience. Genet. Med. 2020, 22, 557–565. [Google Scholar] [CrossRef]
- D’Silva, A.M.; Kariyawasam, D.S.T.; Best, S.; Wiley, V.; Farrar, M.A.; NSW SMA NBS Study Group. Integrating newborn screening for spinal muscular atrophy into health care systems: An Australian pilot programme. Dev. Med. Child. Neurol. 2022, 64, 625–632. [Google Scholar] [CrossRef]
- Boemer, F.; Caberg, J.-H.; Beckers, P.; Dideberg, V.; di Fiore, S.; Bours, V.; Marie, S.; Dewulf, J.; Marcelis, L.; Deconinck, N.; et al. Three years pilot of spinal muscular atrophy newborn screening turned into official program in Southern Belgium. Sci. Rep. 2021, 11, 19922. [Google Scholar] [CrossRef]
- Vill, K.; Kölbel, H.; Schwartz, O.; Blaschek, A.; Olgemöller, B.; Harms, E.; Burggraf, S.; Röschinger, W.; Durner, J.; Gläser, D.; et al. One Year of Newborn Screening for SMA—Results of a German Pilot Project. J. Neuromuscul. Dis. 2019, 6, 503–515. [Google Scholar] [CrossRef]
- Vill, K.; Schwartz, O.; Blaschek, A.; Gläser, D.; Nennstiel, U.; Wirth, B.; Burggraf, S.; Röschinger, W.; Becker, M.; Czibere, L.; et al. Newborn screening for spinal muscular atrophy in Germany: Clinical results after 2 years. Orphanet J. Rare Dis. 2021, 16, 153. [Google Scholar] [CrossRef]
- Shinohara, M.; Niba, E.T.E.; Wijaya, Y.O.S.; Takayama, I.; Mitsuishi, C.; Kumasaka, S.; Kondo, Y.; Takatera, A.; Hokuto, I.; Morioka, I.; et al. A Novel System for Spinal Muscular Atrophy Screening in Newborns: Japanese Pilot Study. Int. J. Neonatal Screen. 2019, 5, 41. [Google Scholar] [CrossRef] [PubMed]
- Sawada, T.; Kido, J.; Sugawara, K.; Yoshida, S.; Ozasa, S.; Nomura, K.; Okada, K.; Fujiyama, N.; Nakamura, K. Newborn screening for spinal muscular atrophy in Japan: One year of experience. Mol. Genet. Metab. Rep. 2022, 32, 100908. [Google Scholar] [CrossRef] [PubMed]
- Chien, Y.-H.; Chiang, S.-C.; Weng, W.-C.; Lee, N.-C.; Lin, C.-J.; Hsieh, W.-S.; Lee, W.-T.; Jong, Y.-J.; Ko, T.-M.; Hwu, W.-L. Presymptomatic Diagnosis of Spinal Muscular Atrophy Through Newborn Screening. J. Pediatr. 2017, 190, 124–129.e1. [Google Scholar] [CrossRef] [PubMed]
- Hale, K.; Ojodu, J.; Singh, S. Landscape of Spinal Muscular Atrophy Newborn Screening in the United States: 2018-2021. Int. J. Neonatal Screen. 2021, 7, 33. [Google Scholar] [CrossRef]
- Pino, M.G.; Rich, K.A.; Kolb, S.J. Update on Biomarkers in Spinal Muscular Atrophy. Biomark. Insights 2021, 16, 11772719211035644. [Google Scholar] [CrossRef]
- Taylor, J.L.; Lee, F.K.; Yazdanpanah, G.K.; Staropoli, J.F.; Liu, M.; Carulli, J.P.; Sun, C.; Dobrowolski, S.F.; Hannon, W.H.; Vogt, R.F. Newborn blood spot screening test using multiplexed real-time PCR to simultaneously screen for spinal muscular atrophy and severe combined immunodeficiency. Clin. Chem. 2015, 61, 412–419. [Google Scholar] [CrossRef]
- Ghetti, G.; Mennini, F.; Marcellusi, A.; Bischof, M.; Pistillo, G.; Pane, M. PCR145 Cost-Effectiveness Analysis of Newborn Screening for Spinal Muscular Atrophy (SMA) in Italy. Value Heal. 2022, 25, S419. [Google Scholar] [CrossRef]
- Bani, M.; Russo, S.; Raggi, E.; Gasperini, S.; Motta, S.; Menni, F.; Furlan, F.; Cefalo, G.; Paci, S.; Banderali, G.; et al. Parents’ experience of the communication process of positivity at newborn screening for metabolic diseases: A qualitative study. Child. Care. Health Dev. 2023. [Google Scholar] [CrossRef]
- Buchbinder, M.; Timmermans, S. Newborn Screening for Metabolic Disorders. Clin. Pediatr. 2012, 51, 739–744. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SMA Type | Age of Onset | Motor Milestones | Other Characteristics |
Life Expectancy | SMN2 Copy Number |
|---|---|---|---|---|---|
| SMA 0 | Pre-natal | None reached |
| <6 months | 1 |
| SMA 1 | 0–6 months | Some head control, sits with support |
| <2 years | 2 |
| SMA 2 | 6–18 months | Sits, never stands independently |
| >2 years | 2–3 |
| SMA 3 | >18 months | Walks |
| Adult | 3–4 |
| SMA 4 | Adulthood | All motor functions |
| Adult | ≥4 |
| Technique | Advantages | Limitations |
|---|---|---|
| multiplex qPCR |
|
|
| MLPA |
|
|
| Therapy | Mechanism of Action | Times and Method of Administration | Adverse Reactions | Age of Administration | Patients for Which It Is Intended |
|---|---|---|---|---|---|
| Evrysdi (Risdiplam) | Small molecule, splicing modifier of the SMN2 gene | Orally, once a day (always at the same time) after eating |
| ≥2 months | Patients with SMA 5q (mutations in the SMN1 gene), 1 to 4 copies of the SMN2 gene SMA 0 and SMA 4 patients excluded Not compatible with patients being treated with nusinersen or onasemnogene abeparvovec |
| Spinraza (Nusinersen) | Antisense oligonucleotide, splicing modifier of the SMN2 gene | Intrathecal injection, on days 0, 14, 28, and 63; followed by a maintenance dose once every 4 months |
| At birth | Patients with a genetically confirmed diagnosis of SMA 5q (mutations in the SMN1 gene) |
| Zolgensma (Onasemnogene abeparvovec) | Non-replicating recombinant adeno-associated virus serotype 9 (AAV9)-based vector containing the cDNA of the human SMN gene | Intravenous injection by slow infusion over about an hour, once in a lifetime |
| At birth | SMA 5q patients up to 13.5 kg:
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Angilletta, I.; Ferrante, R.; Giansante, R.; Lombardi, L.; Babore, A.; Dell’Elice, A.; Alessandrelli, E.; Notarangelo, S.; Ranaudo, M.; Palmarini, C.; et al. Spinal Muscular Atrophy: An Evolving Scenario through New Perspectives in Diagnosis and Advances in Therapies. Int. J. Mol. Sci. 2023, 24, 14873. https://doi.org/10.3390/ijms241914873
Angilletta I, Ferrante R, Giansante R, Lombardi L, Babore A, Dell’Elice A, Alessandrelli E, Notarangelo S, Ranaudo M, Palmarini C, et al. Spinal Muscular Atrophy: An Evolving Scenario through New Perspectives in Diagnosis and Advances in Therapies. International Journal of Molecular Sciences. 2023; 24(19):14873. https://doi.org/10.3390/ijms241914873
Chicago/Turabian StyleAngilletta, Ilaria, Rossella Ferrante, Roberta Giansante, Lucia Lombardi, Alessandra Babore, Anastasia Dell’Elice, Elisa Alessandrelli, Stefania Notarangelo, Marianna Ranaudo, Claudia Palmarini, and et al. 2023. "Spinal Muscular Atrophy: An Evolving Scenario through New Perspectives in Diagnosis and Advances in Therapies" International Journal of Molecular Sciences 24, no. 19: 14873. https://doi.org/10.3390/ijms241914873
APA StyleAngilletta, I., Ferrante, R., Giansante, R., Lombardi, L., Babore, A., Dell’Elice, A., Alessandrelli, E., Notarangelo, S., Ranaudo, M., Palmarini, C., De Laurenzi, V., Stuppia, L., & Rossi, C. (2023). Spinal Muscular Atrophy: An Evolving Scenario through New Perspectives in Diagnosis and Advances in Therapies. International Journal of Molecular Sciences, 24(19), 14873. https://doi.org/10.3390/ijms241914873