
Meeting at the Crossroad between Obesity and Hepatic Carcinogenesis: Unique Pathophysiological Pathways Raise Expectations for Innovative Therapeutic Approaches

 ,
,  ,
,  , , ,
, , ,  , ,
, , 
Abstract
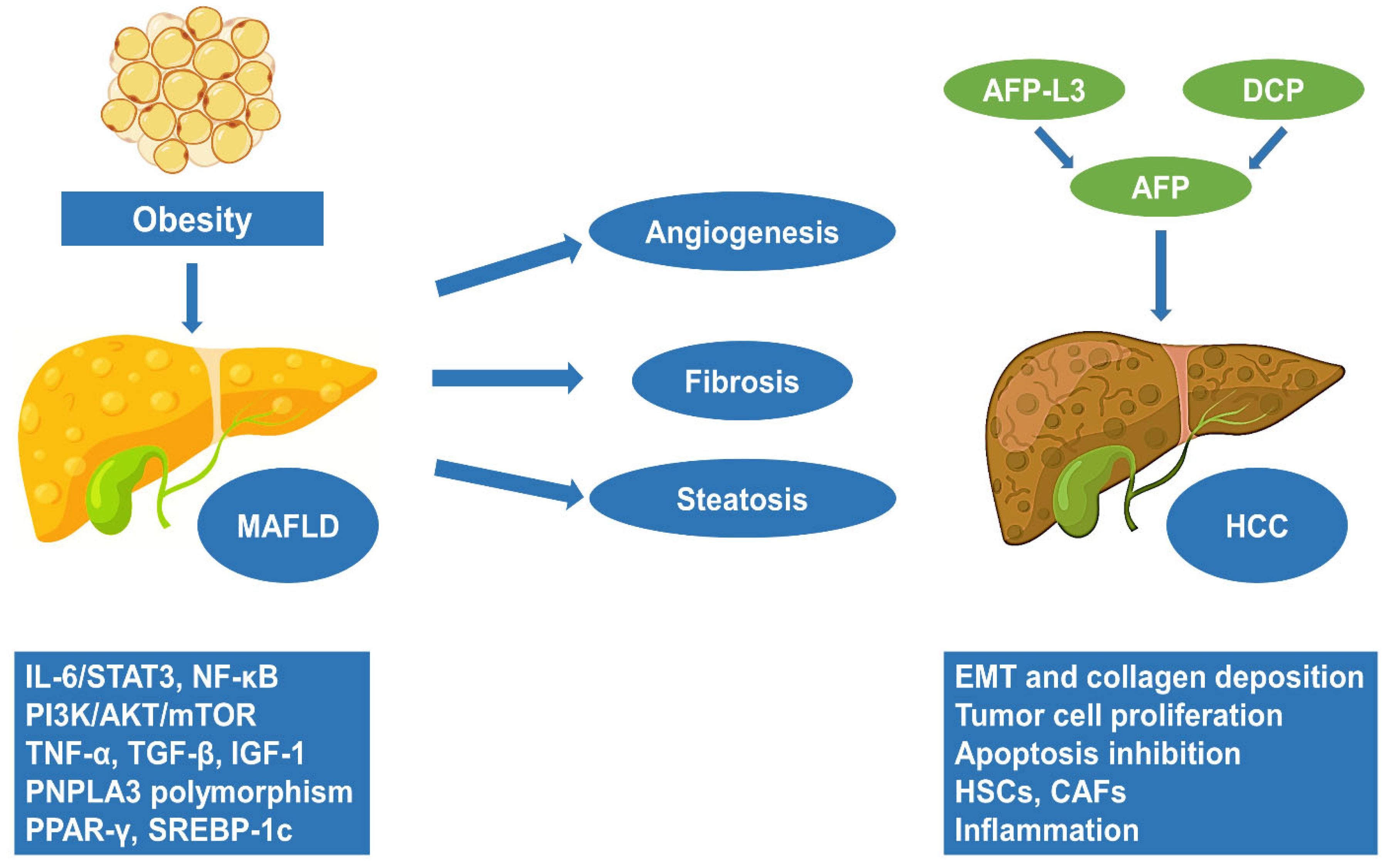
:1. Introduction
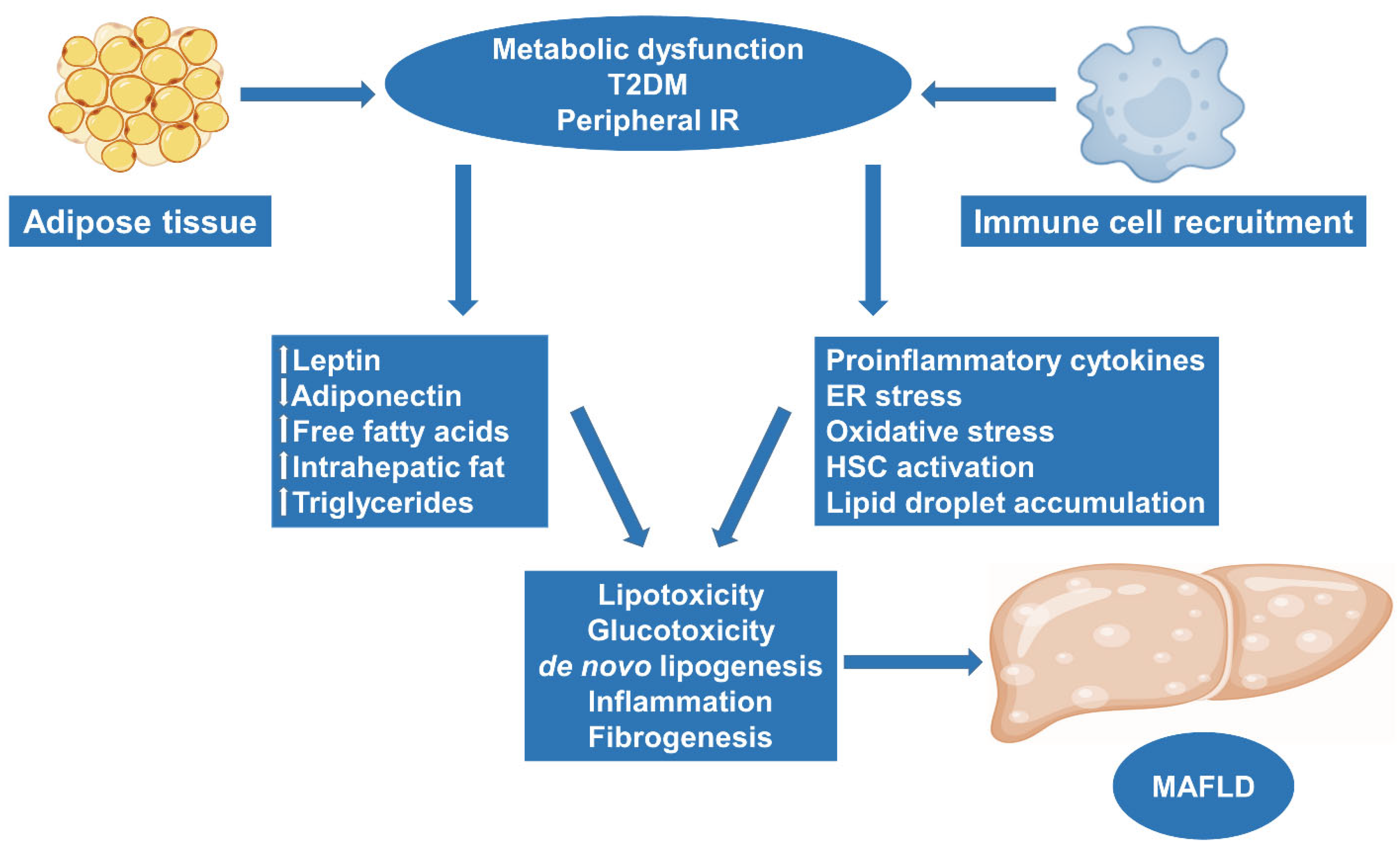
2. Obesity and MAFLD: Molecular Pathways and Pathogenesis
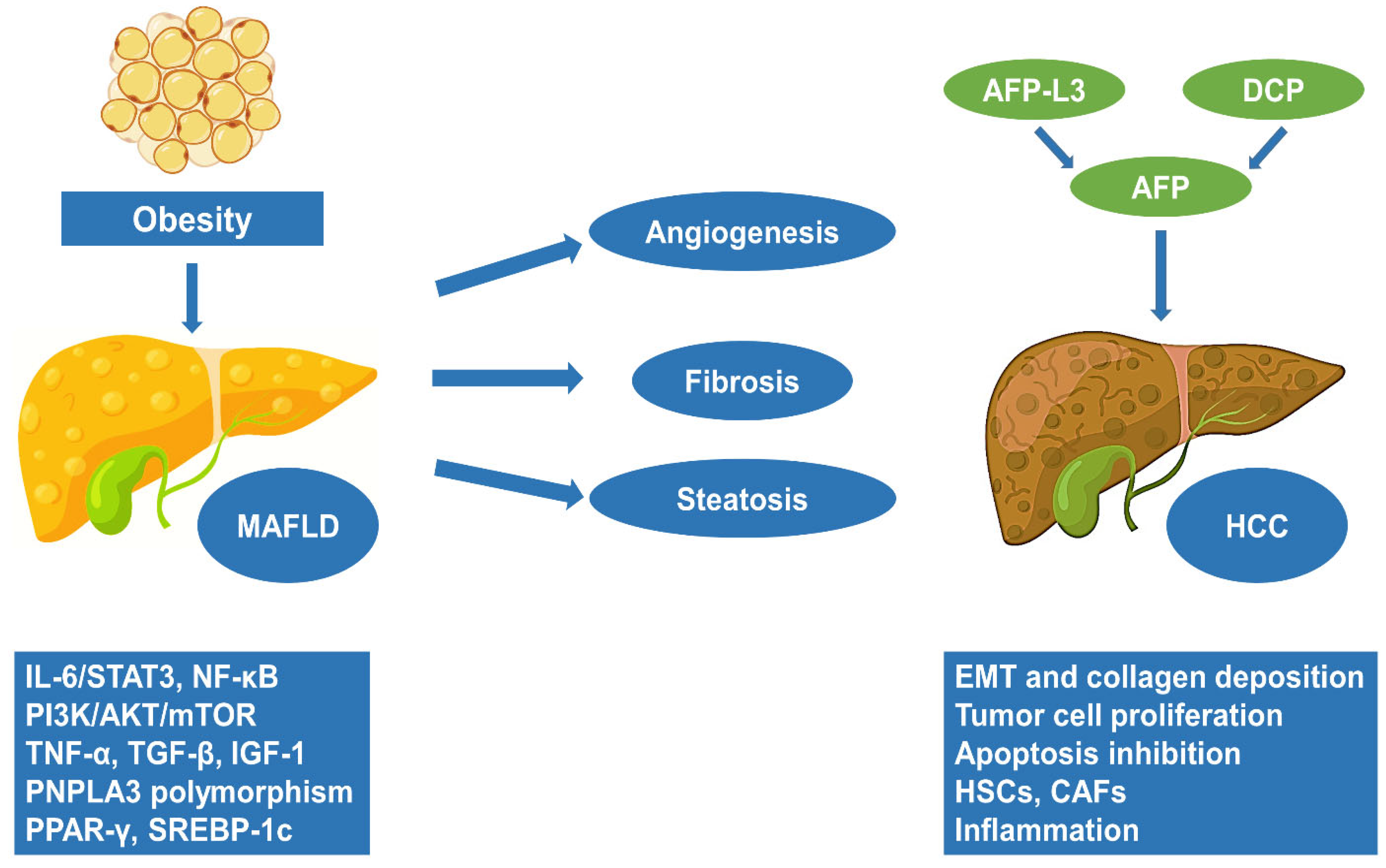
3. The Progression of MAFLD to HCC
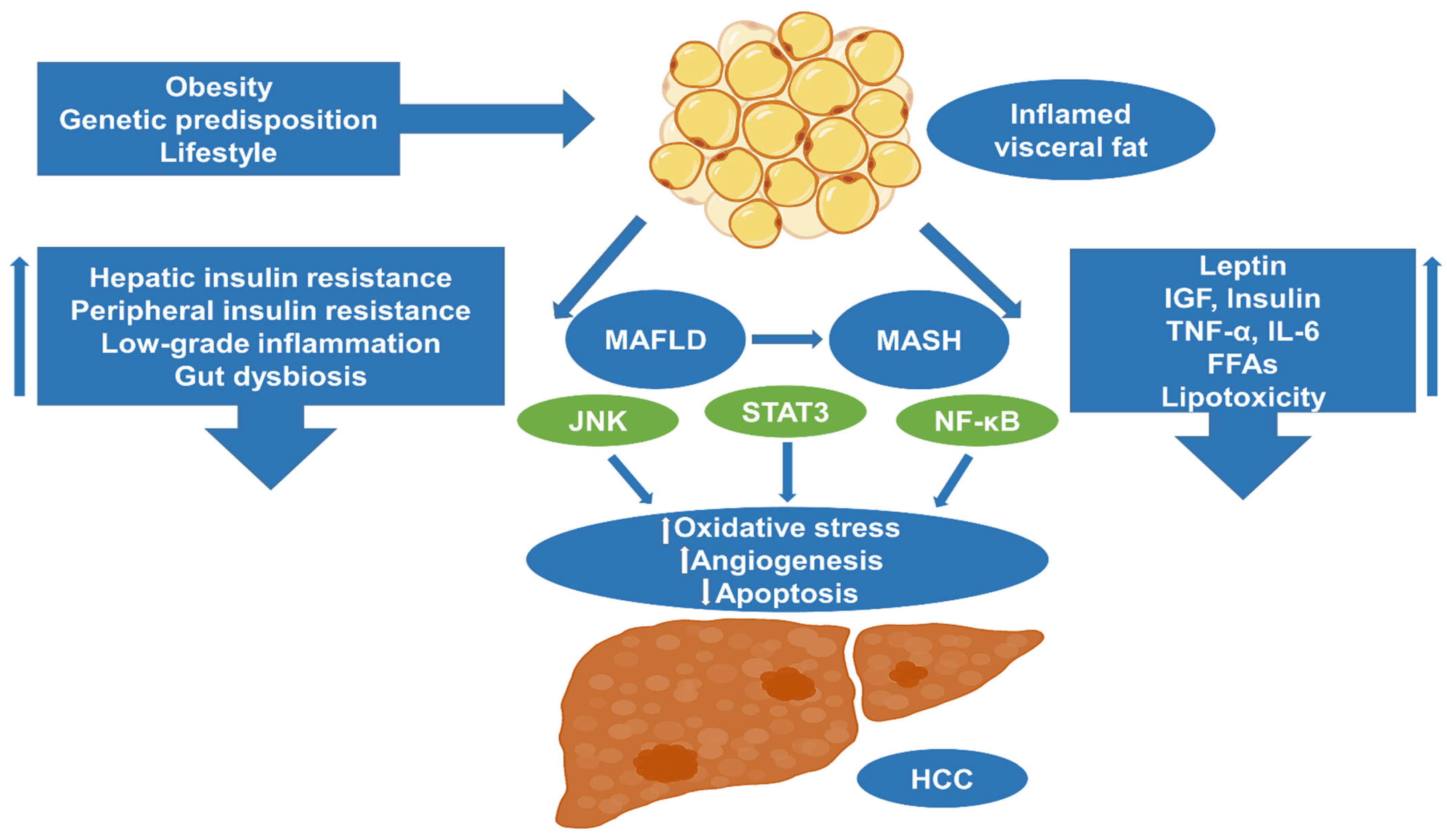
4. Obesity and HCC: Molecular Pathways and Pathogenesis

{kind=link}
{kind=link}
{kind=link}
| Study (Year) | Study Subjects | Outcomes |
|---|---|---|
| Aleffi et al. [70] (2005) | Human | Pro-inflammatory cytokines produced by adipose tissue include IL-6, IL-12, TNF-α, VEGF, CCL2, and CCL5, which regulate immunity and promote neovascularization in human hepatic stellate cells, mediated via the activation of NF-κB |
| Endo et al. [54] (2006) | Animal | The presence of dysfunctional adipose tissue characterized by reduced adiponectin and elevated leptin secretion plays a crucial role in promoting tumor proliferation and angiogenesis, mediated by OB-R/STAT3 signaling |
| Kitade et al. [76] (2006) | Animal | Leptin-mediated neovascularization coordinated with VEGF plays an important role in the development of liver fibrosis and hepatocarcinogenesis in MASH |
| Man et al. [77] (2010) | Animal | Adiponectin reduces HCC growth, hepatic stellate cell and macrophage activation, downregulating angiogenesis via the inhibition of ROCK/CXCL10/VEGF |
| Park et al. [80] (2010) | Animal | Obesity-promoted HCC development is dependent on enhanced production of the tumor-promoting cytokines IL-6 and TNF, which cause hepatic inflammation and activation of the oncogenic transcription factor STAT3 |
| Grohmann et al. [84] (2008) | Animal | Obesity-associated hepatic oxidative stress can independently contribute to the pathogenesis of MASH, fibrosis, and HCC by the inactivation of TCPTP and the upregulation of STAT1 and STAT3 signaling |
| Rensen et al. [103] (2023) | Human | Increased hepatic myeloperoxidase activity in obese subjects is associated with the induction of CXC chemokines and hepatic neutrophil infiltration, contributing to the development of HCC |
| Eferl et al. [86] (2003) | Animal | Activation of TNFR signaling by TNF-α leads to the stimulation of JNK/AP-1 signaling, potentiating hepatocellular proliferation and inhibiting apoptosis |
| Wang et al. [112] (2022) | Animal | Obesity leads to defective macrophage cleavage, while the upregulation of IL-1β and TNF potentiates a TACE-mediated proteolytic cleavage of TREM2, resulting in the aggregation of damaged hepatocytes, stimulating the inflammatory process |
| Kumar et al. [115] (2006) | Animal | HCFD stimulates the TLR4 and the OB-R/phosphoSTAT3 signaling pathways, resulting in liver tumorigenesis via an exaggerated mesenchymal phenotype with prominent Twist1-expressing TICs |
5. Treatment Strategies in Obesity-Related HCC
6. Discussion
7. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- James, W.P.T. WHO recognition of the global obesity epidemic. Int. J. Obes. 2008, 32, S120–S126. [Google Scholar] [CrossRef]
- Hubert, H.B.; Feinleib, M.; McNamara, P.M.; Castelli, W.P. Obesity as an independent risk factor for cardiovascular disease: A 26-year follow-up of participants in the Framingham Heart Study. Circulation 1983, 67, 968–977. [Google Scholar] [CrossRef]
- World Health Organization. Obesity: Preventing and Managing the Global Epidemic: Report of a WHO Consultation; World Health Organization: Geneva, Switzerland, 2000; Volume 894, pp. 1–253. [Google Scholar]
- Jensen, M.D.; Ryan, D.H.; Apovian, C.M.; Ard, J.D.; Comuzzie, A.G.; Donato, K.A.; Hu, F.B.; Hubbard, V.S.; Jakicic, J.M.; Kushner, R.F.; et al. 2013 AHA/ACC/TOS guideline for the management of overweight and obesity in adults: A report of the American College of cardiology/American Heart Association task force on practice guidelines and the obesity society. Circulation 2014, 129, 102–141. [Google Scholar] [CrossRef]
- Finkelstein, E.A.; Trogdon, J.G.; Cohen, J.W.; Dietz, W. Annual medical spending attributable to obesity: Payer-and service-specific estimates. Health Aff. 2009, 28, w822–w831. [Google Scholar] [CrossRef]
- Malkin, J.D.; Baid, D.; Alsukait, R.F.; Alghaith, T.; Alluhidan, M.; Alabdulkarim, H.; Altowaijri, A.; Almalki, Z.S.; Herbst, C.H.; Finkelstein, E.A.; et al. The economic burden of overweight and obesity in Saudi Arabia. PLoS ONE 2022, 17, e0264993. [Google Scholar] [CrossRef]
- D’Errico, M.; Pavlova, M.; Spandonaro, F. The economic burden of obesity in Italy: A cost-of-illness study. Eur. J. Health Econ. 2022, 23, 177–192. [Google Scholar] [CrossRef]
- Bentham, J.; Di Cesare, M.; Bilano, V.; Bixby, H.; Zhou, B.; Stevens, G.A.; Riley, L.M.; Taddei, C.; Hajifathalian, K.; Lu, Y.; et al. Worldwide trends in body-mass index, underweight, overweight, and obesity from 1975 to 2016: A pooled analysis of 2416 population-based measurement studies in 128·9 million children, adolescents, and adults. Lancet 2017, 390, 2627–2642. [Google Scholar] [CrossRef]
- Di Cesare, M.; Bentham, J.; Stevens, G.A.; Zhou, B.; Danaei, G.; Lu, Y.; Bixby, H.; Cowan, M.J.; Riley, L.M.; Hajifathalian, K.; et al. Trends in adult body-mass index in 200 countries from 1975 to 2014: A pooled analysis of 1698 population-based measurement studies with 19.2 million participants. Lancet 2016, 387, 1377–1396. [Google Scholar] [CrossRef]
- Le, M.H.; Le, D.M.; Baez, T.C.; Wu, Y.; Ito, T.; Lee, E.Y.; Lee, K.; Stave, C.D.; Henry, L.; Barnett, S.D.; et al. Global incidence of non-alcoholic fatty liver disease: A systematic review and meta-analysis of 63 studies and 1,201,807 persons. J. Hepatol. 2023, 79, 287–295. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Ioannou, G.N. Epidemiology and risk-stratification of NAFLD-associated HCC. J. Hepatol. 2021, 75, 1476–1484. [Google Scholar] [CrossRef]
- Estes, C.; Razavi, H.; Loomba, R.; Younossi, Z.; Sanyal, A.J. Modeling the epidemic of nonalcoholic fatty liver disease demonstrates an exponential increase in burden of disease. Hepatology 2018, 67, 123–133. [Google Scholar] [CrossRef]
- Eslam, M.; Newsome, P.N.; Sarin, S.K.; Anstee, Q.M.; Targher, G.; Romero-Gomez, M.; Zelber-Sagi, S.; Wong, V.W.-S.; Dufour, J.-F.; Schattenberg, J.M.; et al. A new definition for metabolic dysfunction-associated fatty liver disease: An international expert consensus statement. J. Hepatol. 2020, 73, 202–209. [Google Scholar] [CrossRef]
- Gofton, C.; Upendran, Y.; Zheng, M.-H.; George, J. MAFLD: How is it different from NAFLD? Clin. Mol. Hepatol. 2023, 29, S17–S31. [Google Scholar] [CrossRef]
- Vogel, A.; Meyer, T.; Sapisochin, G.; Salem, R.; Saborowski, A. Hepatocellular carcinoma. Lancet 2022, 400, 1345–1362. [Google Scholar] [CrossRef]
- D’Alessio, A.; Pinato, D.J. Dissecting the Tumor Microenvironment to Predict Immunotherapy Response in Hepatocellular Cancer. Gastroenterology 2022, 163, 1712–1713. [Google Scholar] [CrossRef]
- Soltani, S.; Abdollahi, S.; Aune, D.; Jayedi, A. Body mass index and cancer risk in patients with type 2 diabetes: A dose–response meta-analysis of cohort studies. Sci. Rep. 2021, 11, 2479. [Google Scholar] [CrossRef]
- Cao, Z.; Zheng, X.; Yang, H.; Li, S.; Xu, F.; Yang, X.; Wang, Y. Association of obesity status and metabolic syndrome with site-specific cancers: A population-based cohort study. Br. J. Cancer 2020, 123, 1336–1344. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, X.; Wang, J.; Yan, Z.; Luo, J. Excess body weight and the risk of primary liver cancer: An updated meta-analysis of prospective studies. Eur. J. Cancer 2012, 48, 2137–2145. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, B.; Shen, F.; Fan, J.; Cao, H. Body mass index and risk of primary liver cancer: A meta-analysis of prospective studies. Oncologist 2012, 17, 1461–1468. [Google Scholar] [CrossRef]
- Papazoglou, A.S.; Kartas, A.; Moysidis, D.V.; Tsagkaris, C.; Papadakos, S.P.; Bekiaridou, A.; Samaras, A.; Karagiannidis, E.; Papadakis, M.; Giannakoulas, G. Glycemic control and atrial fibrillation: An intricate relationship, yet under investigation. Cardiovasc. Diabetol. 2022, 21, 39. [Google Scholar] [CrossRef]
- Ajmera, V.; Cepin, S.; Tesfai, K.; Hofflich, H.; Cadman, K.; Lopez, S.; Madamba, E.; Bettencourt, R.; Richards, L.; Behling, C.; et al. A prospective study on the prevalence of NAFLD, advanced fibrosis, cirrhosis and hepatocellular carcinoma in people with type 2 diabetes. J. Hepatol. 2022, 78, 471–478. [Google Scholar] [CrossRef]
- Huang, D.Q.; Noureddin, N.; Ajmera, V.; Amangurbanova, M.; Bettencourt, R.; Truong, E.; Gidener, T.; Siddiqi, H.; Majzoub, A.M.; Nayfeh, T.; et al. Type 2 diabetes, hepatic decompensation, and hepatocellular carcinoma in patients with non-alcoholic fatty liver disease: An individual participant-level data meta-analysis. Lancet Gastroenterol. Hepatol. 2023, 8, 829–836. [Google Scholar] [CrossRef]
- Kanwal, F.; Kramer, J.R.; Mapakshi, S.; Natarajan, Y.; Chayanupatkul, M.; Richardson, P.A.; Li, L.; Desiderio, R.; Thrift, A.P.; Asch, S.M.; et al. Risk of Hepatocellular Cancer in Patients With Non-Alcoholic Fatty Liver Disease. Gastroenterology 2018, 155, 1828–1837.e2. [Google Scholar] [CrossRef]
- White, D.L.; Kanwal, F.; El-Serag, H.B. Association Between Nonalcoholic Fatty Liver Disease and Risk for Hepatocellular Cancer, Based on Systematic Review. Clin. Gastroenterol. Hepatol. 2012, 10, 1342–1359.e2. [Google Scholar] [CrossRef]
- Adams, L.A.; Lymp, J.F.; St. Sauver, J.; Sanderson, S.O.; Lindor, K.D..; Feldstein, A.; Angulo, P. The Natural History of Nonalcoholic Fatty Liver Disease: A Population-Based Cohort Study. Gastroenterology 2005, 129, 113–121. [Google Scholar] [CrossRef]
- Sørensen, H.T.; Mellemkjær, L.; Jepsen, P.; Thulstrup, A.M.; Baron, J.; Olsen, J.H.; Vilstrup, H. Risk of cancer in patients hospitalized with fatty liver: A Danish Cohort Study. J. Clin. Gastroenterol. 2003, 36, 356–359. [Google Scholar] [CrossRef]
- Stine, J.G.; Wentworth, B.J.; Zimmet, A.; Rinella, M.E.; Loomba, R.; Caldwell, S.H.; Argo, C.K. Systematic review with meta-analysis: Risk of hepatocellular carcinoma in non-alcoholic steatohepatitis without cirrhosis compared to other liver diseases. Aliment. Pharmacol. Ther. 2018, 48, 696–703. [Google Scholar] [CrossRef]
- Gawrieh, S.; Dakhoul, L.; Miller, E.; Scanga, A.; DeLemos, A.; Kettler, C.; Burney, H.; Liu, H.; Abu-Sbeih, H.; Chalasani, N.; et al. Characteristics, aetiologies and trends of hepatocellular carcinoma in patients without cirrhosis: A United States multicentre study Funding information Supported in part by. Aliment. Pharmacol. Ther. 2019, 50, 809–821. [Google Scholar] [CrossRef]
- Quek, J.; Chan, K.E.; Wong, Z.Y.; Tan, C.; Tan, B.; Lim, W.H.; Tan, D.J.H.; Tang, A.S.P.; Tay, P.; Xiao, J.; et al. Global prevalence of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis in the overweight and obese population: A systematic review and meta-analysis. Lancet Gastroenterol. Hepatol. 2023, 8, 20–30. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease—Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef]
- Noubiap, J.J.; Nansseu, J.R.; Lontchi-Yimagou, E.; Nkeck, J.R.; Nyaga, U.F.; Ngouo, A.T.; Tounouga, D.N.; Tianyi, F.-L.; Foka, A.J.; Ndoadoumgue, A.L.; et al. Geographic distribution of metabolic syndrome and its components in the general adult population: A meta-analysis of global data from 28 million individuals. Diabetes Res. Clin. Pract. 2022, 188, 109924. [Google Scholar] [CrossRef]
- Longo, M.; Meroni, M.; Paolini, E.; Erconi, V.; Carli, F.; Fortunato, F.; Ronchi, D.; Piciotti, R.; Sabatini, S.; Macchi, C.; et al. TM6SF2/PNPLA3/MBOAT7 Loss-of-Function Genetic Variants Impact on NAFLD Development and Progression Both in Patients and in In Vitro Models. Cell. Mol. Gastroenterol. Hepatol. 2022, 13, 759–788. [Google Scholar] [CrossRef]
- Ponziani, F.R.; De Luca, A.; Picca, A.; Marzetti, E.; Petito, V.; Del Chierico, F.; Reddel, S.; Sterbini, F.P.; Sanguinetti, M.; Putignani, L.; et al. Gut Dysbiosis and Fecal Calprotectin Predict Response to Immune Checkpoint Inhibitors in Patients With Hepatocellular Carcinoma. Hepatol. Commun. 2022, 6, 1492–1501. [Google Scholar] [CrossRef]
- Polyzos, S.A.; Kountouras, J.; Mantzoros, C.S. Obesity and nonalcoholic fatty liver disease: From pathophysiology to therapeutics. Metabolism 2019, 92, 82–97. [Google Scholar] [CrossRef]
- Mendez-Sanchez, N.; Cruz-Ramon, V.; Ramirez-Perez, O.; Hwang, J.; Barranco-Fragoso, B.; Cordova-Gallardo, J. New Aspects of Lipotoxicity in Nonalcoholic Steatohepatitis. Int. J. Mol. Sci. 2018, 19, 2034. [Google Scholar] [CrossRef]
- Rodrigues, R.M.; He, Y.; Hwang, S.; Bertola, A.; Mackowiak, B.; Ahmed, Y.A.; Seo, W.; Ma, J.; Wang, X.; Park, S.H.; et al. E-Selectin-Dependent Inflammation and Lipolysis in Adipose Tissue Exacerbate Steatosis-to-NASH Progression via S100A8/9. Cell. Mol. Gastroenterol. Hepatol. 2022, 13, 151–171. [Google Scholar] [CrossRef]
- Lu, Y.; Yang, A.; Quan, C.; Pan, Y.; Zhang, H.; Li, Y.; Gao, C.; Lu, H.; Wang, X.; Cao, P.; et al. A single-cell atlas of the multicellular ecosystem of primary and metastatic hepatocellular carcinoma. Nat. Commun. 2022, 13, 4594. [Google Scholar] [CrossRef]
- Bellanti, F.; Villani, R.; Facciorusso, A.; Vendemiale, G.; Serviddio, G. Lipid oxidation products in the pathogenesis of non-alcoholic steatohepatitis. Free Radic. Biol. Med. 2017, 111, 173–185. [Google Scholar] [CrossRef]
- Torquato, P.; Bartolini, D.; Giusepponi, D.; Piroddi, M.; Sebastiani, B.; Saluti, G.; Galarini, R.; Galli, F. Increased plasma levels of the lipoperoxyl radical-derived vitamin E metabolite α-tocopheryl quinone are an early indicator of lipotoxicity in fatty liver subjects. Free Radic. Biol. Med. 2019, 131, 115–125. [Google Scholar] [CrossRef]
- Okada, L.S.D.R.R.; Oliveira, C.P.; Stefano, J.T.; Nogueira, M.A.; da Silva, I.D.C.G.; Cordeiro, F.B.; Alves, V.A.F.; Torrinhas, R.S.; Carrilho, F.J.; Puri, P.; et al. Omega-3 PUFA modulate lipogenesis, ER stress, and mitochondrial dysfunction markers in NASH—Proteomic and lipidomic insight. Clin. Nutr. 2018, 37, 1474–1484. [Google Scholar] [CrossRef]
- Murdolo, G.; Piroddi, M.; Luchetti, F.; Tortoioli, C.; Canonico, B.; Zerbinati, C.; Galli, F.; Iuliano, L. Oxidative stress and lipid peroxidation by-products at the crossroad between adipose organ dysregulation and obesity-linked insulin resistance. Biochimie 2013, 95, 585–594. [Google Scholar] [CrossRef]
- Fu, S.; Yang, L.; Li, P.; Hofmann, O.; Dicker, L.; Hide, W.; Lin, X.; Watkins, S.M.; Ivanov, A.R.; Hotamisligil, G.S. Aberrant lipid metabolism disrupts calcium homeostasis causing liver endoplasmic reticulum stress in obesity. Nature 2011, 473, 528–531. [Google Scholar] [CrossRef]
- Paradis, V.; Perlemuter, G.; Bonvoust, F.; Dargere, D.; Parfait, B.; Vidaud, M.; Conti, M.; Huet, S.; Ba, N.; Buffet, C.; et al. High glucose and hyperinsulinemia stimulate connective tissue growth factor expression: A potential mechanism involved in progression to fibrosis in nonalcoholic steatohepatitis. Hepatology 2001, 34, 738–744. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, H.; Meyer, C.; Li, J.; Nadalin, S.; Königsrainer, A.; Weng, H.; Dooley, S.; ten Dijke, P. Transforming Growth Factor-β (TGF-β)-mediated Connective Tissue Growth Factor (CTGF) Expression in Hepatic Stellate Cells Requires Stat3 Signaling Activation. J. Biol. Chem. 2013, 288, 30708–30719. [Google Scholar] [CrossRef]
- Khalid, M.; Petroianu, G.; Adem, A. Advanced Glycation End Products and Diabetes Mellitus: Mechanisms and Perspectives. Biomolecules 2022, 12, 542. [Google Scholar] [CrossRef]
- Jiang, J.X.; Chen, X.; Fukada, H.; Serizawa, N.; Devaraj, S.; Török, N.J. Advanced glycation endproducts induce fibrogenic activity in nonalcoholic steatohepatitis by modulating TNF-α-converting enzyme activity in mice. Hepatology 2013, 58, 1339–1348. [Google Scholar] [CrossRef]
- Šmíd, V.; Dvořák, K.; Šedivý, P.; Kosek, V.; Leníček, M.; Dezortová, M.; Hajšlová, J.; Hájek, M.; Vítek, L.; Bechyňská, K.; et al. Effect of Omega-3 Polyunsaturated Fatty Acids on Lipid Metabolism in Patients With Metabolic Syndrome and NAFLD. Hepatol. Commun. 2022, 6, 1336–1349. [Google Scholar] [CrossRef]
- Garcia-Martinez, I.; Alen, R.; Pereira, L.; Povo-Retana, A.; Astudillo, A.M.; Hitos, A.B.; Gomez-Hurtado, I.; Lopez-Collazo, E.; Boscá, L.; Francés, R.; et al. Saturated fatty acid-enriched small extracellular vesicles mediate a crosstalk inducing liver inflammation and hepatocyte insulin resistance. JHEP Rep. 2023, 5, 100756. [Google Scholar] [CrossRef]
- Polyzos, S.A.; Chrysavgis, L.; Vachliotis, I.D.; Chartampilas, E.; Cholongitas, E. Nonalcoholic fatty liver disease and hepatocellular carcinoma:Insights in epidemiology, pathogenesis, imaging, prevention and therapy. Semin. Cancer Biol. 2023, 93, 20–35. [Google Scholar] [CrossRef]
- Gilbert, C.A.; Slingerland, J.M. Cytokines, Obesity, and Cancer: New Insights on Mechanisms Linking Obesity to Cancer Risk and Progression. Annu. Rev. Med. 2013, 64, 45–57. [Google Scholar] [CrossRef]
- Hui, J.M.; Hodge, A.; Farrell, G.C.; Kench, J.G.; Kriketos, A.; George, J. Beyond insulin resistance in NASH: TNF-? or adiponectin? Hepatology 2004, 40, 46–54. [Google Scholar] [CrossRef]
- Endo, H.; Hosono, K.; Uchiyama, T.; Sakai, E.; Sugiyama, M.; Takahashi, H.; Nakajima, N.; Wada, K.; Takeda, K.; Nakagama, H.; et al. Leptin acts as a growth factor for colorectal tumours at stages subsequent to tumour initiation in murine colon carcinogenesis. Gut 2011, 60, 1363–1371. [Google Scholar] [CrossRef]
- Louis, P.; Hold, G.L.; Flint, H.J. The gut microbiota, bacterial metabolites and colorectal cancer. Nat. Rev. Microbiol. 2014, 12, 661–672. [Google Scholar] [CrossRef]
- Liu, Y.-L.; Patman, G.L.; Leathart, J.B.S.; Piguet, A.-C.; Burt, A.D.; Dufour, J.-F.; Day, C.P.; Daly, A.K.; Reeves, H.L.; Anstee, Q.M. Carriage of the PNPLA3 rs738409 C >G polymorphism confers an increased risk of non-alcoholic fatty liver disease associated hepatocellular carcinoma. J. Hepatol. 2014, 61, 75–81. [Google Scholar] [CrossRef]
- Singal, A.G.; Manjunath, H.; Yopp, A.C.; Beg, M.S.; Marrero, J.A.; Gopal, P.; Waljee, A.K. The Effect of PNPLA3 on Fibrosis Progression and Development of Hepatocellular Carcinoma: A Meta-analysis. Am. J. Gastroenterol. 2014, 109, 325–334. [Google Scholar] [CrossRef]
- Nakagawa, H.; Hayata, Y.; Kawamura, S.; Yamada, T.; Fujiwara, N.; Koike, K. Lipid Metabolic Reprogramming in Hepatocellular Carcinoma. Cancers 2018, 10, 447. [Google Scholar] [CrossRef]
- Montironi, C.; Castet, F.; Haber, P.K.; Pinyol, R.; Torres-Martin, M.; Torrens Fontanals, L.; Mesropian, A.; Wang, H.; Puigvehi, M.; Maeda, M.; et al. Inflamed and non-inflamed classes of HCC: A revised immunogenomic classification. Gut 2022, 72, 129–140. [Google Scholar] [CrossRef]
- Sutti, S.; Albano, E. Adaptive immunity: An emerging player in the progression of NAFLD. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 81–92. [Google Scholar] [CrossRef]
- Ma, C.; Kesarwala, A.H.; Eggert, T.; Medina-Echeverz, J.; Kleiner, D.E.; Jin, P.; Stroncek, D.F.; Terabe, M.; Kapoor, V.; ElGindi, M.; et al. NAFLD causes selective CD4+ T lymphocyte loss and promotes hepatocarcinogenesis. Nature 2016, 531, 253–257. [Google Scholar] [CrossRef]
- Malehmir, M.; Pfister, D.; Gallage, S.; Szydlowska, M.; Inverso, D.; Kotsiliti, E.; Leone, V.; Peiseler, M.; Surewaard, B.G.J.; Rath, D.; et al. Platelet GPIbα is a mediator and potential interventional target for NASH and subsequent liver cancer. Nat. Med. 2019, 25, 641–655. [Google Scholar] [CrossRef]
- Dudek, M.; Pfister, D.; Donakonda, S.; Filpe, P.; Schneider, A.; Laschinger, M.; Hartmann, D.; Hüser, N.; Meiser, P.; Bayerl, F.; et al. Auto-aggressive CXCR6+ CD8 T cells cause liver immune pathology in NASH. Nature 2021, 592, 444–449. [Google Scholar] [CrossRef]
- Pfister, D.; Núñez, N.G.; Pinyol, R.; Govaere, O.; Pinter, M.; Szydlowska, M.; Gupta, R.; Qiu, M.; Deczkowska, A.; Weiner, A.; et al. NASH limits anti-tumour surveillance in immunotherapy-treated HCC. Nature 2021, 592, 450–456. [Google Scholar] [CrossRef]
- Anstee, Q.M.; Reeves, H.L.; Kotsiliti, E.; Govaere, O.; Heikenwalder, M. From NASH to HCC: Current concepts and future challenges. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 411–428. [Google Scholar] [CrossRef]
- Llovet, J.M.; Willoughby, C.E.; Singal, A.G.; Greten, T.F.; Heikenwälder, M.; El-Serag, H.B.; Finn, R.S.; Friedman, S.L. Nonalcoholic steatohepatitis-related hepatocellular carcinoma: Pathogenesis and treatment. Nat. Rev. Gastroenterol. Hepatol. 2023, 20, 487–503. [Google Scholar] [CrossRef]
- Item, F.; Konrad, D. Visceral fat and metabolic inflammation: The portal theory revisited. Obes. Rev. 2012, 13, 30–39. [Google Scholar] [CrossRef]
- Meza-Perez, S.; Randall, T.D. Immunological Functions of the Omentum. Trends Immunol. 2017, 38, 526–536. [Google Scholar] [CrossRef]
- O’Sullivan, J.; Lysaght, J.; Donohoe, C.L.; Reynolds, J.V. Obesity and gastrointestinal cancer: The interrelationship of adipose and tumour microenvironments. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 699–714. [Google Scholar] [CrossRef]
- Aleffi, S.; Petrai, I.; Bertolani, C.; Parola, M.; Colombatto, S.; Novo, E.; Vizzutti, F.; Anania, F.A.; Milani, S.; Rombouts, K.; et al. Upregulation of proinflammatory and proangiogenic cytokines by leptin in human hepatic stellate cells. Hepatology 2005, 42, 1339–1348. [Google Scholar] [CrossRef]
- Pecht, T.; Gutman-Tirosh, A.; Bashan, N.; Rudich, A. Peripheral blood leucocyte subclasses as potential biomarkers of adipose tissue inflammation and obesity subphenotypes in humans. Obes. Rev. 2014, 15, 322–337. [Google Scholar] [CrossRef]
- Peverill, W.; Powell, L.W.; Skoien, R. Evolving concepts in the pathogenesis of NASH: Beyond steatosis and inflammation. Int. J. Mol. Sci. 2014, 15, 8591–8638. [Google Scholar] [CrossRef]
- Yu, J.; Shen, J.; Sun, T.T.; Zhang, X.; Wong, N. Obesity, insulin resistance, NASH and hepatocellular carcinoma. Semin. Cancer Biol. 2013, 23, 483–491. [Google Scholar] [CrossRef]
- Stickel, F.; Hellerbrand, C. Non-alcoholic fatty liver disease as a risk factor for hepatocellular carcinoma: Mechanisms and implications. Gut 2010, 59, 1303–1307. [Google Scholar] [CrossRef]
- Cabia, B.; Andrade, S.; Carreira, M.C.; Casanueva, F.F.; Crujeiras, A.B. A role for novel adipose tissue-secreted factors in obesity-related carcinogenesis. Obes. Rev. 2016, 17, 361–376. [Google Scholar] [CrossRef]
- Kitade, M.; Yoshiji, H.; Kojima, H.; Ikenaka, Y.; Noguchi, R.; Kaji, K.; Yoshii, J.; Yanase, K.; Namisaki, T.; Asada, K.; et al. Leptin-mediated neovascularization is a prerequisite for progression of nonalcoholic steatohepatitis in rats. Hepatology 2006, 44, 983–991. [Google Scholar] [CrossRef]
- Man, K.; Ng, K.T.P.; Xu, A.; Cheng, Q.; Lo, C.M.; Xiao, J.W.; Sun, B.S.; Lim, Z.X.H.; Cheung, J.S.; Wu, E.X.; et al. Suppression of liver tumor growth and metastasis by adiponectin in nude mice through inhibition of tumor angiogenesis and downregulation of rho kinase/IFN-inducible protein 10/matrix metalloproteinase 9 signaling. Clin. Cancer Res. 2010, 16, 967–977. [Google Scholar] [CrossRef]
- Kamada, Y.; Matsumoto, H.; Tamura, S.; Fukushima, J.; Kiso, S.; Fukui, K.; Igura, T.; Maeda, N.; Kihara, S.; Funahashi, T.; et al. Hypoadiponectinemia accelerates hepatic tumor formation in a nonalcoholic steatohepatitis mouse model. J. Hepatol. 2007, 47, 556–564. [Google Scholar] [CrossRef]
- Yamashita, T.; Honda, M.; Takatori, H.; Nishino, R.; Minato, H.; Takamura, H.; Ohta, T.; Kaneko, S. Activation of lipogenic pathway correlates with cell proliferation and poor prognosis in hepatocellular carcinoma. J. Hepatol. 2009, 50, 100–110. [Google Scholar] [CrossRef]
- Park, E.J.; Lee, J.H.; Yu, G.Y.; He, G.; Ali, S.R.; Holzer, R.G.; Österreicher, C.H.; Takahashi, H.; Karin, M. Dietary and Genetic Obesity Promote Liver Inflammation and Tumorigenesis by Enhancing IL-6 and TNF Expression. Cell 2010, 140, 197–208. [Google Scholar] [CrossRef]
- Naugler, W.E.; Sakurai, T.; Kim, S.; Maeda, S.; Kim, K.H.; Elsharkawy, A.M.; Karin, M. Gender disparity in liver cancer due to sex differences in MyD88-dependent IL-6 production. Science 2007, 317, 121–124. [Google Scholar] [CrossRef]
- Rebouissou, S.; Amessou, M.; Couchy, G.; Poussin, K.; Imbeaud, S.; Pilati, C.; Izard, T.; Balabaud, C.; Bioulac-Sage, P.; Zucman-Rossi, J. Frequent in-frame somatic deletions activate gp130 in inflammatory hepatocellular tumours. Nature 2009, 457, 200–204. [Google Scholar] [CrossRef]
- Mehal, W. NASH and HCC Are Driven by Different Signaling Pathways with a Common Regulator. Cell Metab. 2019, 29, 3–4. [Google Scholar] [CrossRef]
- Grohmann, M.; Wiede, F.; Dodd, G.T.; Gurzov, E.N.; Ooi, G.J.; Butt, T.; Rasmiena, A.A.; Kaur, S.; Gulati, T.; Goh, P.K.; et al. Obesity Drives STAT-1-Dependent NASH and STAT-3-Dependent HCC. Cell 2018, 175, 1289–1306.e20. [Google Scholar] [CrossRef]
- Darwish, N.M.; Elshaer, M.M.A.; Almutairi, S.M.; Chen, T.W.; Mohamed, M.O.; Ghaly, W.B.A.; Rasheed, R.A. Omega-3 Polyunsaturated Fatty Acids Provoke Apoptosis in Hepatocellular Carcinoma through Knocking Down the STAT3 Activated Signaling Pathway: In Vivo and In Vitro Study. Molecules 2022, 27, 3032. [Google Scholar] [CrossRef]
- Eferl, R.; Ricci, R.; Kenner, L.; Zenz, R.; David, J.-P.; Rath, M.; Wagner, E.F. Liver tumor development. c-Jun antagonizes the proapoptotic activity of p53. Cell 2003, 112, 181–192. [Google Scholar] [CrossRef]
- Chew, V.; Tow, C.; Teo, M.; Wong, H.L.; Chan, J.; Gehring, A.; Loh, M.; Bolze, A.; Quek, R.; Lee, V.K.M.; et al. Inflammatory tumour microenvironment is associated with superior survival in hepatocellular carcinoma patients. J. Hepatol. 2010, 52, 370–379. [Google Scholar] [CrossRef]
- Starley, B.Q.; Calcagno, C.J.; Harrison, S.A. Nonalcoholic fatty liver disease and hepatocellular carcinoma: A weighty connection. Hepatology 2010, 51, 1820–1832. [Google Scholar] [CrossRef]
- Ish-Shalom, D.; Christoffersen, C.T.; Vorwerk, P.; Sacerdoti-Sierra, N.; Shymko, R.M.; Naor, D.; De Meyts, P. Mitogenic properties of insulin and insulin analogues mediated by the insulin receptor. Diabetologia 1997, 40, S25–S31. [Google Scholar] [CrossRef]
- Tanaka, S.; Mohr, L.; Schmidt, E.V.; Sugimachi, K.; Wands, J.R. Biological effects of human insulin receptor substrate-1 overexpression in hepatocytes. Hepatology 1997, 26, 598–604. [Google Scholar] [CrossRef]
- Page, J.M.; Harrison, S.A. NASH and HCC. Clin. Liver Dis. 2009, 13, 631–647. [Google Scholar] [CrossRef]
- Font-Burgada, J.; Sun, B.; Karin, M. Obesity and Cancer: The Oil that Feeds the Flame. Cell Metab. 2016, 23, 48–62. [Google Scholar] [CrossRef]
- Jee, S.H.; Ohrr, H.; Sull, J.W.; Yun, J.E.; Ji, M.; Samet, J.M. Fasting serum glucose level and cancer risk in Korean men and women. JAMA 2005, 293, 194–202. [Google Scholar] [CrossRef]
- Papadakos, S.P.; Dedes, N.; Kouroumalis, E.; Theocharis, S. The Role of the NLRP3 Inflammasome in HCC Carcinogenesis and Treatment: Harnessing Innate Immunity. Cancers 2022, 14, 3150. [Google Scholar] [CrossRef]
- Papadakos, S.P.; Stergiou, I.E.; Gkolemi, N.; Arvanitakis, K.; Theocharis, S. Unraveling the Significance of EPH/Ephrin Signaling in Liver Cancer: Insights into Tumor Progression and Therapeutic Implications. Cancers 2023, 15, 3434. [Google Scholar] [CrossRef]
- Papadakos, S.P.; Arvanitakis, K.; Stergiou, I.E.; Lekakis, V.; Davakis, S.; Christodoulou, M.-I.; Germanidis, G.; Theocharis, S. The Role of TLR4 in the Immunotherapy of Hepatocellular Carcinoma: Can We Teach an Old Dog New Tricks? Cancers 2023, 15, 2795. [Google Scholar] [CrossRef]
- Arvanitakis, K.; Mitroulis, I.; Chatzigeorgiou, A.; Elefsiniotis, I.; Germanidis, G. The Liver Cancer Immune Microenvironment: Emerging Concepts for Myeloid Cell Profiling with Diagnostic and Therapeutic Implications. Cancers 2023, 15, 1522. [Google Scholar] [CrossRef]
- Arvanitakis, K.; Koletsa, T.; Mitroulis, I.; Germanidis, G. Tumor-Associated Macrophages in Hepatocellular Carcinoma Pathogenesis, Prognosis and Therapy. Cancers 2022, 14, 226. [Google Scholar] [CrossRef]
- Arvanitakis, K.; Mitroulis, I.; Germanidis, G. Tumor-associated neutrophils in hepatocellular carcinoma pathogenesis, prognosis, and therapy. Cancers 2021, 13, 2899. [Google Scholar] [CrossRef]
- Anderson, N.M.; Simon, M.C. The tumor microenvironment. Curr. Biol. 2020, 30, R921–R925. [Google Scholar] [CrossRef]
- Yahoo, N.; Dudek, M.; Knolle, P.; Heikenwälder, M. Role of immune responses in the development of NAFLD-associated liver cancer and prospects for therapeutic modulation. J. Hepatol. 2023, 79, 538–551. [Google Scholar] [CrossRef]
- Ogawa, Y.; Imajo, K.; Yoneda, M.; Kessoku, T.; Tomeno, W.; Shinohara, Y.; Kato, S.; Mawatari, H.; Nozaki, Y.; Fujita, K.; et al. Soluble CD14 levels reflect liver inflammation in patients with nonalcoholic steatohepatitis. PLoS ONE 2013, 8, e65211. [Google Scholar] [CrossRef]
- Rensen, S.S.; Slaats, Y.; Nijhuis, J.; Jans, A.; Bieghs, V.; Driessen, A.; Malle, E.; Greve, J.W.; Buurman, W.A. Increased hepatic myeloperoxidase activity in obese subjects with nonalcoholic steatohepatitis. Am. J. Pathol. 2009, 175, 1473–1482. [Google Scholar] [CrossRef]
- Ibrahim, J.; Nguyen, A.H.; Rehman, A.; Ochi, A.; Jamal, M.; Graffeo, C.S.; Henning, J.R.; Zambirinis, C.P.; Fallon, N.C.; Barilla, R.; et al. Dendritic cell populations with different concentrations of lipid regulate tolerance and immunity in mouse and human liver. Gastroenterology 2012, 143, 1061–1072. [Google Scholar] [CrossRef]
- Rau, M.; Schilling, A.-K.; Meertens, J.; Hering, I.; Weiss, J.; Jurowich, C.; Kudlich, T.; Hermanns, H.M.; Bantel, H.; Beyersdorf, N.; et al. Progression from Nonalcoholic Fatty Liver to Nonalcoholic Steatohepatitis Is Marked by a Higher Frequency of Th17 Cells in the Liver and an Increased Th17/Resting Regulatory T Cell Ratio in Peripheral Blood and in the Liver. J. Immunol. 2016, 196, 97–105. [Google Scholar] [CrossRef]
- Gadd, V.L.; Skoien, R.; Powell, E.E.; Fagan, K.J.; Winterford, C.; Horsfall, L.; Irvine, K.; Clouston, A.D. The portal inflammatory infiltrate and ductular reaction in human nonalcoholic fatty liver disease. Hepatology 2014, 59, 1393–1405. [Google Scholar] [CrossRef]
- Tajiri, K.; Shimizu, Y.; Tsuneyama, K.; Sugiyama, T. Role of liver-infiltrating CD3+CD56+ natural killer T cells in the pathogenesis of nonalcoholic fatty liver disease. Eur. J. Gastroenterol. Hepatol. 2009, 21, 673–680. [Google Scholar] [CrossRef]
- Ringel, A.E.; Drijvers, J.M.; Baker, G.J.; Catozzi, A.; García-Cañaveras, J.C.; Gassaway, B.M.; Miller, B.C.; Juneja, V.R.; Nguyen, T.H.; Joshi, S.; et al. Obesity Shapes Metabolism in the Tumor Microenvironment to Suppress Anti-Tumor Immunity. Cell 2020, 183, 1848–1866.e26. [Google Scholar] [CrossRef]
- Dyck, L.; Prendeville, H.; Raverdeau, M.; Wilk, M.M.; Loftus, R.M.; Douglas, A.; McCormack, J.; Moran, B.; Wilkinson, M.; Mills, E.L.; et al. Suppressive effects of the obese tumor microenvironment on CD8 T cell infiltration and effector function. J. Exp. Med. 2022, 219, e20210042. [Google Scholar] [CrossRef]
- Zhang, C.-Y.; Liu, S.; Yang, M. Nutrition deprivation affects the cytotoxic effect of CD8 T cells in hepatocellular carcinoma. World J. Gastrointest. Oncol. 2022, 14, 1887–1891. [Google Scholar] [CrossRef]
- Tang, W.; Zhou, J.; Yang, W.; Feng, Y.; Wu, H.; Mok, M.T.S.; Zhang, L.; Liang, Z.; Liu, X.; Xiong, Z.; et al. Aberrant cholesterol metabolic signaling impairs antitumor immunosurveillance through natural killer T cell dysfunction in obese liver. Cell. Mol. Immunol. 2022, 19, 834–847. [Google Scholar] [CrossRef]
- Wang, X.; He, Q.; Zhou, C.; Xu, Y.; Liu, D.; Fujiwara, N.; Kubota, N.; Click, A.; Henderson, P.; Vancil, J.; et al. Prolonged hypernutrition impairs TREM2-dependent efferocytosis to license chronic liver inflammation and NASH development. Immunity 2022, 56, 58–77.e11. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Ricciardi, M.; Zanotto, M.; Malpeli, G.; Bassi, G.; Perbellini, O.; Chilosi, M.; Bifari, F.; Krampera, M. Epithelial-to-mesenchymal transition (EMT) induced by inflammatory priming elicits mesenchymal stromal cell-like immune-modulatory properties in cancer cells. Br. J. Cancer 2015, 112, 1067–1075. [Google Scholar] [CrossRef]
- Kumar, D.B.U.; Chen, C.L.; Liu, J.C.; Feldman, D.E.; Sher, L.S.; French, S.; Dinorcia, J.; French, S.W.; Naini, B.V.; Junrungsee, S.; et al. TLR4 Signaling via NANOG Cooperates with STAT3 to Activate Twist1 and Promote Formation of Tumor-Initiating Stem-Like Cells in Livers of Mice. Gastroenterology 2016, 150, 707–719. [Google Scholar] [CrossRef]
- Zheng, X.; Xu, M.; Yao, B.; Wang, C.; Jia, Y.; Liu, Q. IL-6/STAT3 axis initiated CAFs via up-regulating TIMP-1 which was attenuated by acetylation of STAT3 induced by PCAF in HCC microenvironment. Cell. Signal. 2016, 28, 1314–1324. [Google Scholar] [CrossRef]
- Jia, C.; Wang, G.; Wang, T.; Fu, B.; Zhang, Y.; Huang, L.; Deng, Y.; Chen, G.; Wu, X.; Chen, J.; et al. Cancer-associated fibroblasts induce epithelial-mesenchymal transition via the transglutaminase 2-dependent il-6/il6r/stat3 axis in hepatocellular carcinoma. Int. J. Biol. Sci. 2020, 16, 2542–2558. [Google Scholar] [CrossRef]
- Orel, V.B.; Papazoglou, A.S.; Tsagkaris, C.; Moysidis, D.V.; Papadakos, S.; Galkin, O.Y.; Orel, V.E.; Syvak, L.A. Nanotherapy based on magneto-mechanochemical modulation of tumor redox state. WIREs Nanomed. Nanobiotechnol. 2022, 15, e1868. [Google Scholar] [CrossRef]
- Vogel, A.; Martinelli, E.; Vogel, A.; Cervantes, A.; Chau, I.; Daniele, B.; Llovet, J.M.; Meyer, T.; Nault, J.-C.; Neumann, U.; et al. Updated treatment recommendations for hepatocellular carcinoma (HCC) from the ESMO Clinical Practice Guidelines. Ann. Oncol. 2021, 32, 801–805. [Google Scholar] [CrossRef]
- Bruix, J.; Chan, S.L.; Galle, P.R.; Rimassa, L.; Sangro, B. Systemic treatment of hepatocellular carcinoma: An EASL position paper. J. Hepatol. 2021, 75, 960–974. [Google Scholar] [CrossRef]
- Reig, M.; Forner, A.; Rimola, J.; Ferrer-Fàbrega, J.; Burrel, M.; Garcia-Criado, Á.; Kelley, R.K.; Galle, P.R.; Mazzaferro, V.; Salem, R.; et al. BCLC strategy for prognosis prediction and treatment recommendation: The 2022 update. J. Hepatol. 2022, 76, 681–693. [Google Scholar] [CrossRef]
- Lempesis, I.G.; Meijel, R.L.J.; Manolopoulos, K.N.; Goossens, G.H. Oxygenation of adipose tissue: A human perspective. Acta Physiol. 2020, 228, e13298. [Google Scholar] [CrossRef]
- Lempesis, I.G.; Tsilingiris, D.; Liu, J.; Dalamaga, M. Of mice and men: Considerations on adipose tissue physiology in animal models of obesity and human studies. Metab. Open 2022, 15, 100208. [Google Scholar] [CrossRef]
- Vallianou, N.G.; Tsilingiris, D.; Kounatidis, D.; Lempesis, I.G.; Karampela, I.; Dalamaga, M. SGLT-2 inhibitors in obesity and its associated cardiometabolic disorders: Where do we stand? Pol. Arch. Intern. Med. 2022, 132, 16342. [Google Scholar] [CrossRef]
- Lempesis, I.G.; Hoebers, N.; Essers, Y.; Jocken, J.W.E.; Rouschop, K.M.A.; Blaak, E.E.; Manolopoulos, K.N.; Goossens, G.H. Physiological Oxygen Levels Differentially Regulate Adipokine Production in Abdominal and Femoral Adipocytes from Individuals with Obesity Versus Normal Weight. Cells 2022, 11, 3532. [Google Scholar] [CrossRef]
- Dyson, J.; Jaques, B.; Chattopadyhay, D.; Lochan, R.; Graham, J.; Das, D.; Aslam, T.; Patanwala, I.; Gaggar, S.; Cole, M.; et al. Hepatocellular cancer: The impact of obesity, type 2 diabetes and a multidisciplinary team. J. Hepatol. 2014, 60, 110–117. [Google Scholar] [CrossRef]
- Sagris, M.; Kokkinidis, D.G.; Lempesis, I.G.; Giannopoulos, S.; Rallidis, L.; Mena-Hurtado, C.; Bakoyiannis, C. Nutrition, dietary habits, and weight management to prevent and treat patients with peripheral artery disease. Rev. Cardiovasc. Med. 2020, 21, 565. [Google Scholar] [CrossRef]
- Stefan, N. Causes, consequences, and treatment of metabolically unhealthy fat distribution. Lancet Diabetes Endocrinol. 2020, 8, 616–627. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, S.; Yang, M. Hepatocellular Carcinoma and Obesity, Type 2 Diabetes Mellitus, Cardiovascular Disease: Causing Factors, Molecular Links, and Treatment Options. Front. Endocrinol. 2021, 12, 808526. [Google Scholar] [CrossRef]
- Romero-Gómez, M.; Zelber-Sagi, S.; Trenell, M. Treatment of NAFLD with diet, physical activity and exercise. J. Hepatol. 2017, 67, 829–846. [Google Scholar] [CrossRef]
- Tangjarusritaratorn, T.; Tangjittipokin, W.; Kunavisarut, T. Incidence and Survival of Hepatocellular Carcinoma in Type 2 Diabetes Patients with Cirrhosis Who Were Treated with and without Metformin. Diabetes Metab. Syndr. Obes. Targets Ther. 2021, 14, 1563–1574. [Google Scholar] [CrossRef]
- Zhou, J.; Ke, Y.; Lei, X.; Wu, T.; Li, Y.; Bao, T.; Tang, H.; Zhang, C.; Wu, X.; Wang, G.; et al. Meta-analysis: The efficacy of metformin and other anti-hyperglycemic agents in prolonging the survival of hepatocellular carcinoma patients with type 2 diabetes. Ann. Hepatol. 2020, 19, 320–328. [Google Scholar] [CrossRef]
- Wang, C.-P.; Kuhn, J.; Shah, D.P.; Schmidt, S.; Lam, Y.-W.F.; MacCarthy, D.; Tenner, L.; Ramirez, A.G. Metformin modifies disparity in hepatocellular carcinoma incidence in men with type 2 diabetes but without chronic liver diseases. Cancer Med. 2019, 8, 3206–3215. [Google Scholar] [CrossRef]
- Hendryx, M.; Dong, Y.; Ndeke, J.M.; Luo, J. Sodium-glucose cotransporter 2 (SGLT2) inhibitor initiation and hepatocellular carcinoma prognosis. PLoS ONE 2022, 17, e0274519. [Google Scholar] [CrossRef]
- Arvanitakis, K.; Koufakis, T.; Kotsa, K.; Germanidis, G. The effects of sodium-glucose cotransporter 2 inhibitors on hepatocellular carcinoma: From molecular mechanisms to potential clinical implications. Pharmacol. Res. 2022, 181, 106261. [Google Scholar] [CrossRef]
- Lee, H.A.; Kim, H.Y. Therapeutic Mechanisms and Clinical Effects of Glucagon-like Peptide 1 Receptor Agonists in Nonalcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2023, 24, 9324. [Google Scholar] [CrossRef]
- Hartman, M.L.; Sanyal, A.J.; Loomba, R.; Wilson, J.M.; Nikooienejad, A.; Bray, R.; Karanikas, C.A.; Duffin, K.L.; Robins, D.A.; Haupt, A. Effects of novel dual GIP and GLP-1 receptor agonist tirzepatide on biomarkers of nonalcoholic steatohepatitis in patients with type 2 diabetes. Diabetes Care 2020, 43, 1352–1355. [Google Scholar] [CrossRef]
- Arvanitakis, K.; Koufakis, T.; Kotsa, K.; Germanidis, G. How Far beyond Diabetes Can the Benefits of Glucagon-like Peptide-1 Receptor Agonists Go? A Review of the Evidence on Their Effects on Hepatocellular Carcinoma. Cancers 2022, 14, 4651. [Google Scholar] [CrossRef]
- Lassailly, G.; Caiazzo, R.; Buob, D.; Pigeyre, M.; Verkindt, H.; Labreuche, J.; Raverdy, V.; Leteurtre, E.; Dharancy, S.; Louvet, A.; et al. Bariatric Surgery Reduces Features of Nonalcoholic Steatohepatitis in Morbidly Obese Patients. Gastroenterology 2015, 149, 379–388. [Google Scholar] [CrossRef]
- Kwak, M.; Mehaffey, J.H.; Hsu, A.; Hawkins, R.B.; Schirmer, B.; Hallowell, P.T. Discussion on: Bariatric surgery is associated with reduction in non-alcoholic steatohepatitis and hepatocellular carcinoma: A propensity matched analysis. Am. J. Surg. 2020, 219, 508. [Google Scholar] [CrossRef]
- Ramai, D.; Singh, J.; Lester, J.; Khan, S.R.; Chandan, S.; Tartaglia, N.; Ambrosi, A.; Serviddio, G.; Facciorusso, A. Systematic review with meta-analysis: Bariatric surgery reduces the incidence of hepatocellular carcinoma. Aliment. Pharmacol. Ther. 2021, 53, 977–984. [Google Scholar] [CrossRef]
- Kim, G.; Jang, S.-Y.; Han, E.; Lee, Y.-H.; Park, S.-Y.; Nam, C.M.; Kang, E.S. Effect of statin on hepatocellular carcinoma in patients with type 2 diabetes: A nationwide nested case-control study. Int. J. Cancer 2017, 140, 798–806. [Google Scholar] [CrossRef]
- Yang, S.-Y.; Wang, C.-C.; Chen, K.-D.; Liu, Y.-W.; Lin, C.-C.; Chuang, C.-H.; Tsai, Y.-C.; Yao, C.-C.; Yen, Y.-H.; Hsiao, C.-C.; et al. Statin use is associated with a lower risk of recurrence after curative resection in BCLC stage 0-A hepatocellular carcinoma. BMC Cancer 2021, 21, 70. [Google Scholar] [CrossRef]
- Simon, T.G.; Henson, J.; Osganian, S.; Masia, R.; Chan, A.T.; Chung, R.T.; Corey, K.E. Daily Aspirin Use Associated With Reduced Risk For Fibrosis Progression In Patients With Nonalcoholic Fatty Liver Disease. Clin. Gastroenterol. Hepatol. 2019, 17, 2776–2784.e4. [Google Scholar] [CrossRef]
- Ricciotti, E.; Wangensteen, K.J.; FitzGerald, G.A. Aspirin in Hepatocellular Carcinoma. Cancer Res. 2021, 81, 3751–3761. [Google Scholar] [CrossRef]
- Dhir, M.; Melin, A.A.; Douaiher, J.; Lin, C.; Zhen, W.K.; Hussain, S.M.; Geschwind, J.-F.H.; Doyle, M.B.M.; Abou-Alfa, G.K.; Are, C. A Review and Update of Treatment Options and Controversies in the Management of Hepatocellular Carcinoma. Ann. Surg. 2016, 263, 1112–1125. [Google Scholar] [CrossRef]
- Wang, M.-D.; Tang, S.-C.; Li, C.; Sun, L.-Y.; Xu, X.; Liang, Y.-J.; Liu, F.-B.; Gu, W.-M.; Wang, X.-M.; Zhou, Y.-H.; et al. Association of Concurrent Metabolic Syndrome with Long-term Oncological Prognosis Following Liver Resection for Hepatocellular Carcinoma Among Patients with Chronic Hepatitis B Virus Infection: A Multicenter Study of 1753 Patients. Ann. Surg. Oncol. 2023, 30, 346–358. [Google Scholar] [CrossRef]
- Grandhi, M.S.; Kim, A.K.; Ronnekleiv-Kelly, S.M.; Kamel, I.R.; Ghasebeh, M.A.; Pawlik, T.M. Hepatocellular carcinoma: From diagnosis to treatment. Surg. Oncol. 2016, 25, 74–85. [Google Scholar] [CrossRef]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.-F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.-L.; Forner, A.; et al. Sorafenib in Advanced Hepatocellular Carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef]
- Papadakos, S.P.; Ferraro, D.; Carbone, G.; Frampton, A.E.; Vennarecci, G.; Kykalos, S.; Schizas, D.; Theocharis, S.; Machairas, N. The Emerging Role of Metformin in the Treatment of Hepatocellular Carcinoma: Is There Any Value in Repurposing Metformin for HCC Immunotherapy? Cancers 2023, 15, 3161. [Google Scholar] [CrossRef]
- Papadakos, S.P.; Machairas, N.; Stergiou, I.E.; Arvanitakis, K.; Germanidis, G.; Frampton, A.E.; Theocharis, S. Unveiling the Yin-Yang Balance of M1 and M2 Macrophages in Hepatocellular Carcinoma: Role of Exosomes in Tumor Microenvironment and Immune Modulation. Cells 2023, 12, 2036. [Google Scholar] [CrossRef]
- Kawai, T.; Autieri, M.V.; Scalia, R. Adipose tissue inflammation and metabolic dysfunction in obesity. Am. J. Physiol. Cell Physiol. 2021, 320, C375–C391. [Google Scholar] [CrossRef]
- Locatelli, I.; Sutti, S.; Vacchiano, M.; Bozzola, C.; Albano, E. NF-κB1 deficiency stimulates the progression of non-alcoholic steatohepatitis (NASH) in mice by promoting NKT-cell-mediated responses. Clin. Sci. 2013, 124, 279–287. [Google Scholar] [CrossRef]
- Kaseb, A.O.; Morris, J.S.; Hassan, M.M.; Siddiqui, A.M.; Lin, E.; Xiao, L.; Abdalla, E.K.; Vauthey, J.-N.; Aloia, T.A.; Krishnan, S.; et al. Clinical and Prognostic Implications of Plasma Insulin-Like Growth Factor-1 and Vascular Endothelial Growth Factor in Patients With Hepatocellular Carcinoma. J. Clin. Oncol. 2011, 29, 3892–3899. [Google Scholar] [CrossRef]
- Wang, C.; Yu, S.; Qian, R.; Chen, S.; Dai, C.; Shan, X. Prognostic and immunological significance of peroxisome proliferator-activated receptor gamma in hepatocellular carcinoma based on multiple databases. Transl. Cancer Res. 2022, 11, 1938–1953. [Google Scholar] [CrossRef]
- Zhang, L.; Yuan, Q.; Li, M.; Chai, D.; Deng, W.; Wang, W. The association of leptin and adiponectin with hepatocellular carcinoma risk and prognosis: A combination of traditional, survival, and dose-response meta-analysis. BMC Cancer 2020, 20, 1167. [Google Scholar] [CrossRef]
- Ponziani, F.R.; Bhoori, S.; Castelli, C.; Putignani, L.; Rivoltini, L.; Del Chierico, F.; Sanguinetti, M.; Morelli, D.; Sterbini, F.P.; Petito, V.; et al. Hepatocellular Carcinoma Is Associated With Gut Microbiota Profile and Inflammation in Nonalcoholic Fatty Liver Disease. Hepatology 2019, 69, 107–120. [Google Scholar] [CrossRef]
- Alharthi, J.; Eslam, M. Biomarkers of Metabolic (Dysfunction)-associated Fatty Liver Disease: An Update. J. Clin. Transl. Hepatol. 2022, 10, 134–139. [Google Scholar] [CrossRef]
- Zhu, M.; Guo, J.; Xia, H.; Li, W.; Lu, Y.; Dong, X.; Chen, Y.; Xie, X.; Fu, S.; Li, M. Alpha-fetoprotein activates AKT/mTOR signaling to promote CXCR4 expression and migration of hepatoma cells. Oncoscience 2015, 2, 59–70. [Google Scholar] [CrossRef]
- Moldogazieva, N.; Mokhosoev, I.; Zavadskiy, S.; Terentiev, A. Proteomic Profiling and Artificial Intelligence for Hepatocellular Carcinoma Translational Medicine. Biomedicines 2021, 9, 159. [Google Scholar] [CrossRef]
- Yamao, T.; Yamashita, Y.-I.; Imai, K.; Umezaki, N.; Tsukamoto, M.; Kitano, Y.; Arima, K.; Miyata, T.; Nakagawa, S.; Okabe, H.; et al. Clinical Significance of Preoperative Hepatocellular Carcinoma With High Lens culinaris Agglutinin-reactive Fraction of Alpha-Fetoprotein, But Low Alpha-Fetoprotein. Anticancer Res. 2019, 39, 883–889. [Google Scholar] [CrossRef]
- Cui, S.-X.; Yu, X.-F.; Qu, X.-J. Roles and Signaling Pathways of Des-γ-Carboxyprothrombin in the Progression of Hepatocellular Carcinoma. Cancer Investig. 2016, 34, 459–464. [Google Scholar] [CrossRef]
- Cao, L.; Fan, X.; Jing, W.; Liang, Y.; Chen, R.; Liu, Y.; Zhu, M.; Jia, R.; Wang, H.; Zhang, X.; et al. Osteopontin promotes a cancer stem cell-like phenotype in hepatocellular carcinoma cells via an integrin-NF-κB–HIF-1α pathway. Oncotarget 2015, 6, 6627–6640. [Google Scholar] [CrossRef]
- Yu, X.; Zheng, Y.; Zhu, X.; Gao, X.; Wang, C.; Sheng, Y.; Cheng, W.; Qin, L.; Ren, N.; Jia, H.; et al. Osteopontin promotes hepatocellular carcinoma progression via the PI3K/AKT/Twist signaling pathway. Oncol. Lett. 2018, 16, 5299–5308. [Google Scholar] [CrossRef]
- Yao, M.; Yao, D.-F.; Bian, Y.-Z.; Zhang, C.-G.; Qiu, L.-W.; Wu, W.; Sai, W.-L.; Yang, J.-L.; Zhang, H.-J. Oncofetal antigen glypican-3 as a promising early diagnostic marker for hepatocellular carcinoma. Hepatobiliary Pancreat. Dis. Int. 2011, 10, 289–294. [Google Scholar] [CrossRef]
- Duan, R.; Li, C.; Wang, F.; Han, F.; Zhu, L. The Long Noncoding RNA ZFAS1 Potentiates the Development of Hepatocellular Carcinoma via the microRNA-624/MDK/ERK/JNK/P38 Signaling Pathway. Onco Targets Ther. 2020, 13, 4431–4444. [Google Scholar] [CrossRef]
- Vongsuvanh, R.; van der Poorten, D.; Iseli, T.; Strasser, S.I.; McCaughan, G.W.; George, J. Midkine Increases Diagnostic Yield in AFP Negative and NASH-Related Hepatocellular Carcinoma. PLoS ONE 2016, 11, e0155800. [Google Scholar] [CrossRef]
- Shen, Q.; Fan, J.; Yang, X.-R.; Tan, Y.; Zhao, W.; Xu, Y.; Wang, N.; Niu, Y.; Wu, Z.; Zhou, J.; et al. Serum DKK1 as a protein biomarker for the diagnosis of hepatocellular carcinoma: A large-scale, multicentre study. Lancet Oncol. 2012, 13, 817–826. [Google Scholar] [CrossRef]
- Fezza, M.; Moussa, M.; Aoun, R.; Haber, R.; Hilal, G. DKK1 promotes hepatocellular carcinoma inflammation, migration and invasion: Implication of TGF-β1. PLoS ONE 2019, 14, e0223252. [Google Scholar] [CrossRef]
- Bertino, G.; Neri, S.; Bruno, C.M.; Ardiri, A.M.; Calvagno, G.S.; Malaguarnera, M.; Toro, A.; Malaguarnera, M.; Clementi, S.; Bertino, N.; et al. Diagnostic and prognostic value of alpha-fetoprotein, des-γ-carboxy prothrombin and squamous cell carcinoma antigen immunoglobulin M complexes in hepatocellular carcinoma. Minerva Med. 2011, 102, 363–371. [Google Scholar]
- Liu, C.-H.; Gil-Gómez, A.; Ampuero, J.; Romero-Gómez, M. Diagnostic accuracy of SCCA and SCCA-IgM for hepatocellular carcinoma: A meta-analysis. Liver Int. 2018, 38, 1820–1831. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arvanitakis, K.; Papadakos, S.P.; Lekakis, V.; Koufakis, T.; Lempesis, I.G.; Papantoniou, E.; Kalopitas, G.; Georgakopoulou, V.E.; Stergiou, I.E.; Theocharis, S.; et al. Meeting at the Crossroad between Obesity and Hepatic Carcinogenesis: Unique Pathophysiological Pathways Raise Expectations for Innovative Therapeutic Approaches. Int. J. Mol. Sci. 2023, 24, 14704. https://doi.org/10.3390/ijms241914704
Arvanitakis K, Papadakos SP, Lekakis V, Koufakis T, Lempesis IG, Papantoniou E, Kalopitas G, Georgakopoulou VE, Stergiou IE, Theocharis S, et al. Meeting at the Crossroad between Obesity and Hepatic Carcinogenesis: Unique Pathophysiological Pathways Raise Expectations for Innovative Therapeutic Approaches. International Journal of Molecular Sciences. 2023; 24(19):14704. https://doi.org/10.3390/ijms241914704
Chicago/Turabian StyleArvanitakis, Konstantinos, Stavros P. Papadakos, Vasileios Lekakis, Theocharis Koufakis, Ioannis G. Lempesis, Eleni Papantoniou, Georgios Kalopitas, Vasiliki E. Georgakopoulou, Ioanna E. Stergiou, Stamatios Theocharis, and et al. 2023. "Meeting at the Crossroad between Obesity and Hepatic Carcinogenesis: Unique Pathophysiological Pathways Raise Expectations for Innovative Therapeutic Approaches" International Journal of Molecular Sciences 24, no. 19: 14704. https://doi.org/10.3390/ijms241914704
APA StyleArvanitakis, K., Papadakos, S. P., Lekakis, V., Koufakis, T., Lempesis, I. G., Papantoniou, E., Kalopitas, G., Georgakopoulou, V. E., Stergiou, I. E., Theocharis, S., & Germanidis, G. (2023). Meeting at the Crossroad between Obesity and Hepatic Carcinogenesis: Unique Pathophysiological Pathways Raise Expectations for Innovative Therapeutic Approaches. International Journal of Molecular Sciences, 24(19), 14704. https://doi.org/10.3390/ijms241914704