
Annexin A2 Stabilizes Oncogenic JAG1 Intracellular Domain by Inhibiting Proteasomal Degradation in Glioblastoma Cells
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. ANXA2 Binds to JICD1 to Regulate JICD1-Driven CSC Properties
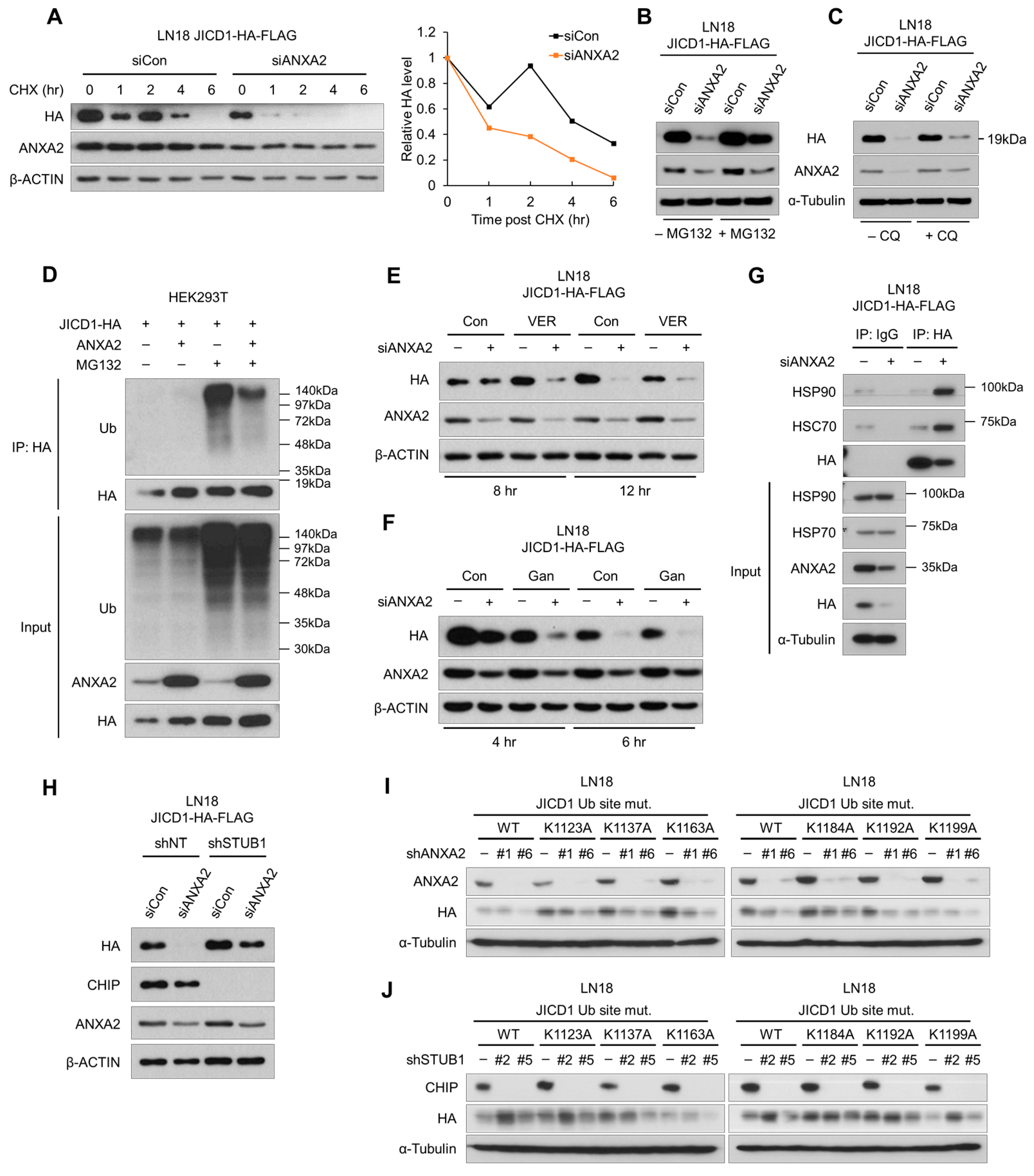
2.2. ANXA2 Inhibits Heat Shock Protein 70/90-Mediated Degradation of JICD1
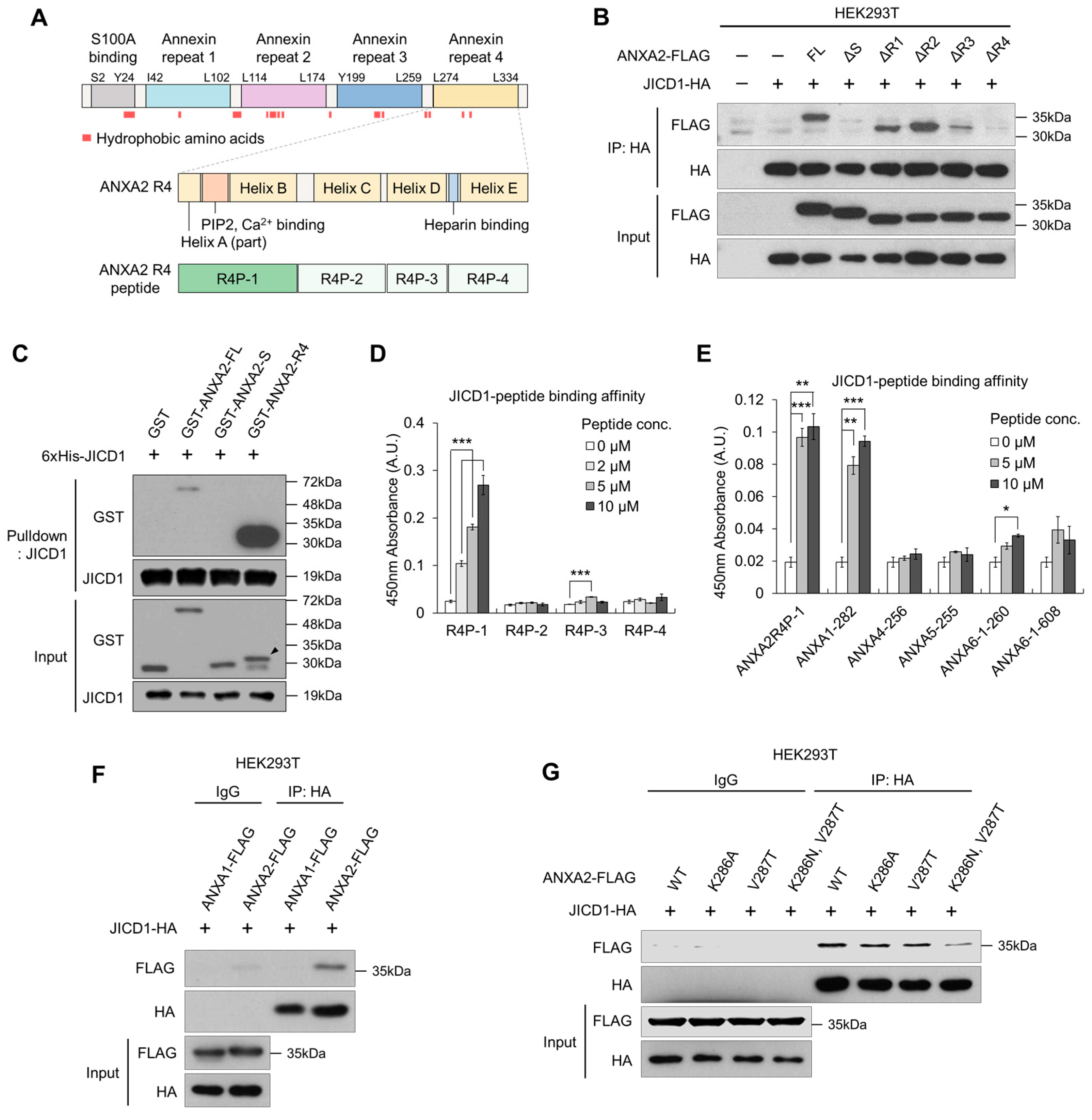
2.3. JICD1 Interacts with the Annexin Repeat 4 Domain within ANXA2
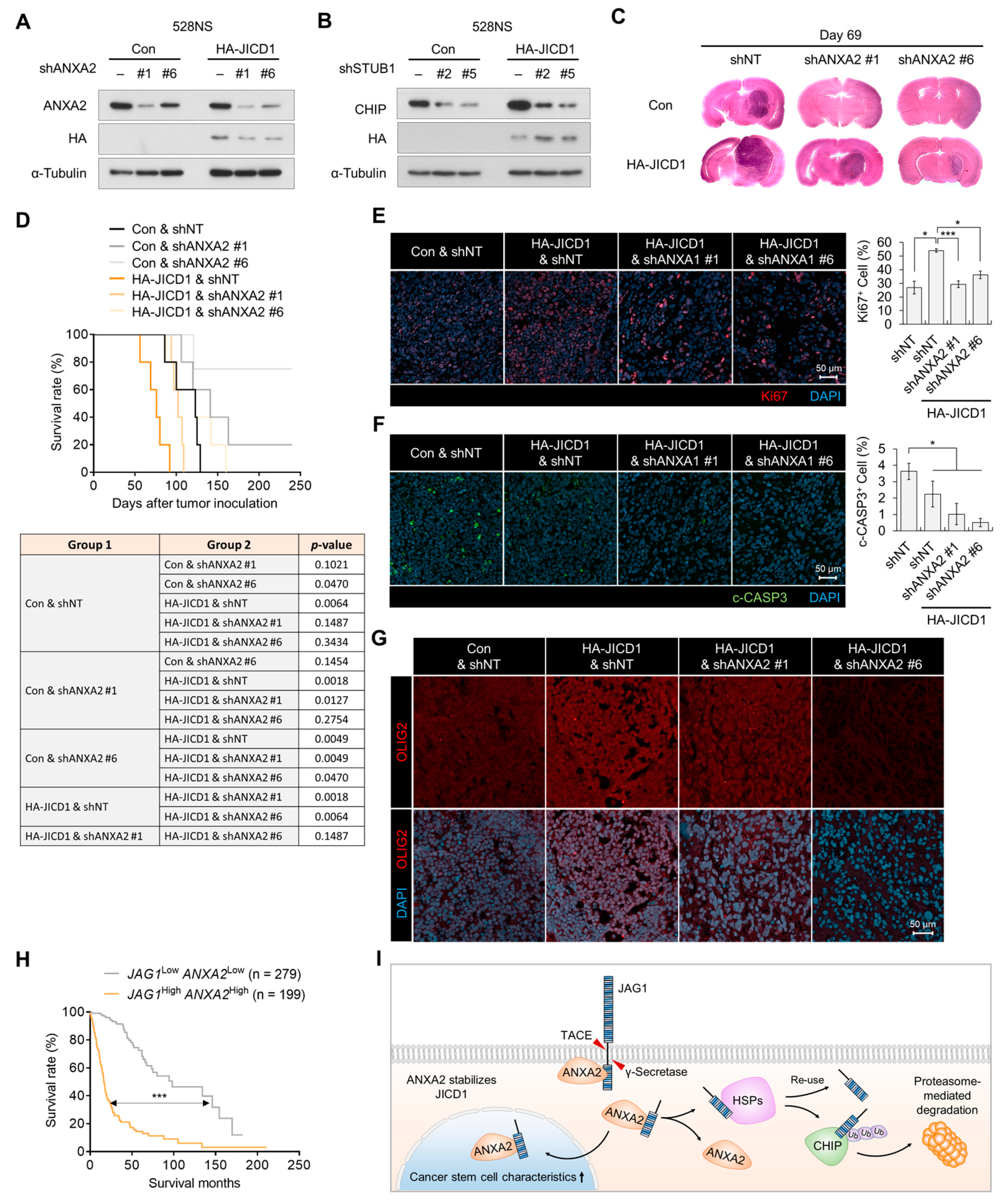
2.4. JICD1–ANXA2 Interaction Promotes Tumor Proliferation and Aggressiveness
3. Discussion
4. Methods
4.1. Cell Lines and Cell Culture
4.2. Overexpression and Knockdown of Genes
4.3. LDA
4.4. Antibodies
4.5. IP
4.6. Western Blotting
4.7. Immunofluorescence Imaging
4.8. PLA
4.9. Purification of the Recombinant Proteins
4.10. IVB
4.11. Quantitative Reverse Transcription Polymerase Chain Reaction
4.12. JICD1-Peptide Binding Assay
4.13. Peptide Sequence Clustering
4.14. Protein Structure Presentation
4.15. Intracranial Xenograft Model
4.16. Hematoxylin–Eosin Staining
4.17. In Silico Analysis
4.18. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ostrom, Q.T.; Price, M.; Neff, C.; Cioffi, G.; Waite, K.A.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2015–2019. Neuro-Oncol. 2022, 24, v1–v95. [Google Scholar] [CrossRef] [PubMed]
- Lukas, R.V.; Wainwright, D.A.; Ladomersky, E.; Sachdev, S.; Sonabend, A.M.; Stupp, R. Newly Diagnosed Glioblastoma: A Review on Clinical Management. Oncology 2019, 33, 91–100. [Google Scholar] [PubMed]
- Keime-Guibert, F.; Chinot, O.; Taillandier, L.; Cartalat-Carel, S.; Frenay, M.; Kantor, G.; Guillamo, J.S.; Jadaud, E.; Colin, P.; Bondiau, P.Y.; et al. Radiotherapy for glioblastoma in the elderly. N. Engl. J. Med. 2007, 356, 1527–1535. [Google Scholar] [CrossRef] [PubMed]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef]
- Eun, K.; Ham, S.W.; Kim, H. Cancer stem cell heterogeneity: Origin and new perspectives on CSC targeting. BMB Rep. 2017, 50, 117–125. [Google Scholar] [CrossRef]
- Prager, B.C.; Bhargava, S.; Mahadev, V.; Hubert, C.G.; Rich, J.N. Glioblastoma Stem Cells: Driving Resilience through Chaos. Trends Cancer 2020, 6, 223–235. [Google Scholar] [CrossRef]
- Bray, S.J. Notch signalling in context. Nat. Rev. Mol. Cell. Biol. 2016, 17, 722–735. [Google Scholar] [CrossRef]
- Nowell, C.S.; Radtke, F. Notch as a tumour suppressor. Nat. Rev. Cancer 2017, 17, 145–159. [Google Scholar] [CrossRef]
- Moore, G.; Annett, S.; McClements, L.; Robson, T. Top Notch Targeting Strategies in Cancer: A Detailed Overview of Recent Insights and Current Perspectives. Cells 2020, 9, 1503. [Google Scholar] [CrossRef]
- LaVoie, M.J.; Selkoe, D.J. The Notch ligands, Jagged and Delta, are sequentially processed by alpha-secretase and presenilin/gamma-secretase and release signaling fragments. J. Biol. Chem. 2003, 278, 34427–34437. [Google Scholar] [CrossRef]
- Kim, E.J.; Kim, J.Y.; Kim, S.O.; Hong, N.; Choi, S.H.; Park, M.G.; Jang, J.; Ham, S.W.; Seo, S.; Lee, S.Y.; et al. The oncogenic JAG1 intracellular domain is a transcriptional cofactor that acts in concert with DDX17/SMAD3/TGIF2. Cell Rep. 2022, 41, 111626. [Google Scholar] [CrossRef] [PubMed]
- De Biasio, A.; Guarnaccia, C.; Popovic, M.; Uversky, V.N.; Pintar, A.; Pongor, S. Prevalence of intrinsic disorder in the intracellular region of human single-pass type I proteins: The case of the notch ligand Delta-4. J. Proteome Res. 2008, 7, 2496–2506. [Google Scholar] [CrossRef] [PubMed]
- Dunker, A.K.; Lawson, J.D.; Brown, C.J.; Williams, R.M.; Romero, P.; Oh, J.S.; Oldfield, C.J.; Campen, A.M.; Ratliff, C.M.; Hipps, K.W.; et al. Intrinsically disordered protein. J. Mol. Graph. Model. 2001, 19, 26–59. [Google Scholar] [CrossRef] [PubMed]
- Van der Lee, R.; Buljan, M.; Lang, B.; Weatheritt, R.J.; Daughdrill, G.W.; Dunker, A.K.; Fuxreiter, M.; Gough, J.; Gsponer, J.; Jones, D.T.; et al. Classification of intrinsically disordered regions and proteins. Chem. Rev. 2014, 114, 6589–6631. [Google Scholar] [CrossRef] [PubMed]
- Wright, P.E.; Dyson, H.J. Intrinsically disordered proteins in cellular signalling and regulation. Nat. Rev. Mol. Cell Biol. 2015, 16, 18–29. [Google Scholar] [CrossRef]
- Von Deimling, A.; Louis, D.N.; von Ammon, K.; Petersen, I.; Wiestler, O.D.; Seizinger, B.R. Evidence for a tumor suppressor gene on chromosome 19q associated with human astrocytomas, oligodendrogliomas, and mixed gliomas. Cancer Res. 1992, 52, 4277–4279. [Google Scholar]
- Darmanis, S.; Sloan, S.A.; Croote, D.; Mignardi, M.; Chernikova, S.; Samghababi, P.; Zhang, Y.; Neff, N.; Kowarsky, M.; Caneda, C.; et al. Single-Cell RNA-Seq Analysis of Infiltrating Neoplastic Cells at the Migrating Front of Human Glioblastoma. Cell Rep. 2017, 21, 1399–1410. [Google Scholar] [CrossRef]
- Bharadwaj, A.; Bydoun, M.; Holloway, R.; Waisman, D. Annexin A2 heterotetramer: Structure and function. Int. J. Mol. Sci. 2013, 14, 6259–6305. [Google Scholar] [CrossRef]
- Gerke, V.; Creutz, C.E.; Moss, S.E. Annexins: Linking Ca2+ signalling to membrane dynamics. Nat. Rev. Mol. Cell Biol. 2005, 6, 449–461. [Google Scholar] [CrossRef]
- Morel, E.; Gruenberg, J. Annexin A2 binding to endosomes and functions in endosomal transport are regulated by tyrosine 23 phosphorylation. J. Biol. Chem. 2009, 284, 1604–1611. [Google Scholar] [CrossRef]
- Kim, J.; Hajjar, K.A. Annexin II: A plasminogen-plasminogen activator co-receptor. Front. Biosci. 2002, 7, d341–d348. [Google Scholar] [CrossRef]
- Wang, C.Y.; Lin, C.F. Annexin A2: Its molecular regulation and cellular expression in cancer development. Dis. Markers 2014, 2014, 308976. [Google Scholar] [CrossRef]
- Chen, L.; Lin, L.; Xian, N.; Zheng, Z. Annexin A2 regulates glioma cell proliferation through the STAT3-cyclin D1 pathway. Oncol. Rep. 2019, 42, 399–413. [Google Scholar] [CrossRef] [PubMed]
- Buchberger, A.; Bukau, B.; Sommer, T. Protein quality control in the cytosol and the endoplasmic reticulum: Brothers in arms. Mol. Cell. 2010, 40, 238–252. [Google Scholar] [CrossRef] [PubMed]
- Arndt, V.; Rogon, C.; Höhfeld, J. To be, or not to be–Molecular chaperones in protein degradation. Cell. Mol. Life Sci. 2007, 64, 2525–2541. [Google Scholar] [CrossRef] [PubMed]
- D’Amico, M.; De Amicis, F. Aberrant Notch signaling in gliomas: A potential landscape of actionable converging targets for combination approach in therapies resistance. Cancer Drug Resist. 2022, 5, 939–953. [Google Scholar] [CrossRef]
- Unger, F.T.; Witte, I.; David, K.A. Prediction of individual response to anticancer therapy: Historical and future perspectives. Cell. Mol. Life Sci. 2015, 72, 729–757. [Google Scholar] [CrossRef]
- Hoadley, K.A.; Yau, C.; Wolf, D.M.; Cherniack, A.D.; Tamborero, D.; Ng, S.; Leiserson, M.D.M.; Niu, B.; McLellan, M.D.; Uzunangelov, V.; et al. Multiplatform analysis of 12 cancer types reveals molecular classification within and across tissues of origin. Cell 2014, 158, 929–944. [Google Scholar] [CrossRef]
- Nandagopal, N.; Santat, L.A.; LeBon, L.; Sprinzak, D.; Bronner, M.E.; Elowitz, M.B. Dynamic Ligand Discrimination in the Notch Signaling Pathway. Cell 2018, 172, 869–880. [Google Scholar] [CrossRef]
- Lavecchia, A.; Di Giovanni, C. Virtual screening strategies in drug discovery: A critical review. Curr. Med. Chem. 2013, 20, 2839–2860. [Google Scholar] [CrossRef]
- Zinzalla, G.; Thurston, D.E. Targeting protein-protein interactions for therapeutic intervention: A challenge for the future. Future Med. Chem. 2009, 1, 65–93. [Google Scholar] [CrossRef]
- Nusinow, D.P.; Szpyt, J.; Ghandi, M.; Rose, C.M.; McDonald, E.R., 3rd; Kalocsay, M.; Jané-Valbuena, J.; Gelfand, E.; Schweppe, D.K.; Jedrychowski, M.; et al. Quantitative Proteomics of the Cancer Cell Line Encyclopedia. Cell 2020, 180, 387–402.e316. [Google Scholar] [CrossRef]
- Madhavan, S.; Zenklusen, J.C.; Kotliarov, Y.; Sahni, H.; Fine, H.A.; Buetow, K. Rembrandt: Helping personalized medicine become a reality through integrative translational research. Mol. Cancer Res. 2009, 7, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Gravendeel, L.A.; Kouwenhoven, M.C.; Gevaert, O.; de Rooi, J.J.; Stubbs, A.P.; Duijm, J.E.; Daemen, A.; Bleeker, F.E.; Bralten, L.B.; Kloosterhof, N.K.; et al. Intrinsic gene expression profiles of gliomas are a better predictor of survival than histology. Cancer Res. 2009, 69, 9065–9072. [Google Scholar] [CrossRef] [PubMed]
- Ceccarelli, M.; Barthel, F.P.; Malta, T.M.; Sabedot, T.S.; Salama, S.R.; Murray, B.A.; Morozova, O.; Newton, Y.; Radenbaugh, A.; Pagnotta, S.M.; et al. Molecular Profiling Reveals Biologically Discrete Subsets and Pathways of Progression in Diffuse Glioma. Cell 2016, 164, 550–563. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ham, S.W.; Kim, J.Y.; Seo, S.; Hong, N.; Park, M.J.; Kim, Y.; Jang, J.; Park, S.; Lee, S.J.; Kim, J.-K.; et al. Annexin A2 Stabilizes Oncogenic JAG1 Intracellular Domain by Inhibiting Proteasomal Degradation in Glioblastoma Cells. Int. J. Mol. Sci. 2023, 24, 14776. https://doi.org/10.3390/ijms241914776
Ham SW, Kim JY, Seo S, Hong N, Park MJ, Kim Y, Jang J, Park S, Lee SJ, Kim J-K, et al. Annexin A2 Stabilizes Oncogenic JAG1 Intracellular Domain by Inhibiting Proteasomal Degradation in Glioblastoma Cells. International Journal of Molecular Sciences. 2023; 24(19):14776. https://doi.org/10.3390/ijms241914776
Chicago/Turabian StyleHam, Seok Won, Jung Yun Kim, Sunyoung Seo, Nayoung Hong, Min Ji Park, Yoonji Kim, Junseok Jang, Sehyeon Park, Silvee Jisoo Lee, Jun-Kyum Kim, and et al. 2023. "Annexin A2 Stabilizes Oncogenic JAG1 Intracellular Domain by Inhibiting Proteasomal Degradation in Glioblastoma Cells" International Journal of Molecular Sciences 24, no. 19: 14776. https://doi.org/10.3390/ijms241914776
APA StyleHam, S. W., Kim, J. Y., Seo, S., Hong, N., Park, M. J., Kim, Y., Jang, J., Park, S., Lee, S. J., Kim, J.-K., Kim, E.-J., Kim, S.-O., Kim, S.-C., Park, J.-W., & Kim, H. (2023). Annexin A2 Stabilizes Oncogenic JAG1 Intracellular Domain by Inhibiting Proteasomal Degradation in Glioblastoma Cells. International Journal of Molecular Sciences, 24(19), 14776. https://doi.org/10.3390/ijms241914776