

Antioxidants Prevent Iron Accumulation and Lipid Peroxidation, but Do Not Correct Autophagy Dysfunction or Mitochondrial Bioenergetics in Cellular Models of BPAN


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
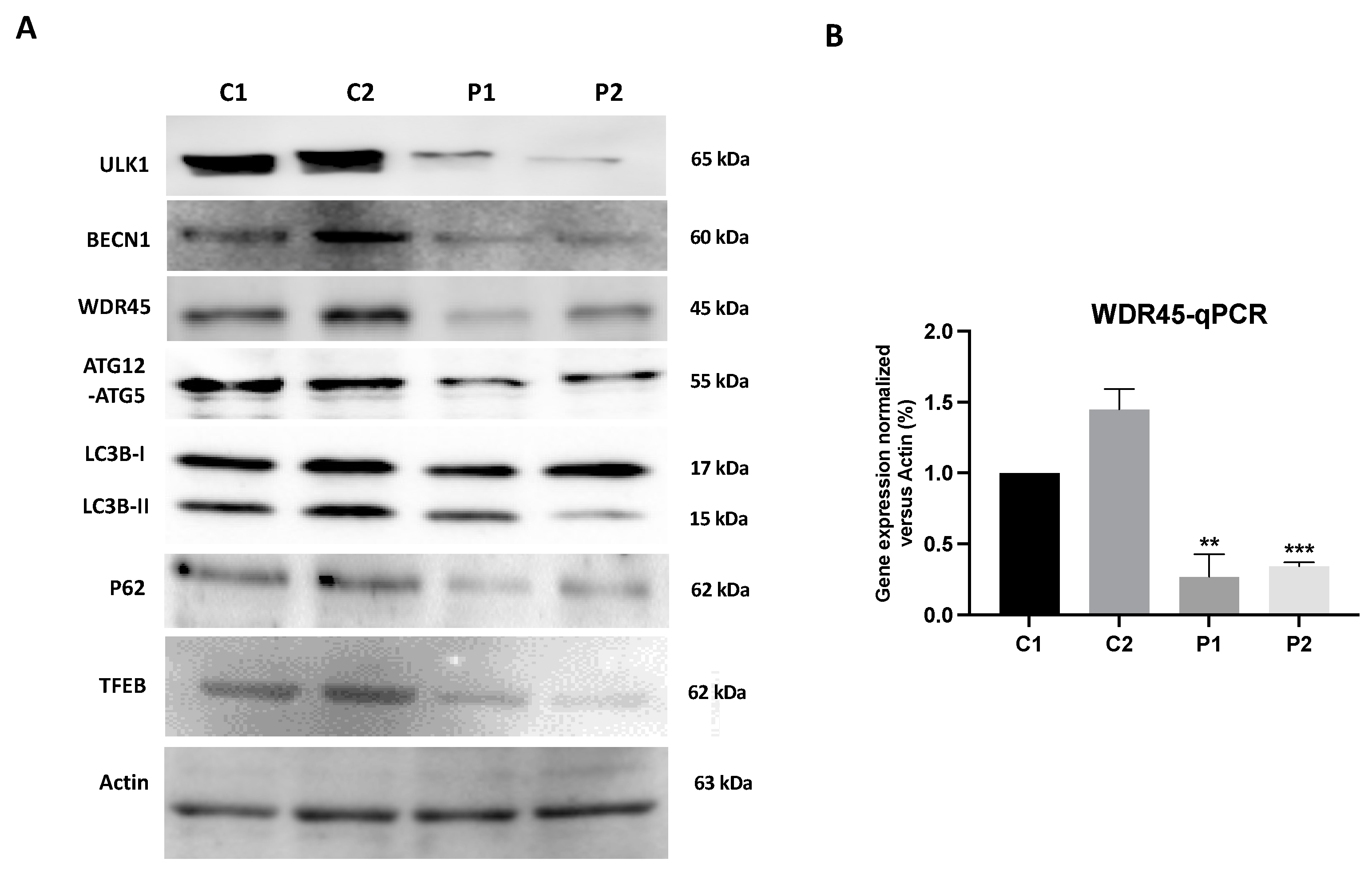
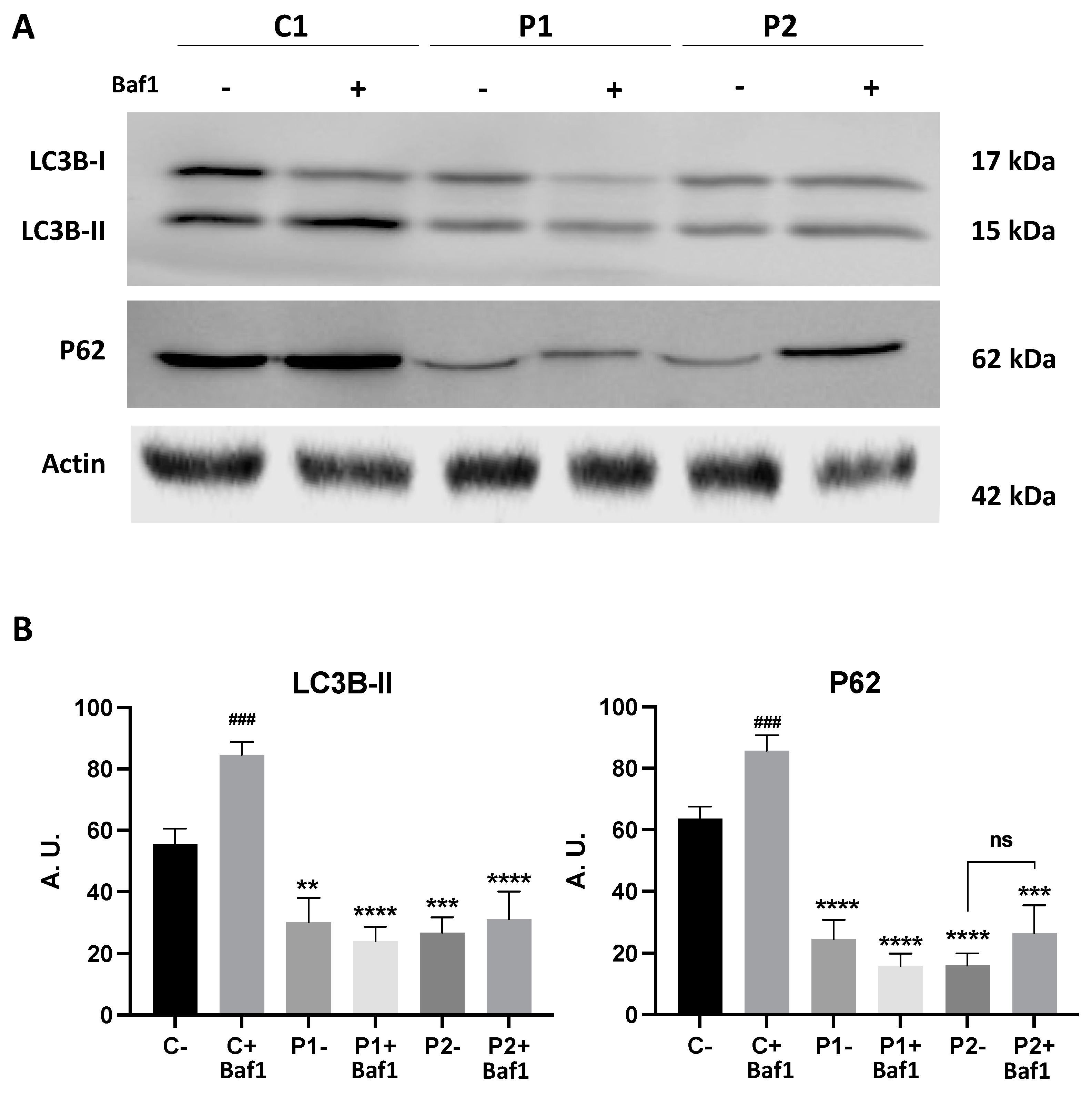
2.1. WDR45 Deficiency Leads to Autophagy Dysfunction in BPAN Cells
2.2. WDR45 Mutant Cells Show a Deficient Autophagic Flux
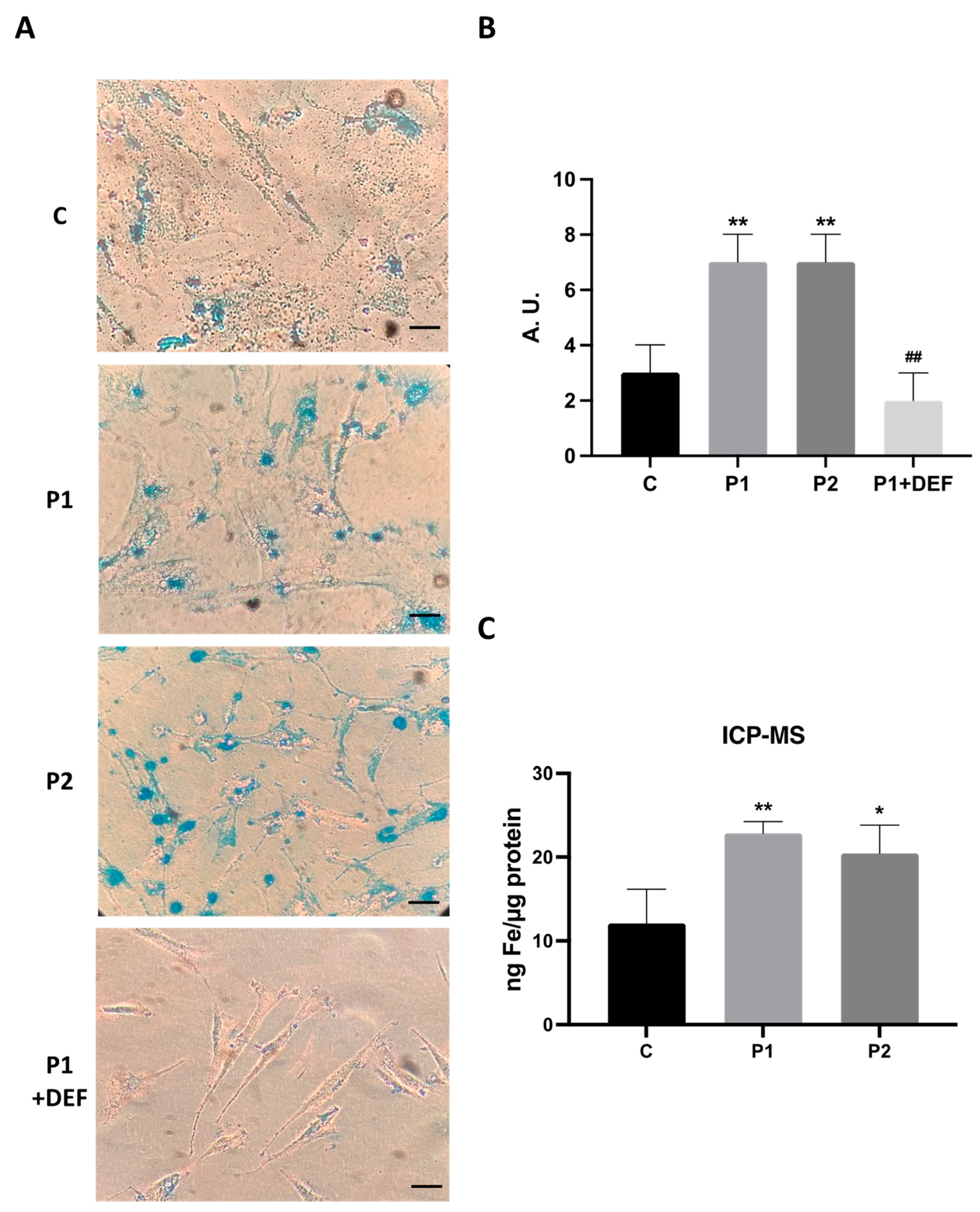
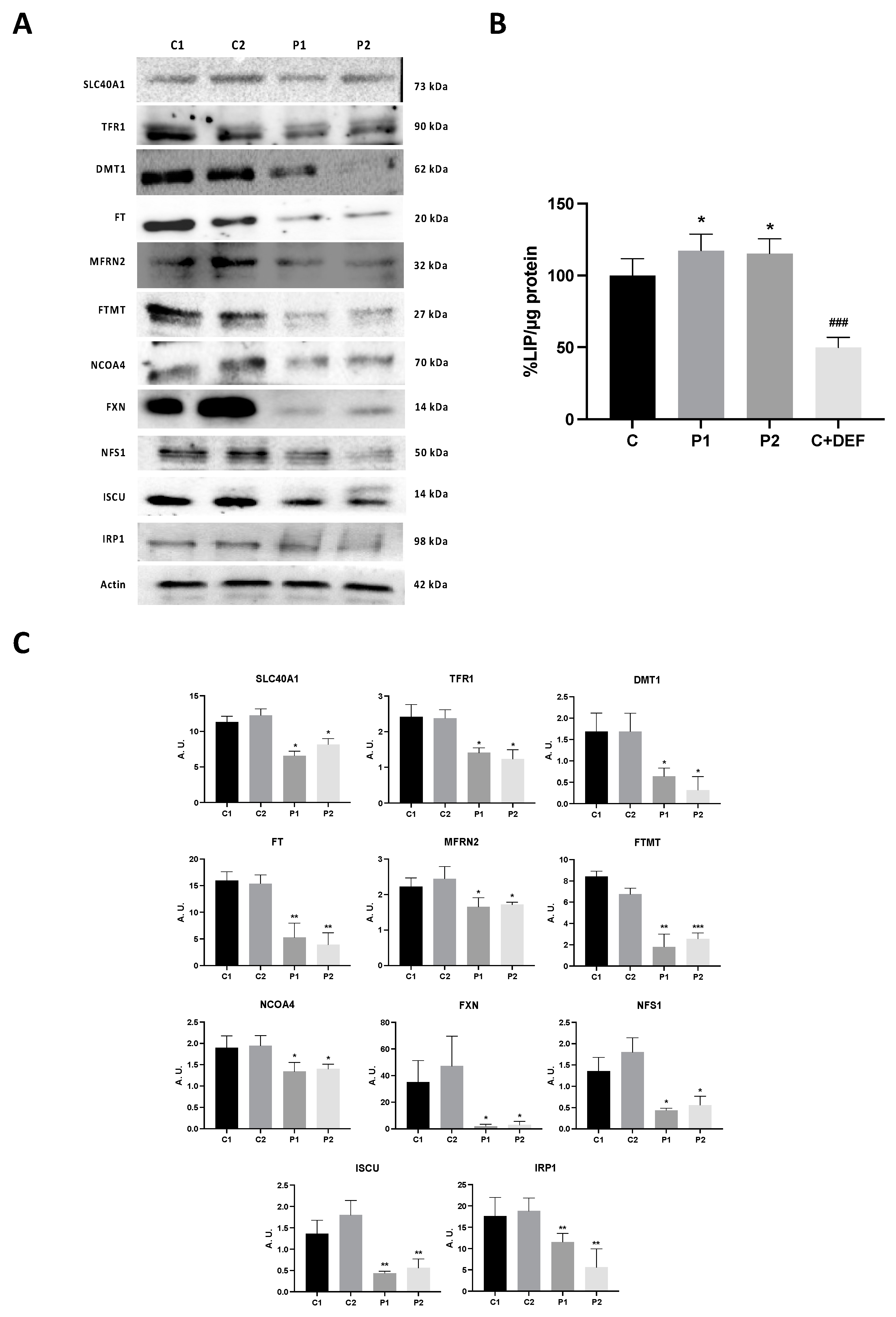
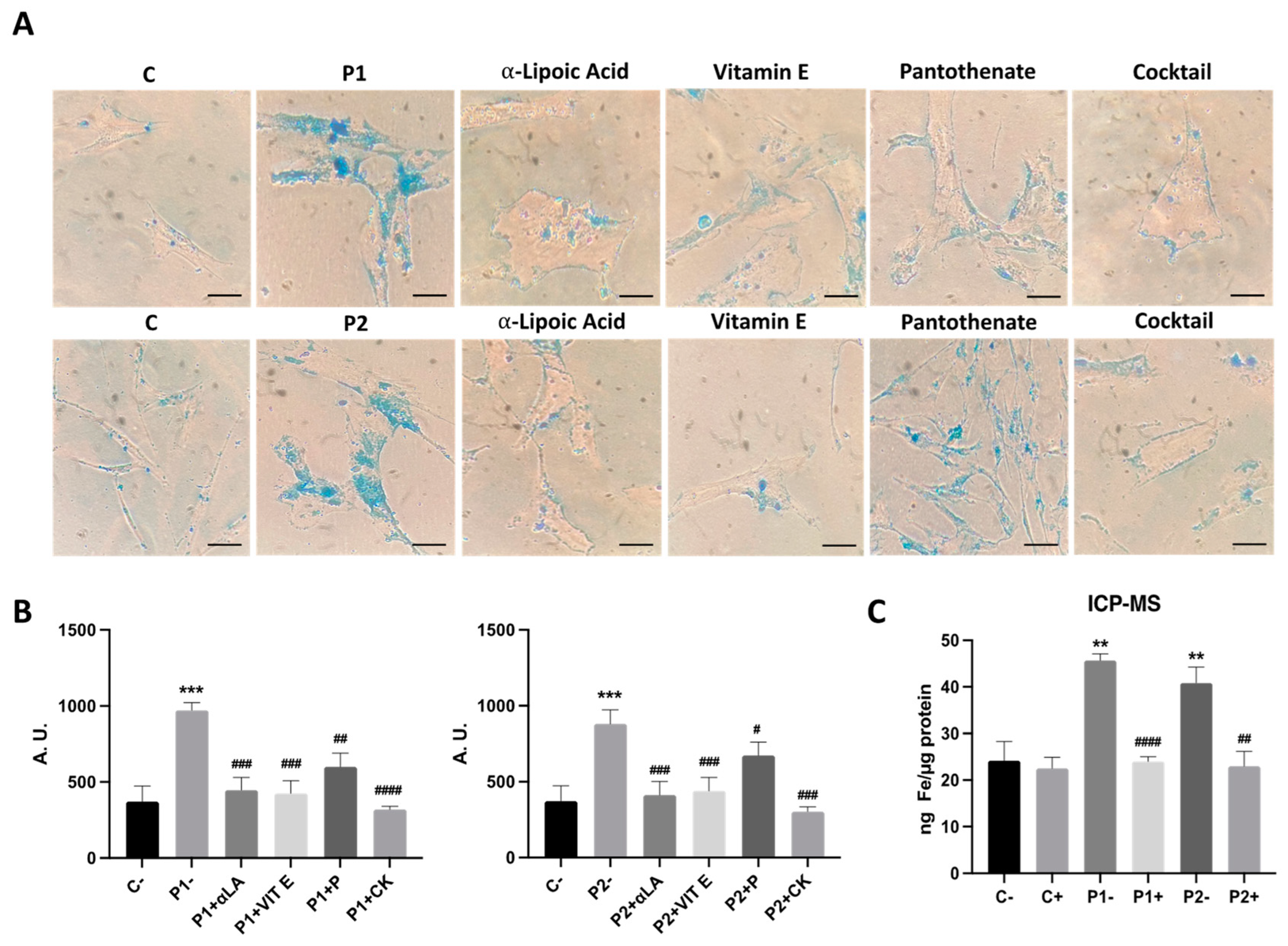
2.3. WDR45 Mutations Increase Intracellular Iron Levels
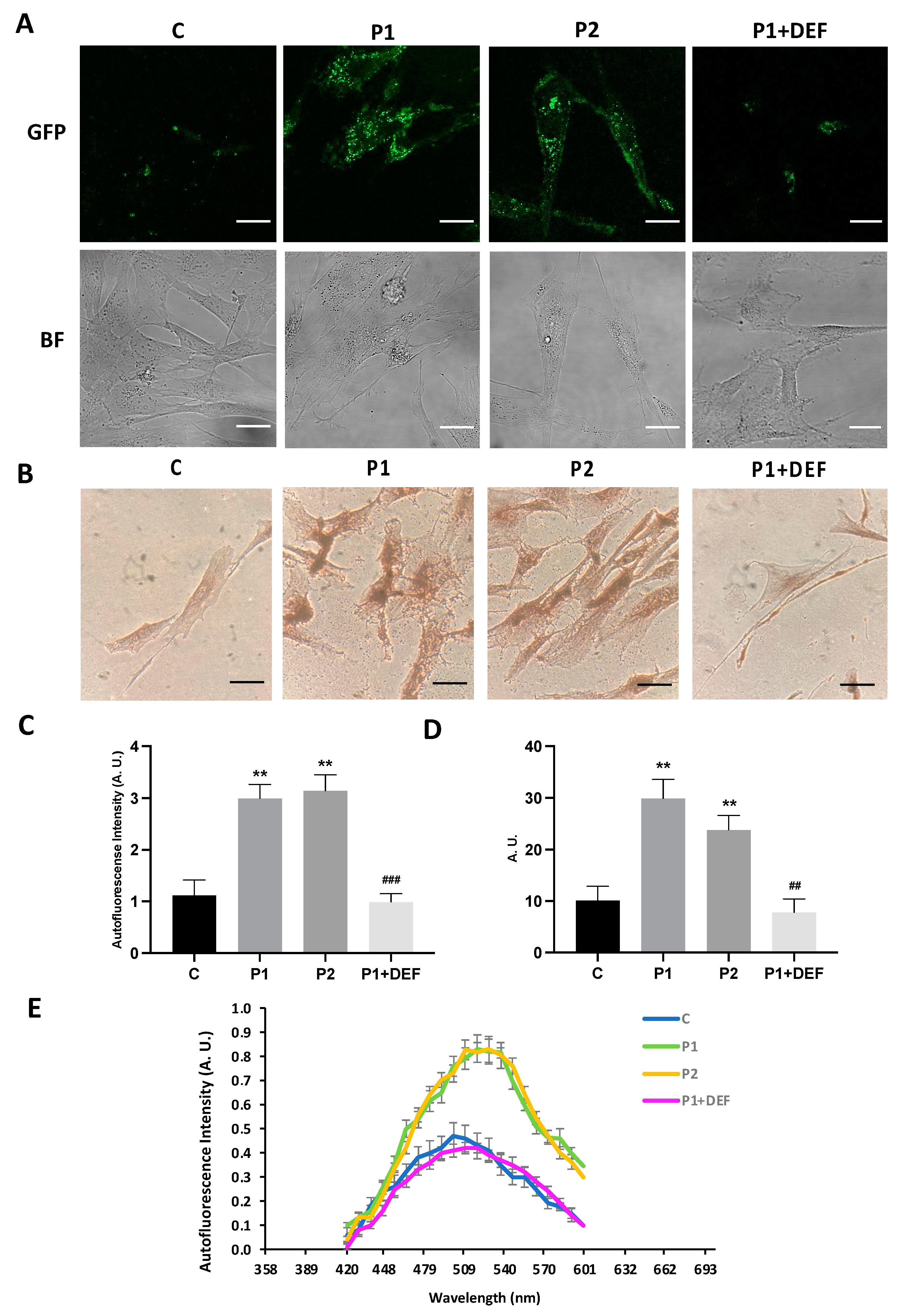
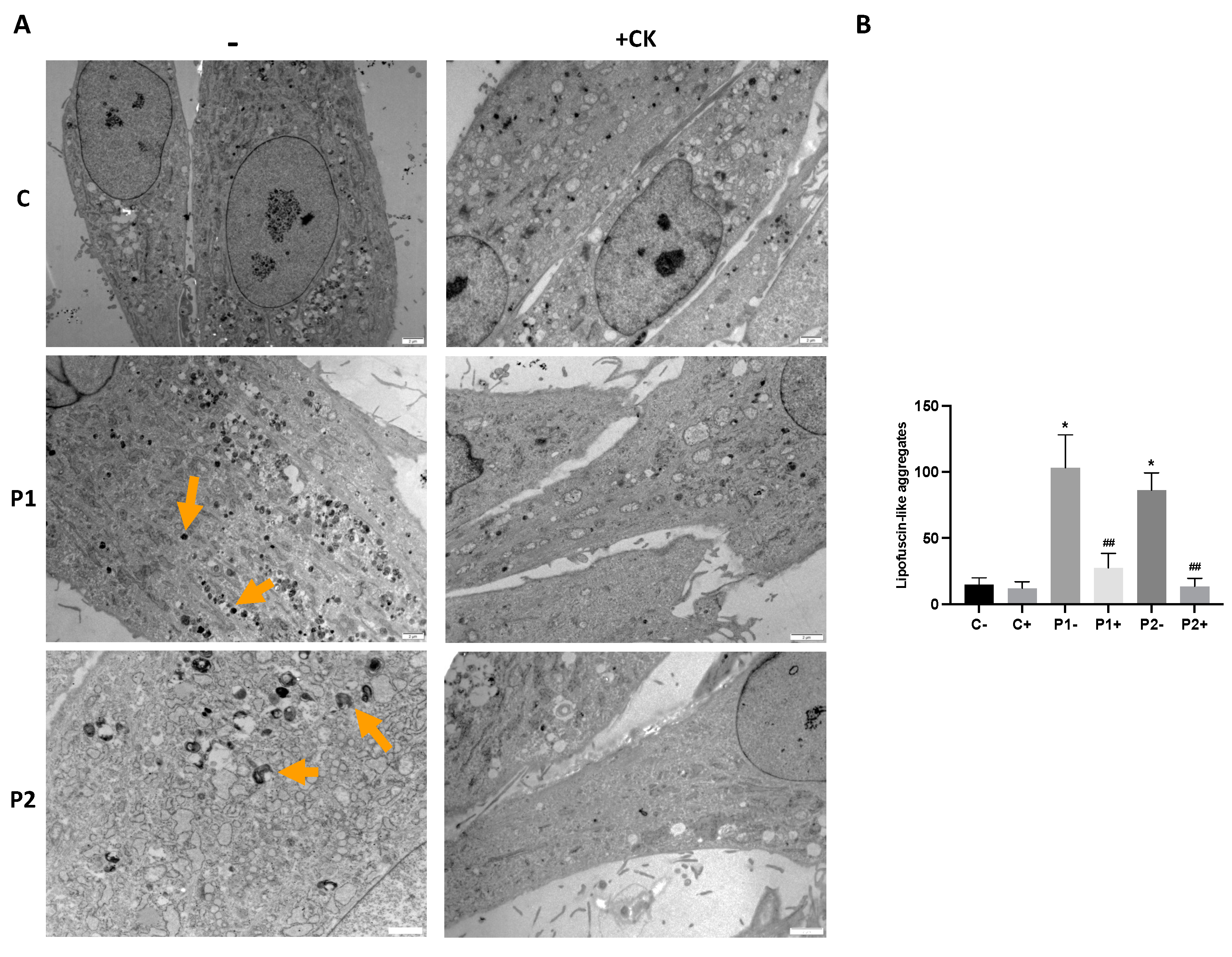
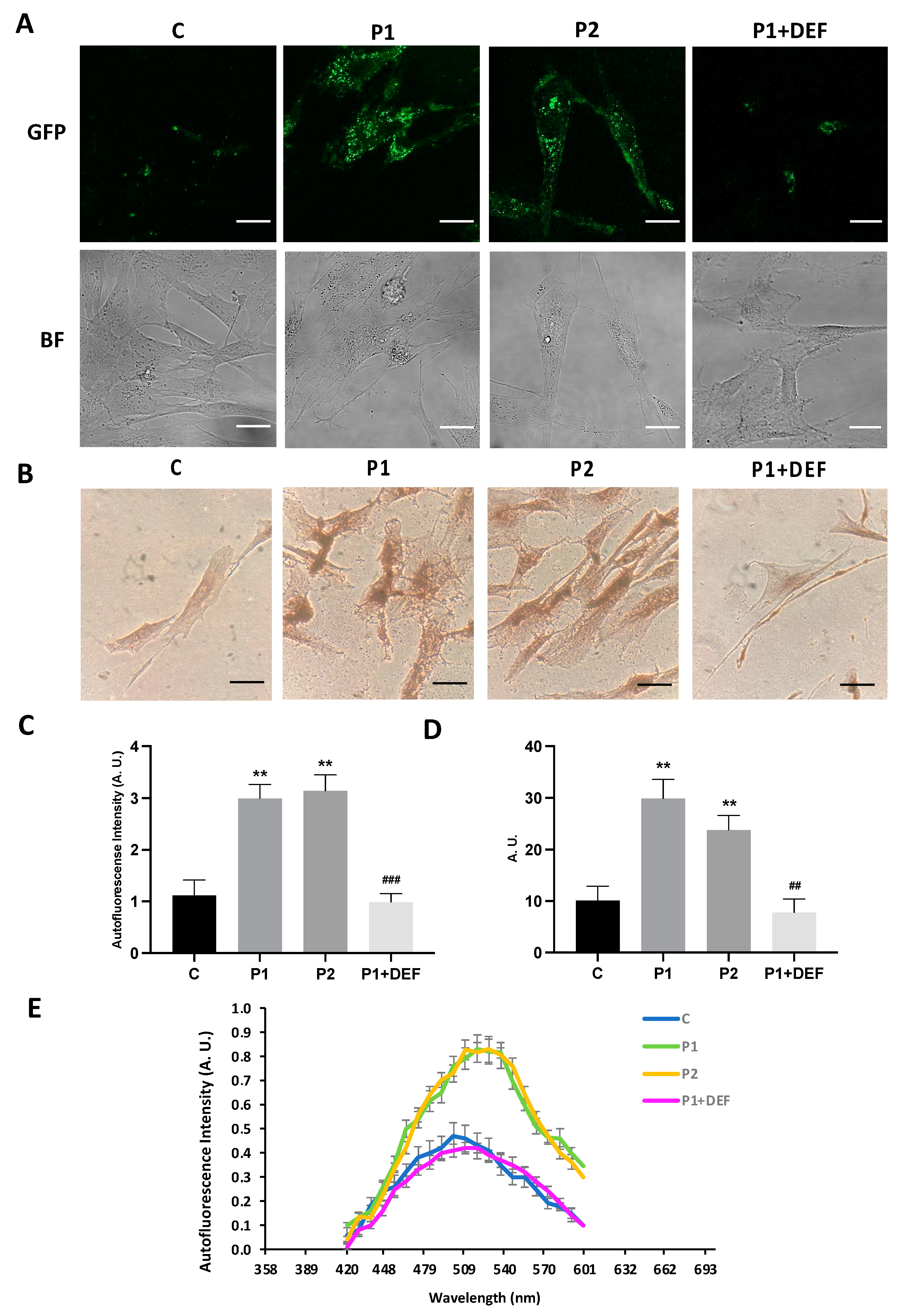
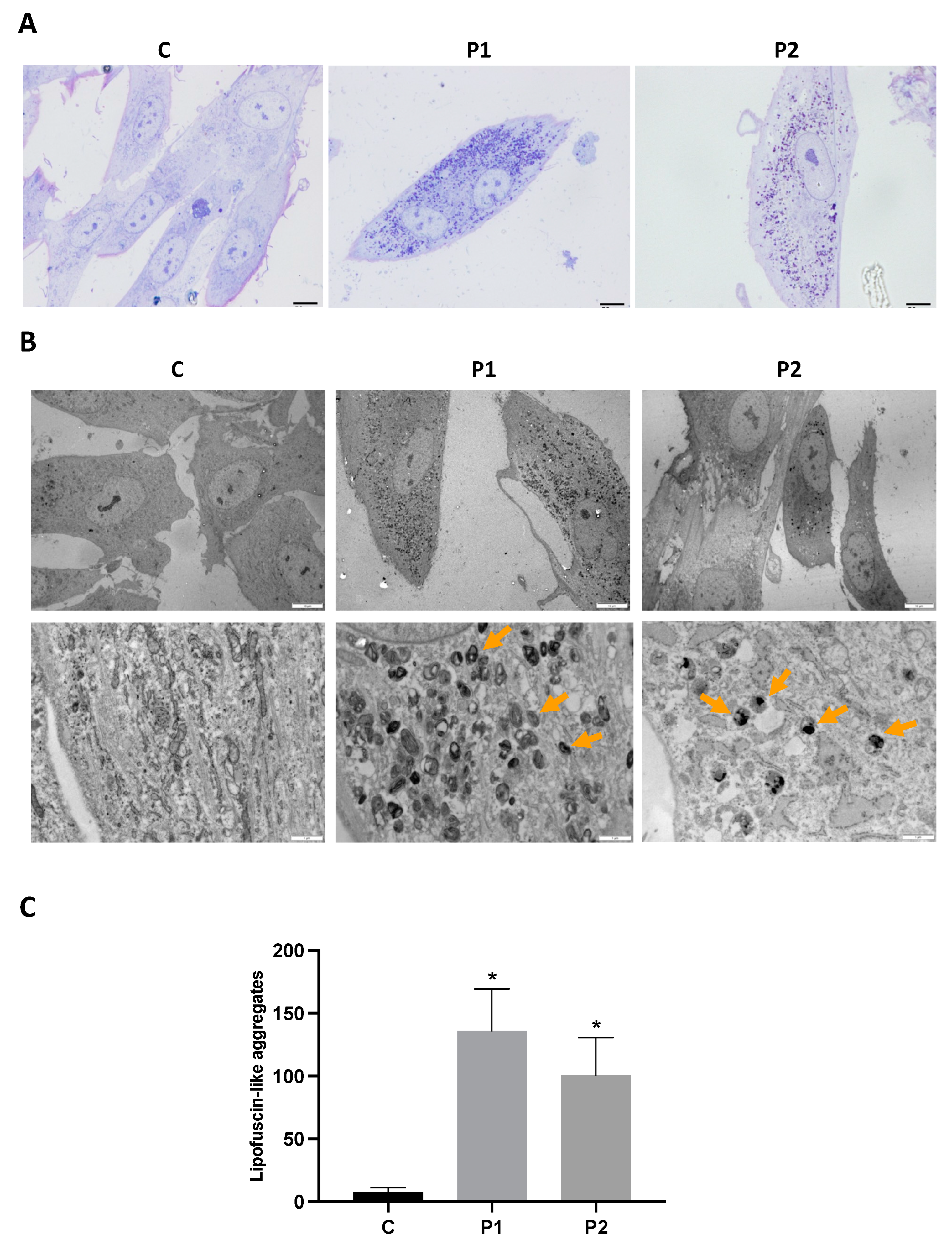
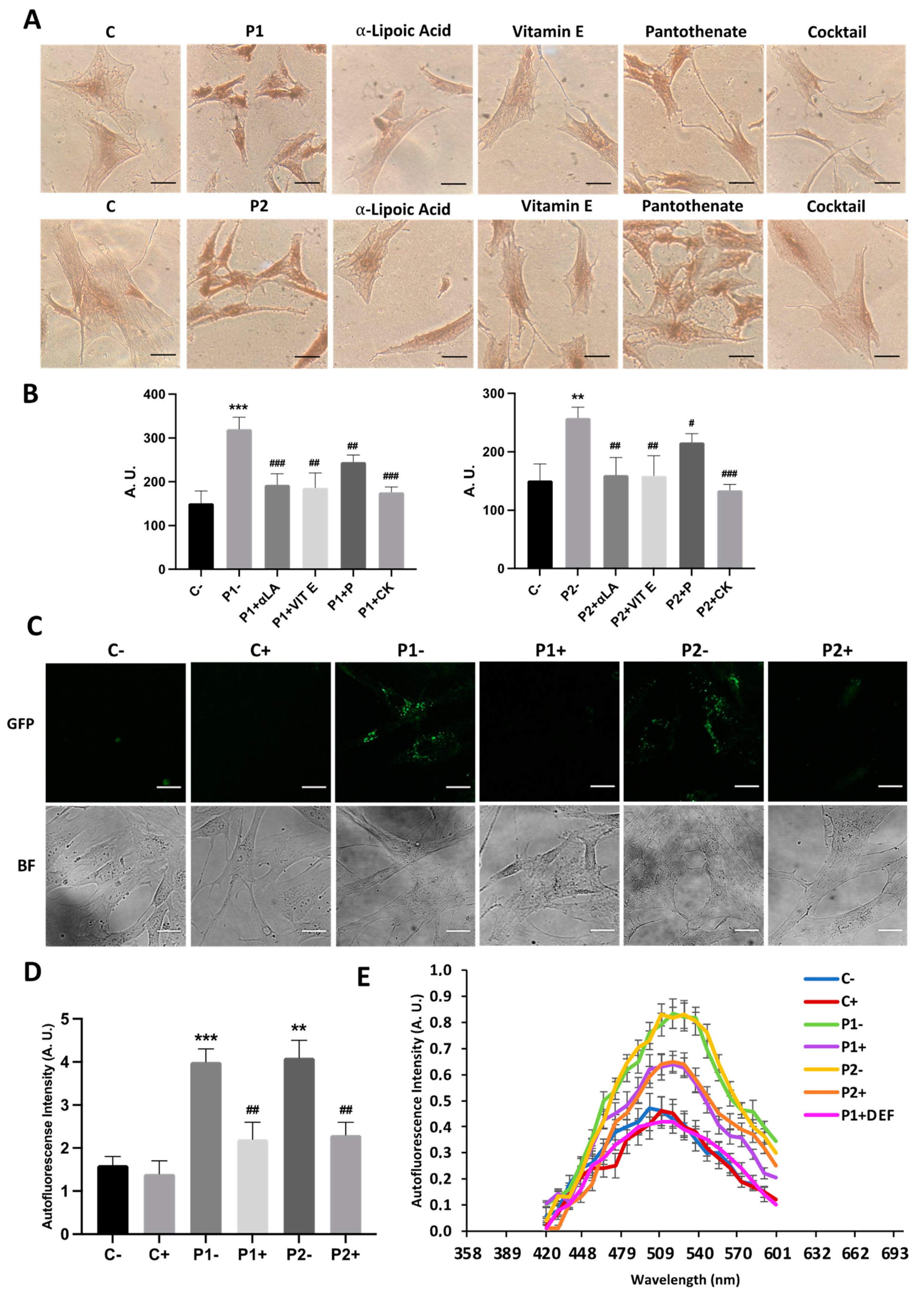
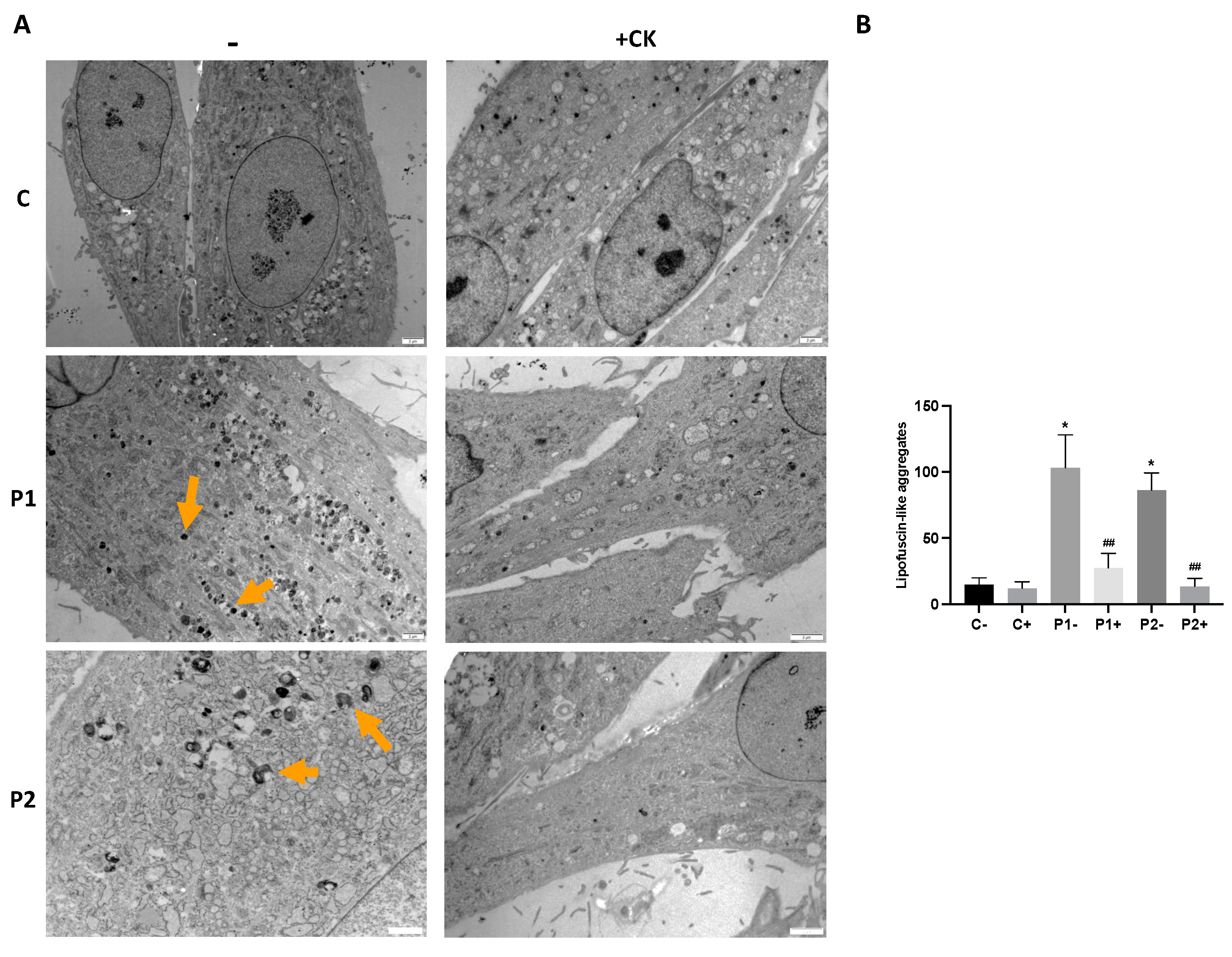
2.4. WDR45 Mutant Cells Accumulate Aberrant Lipofuscin-like Aggregates
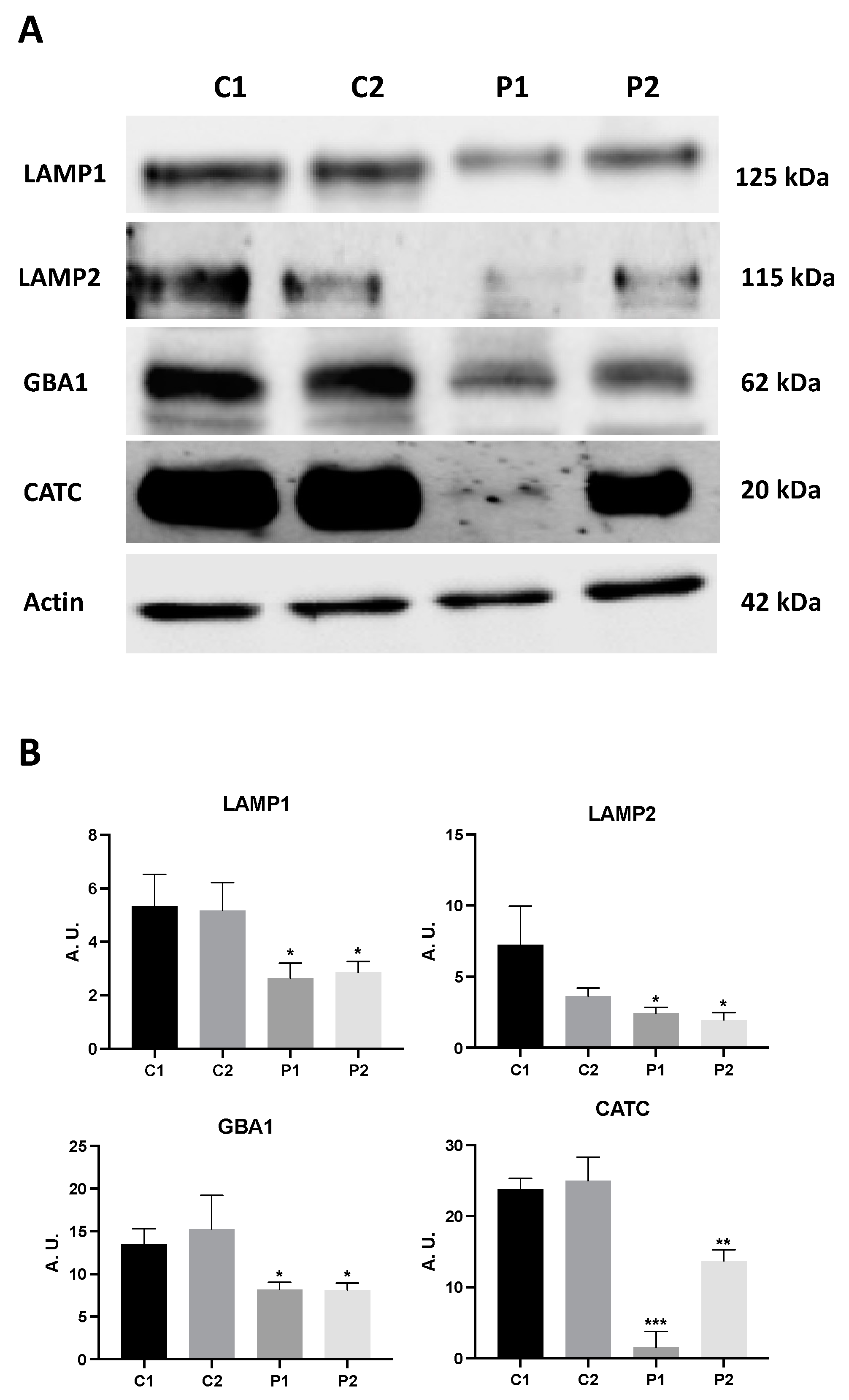
2.5. WDR45 Deficiency Alters the Expression Levels of Proteins Involved in Iron Metabolism
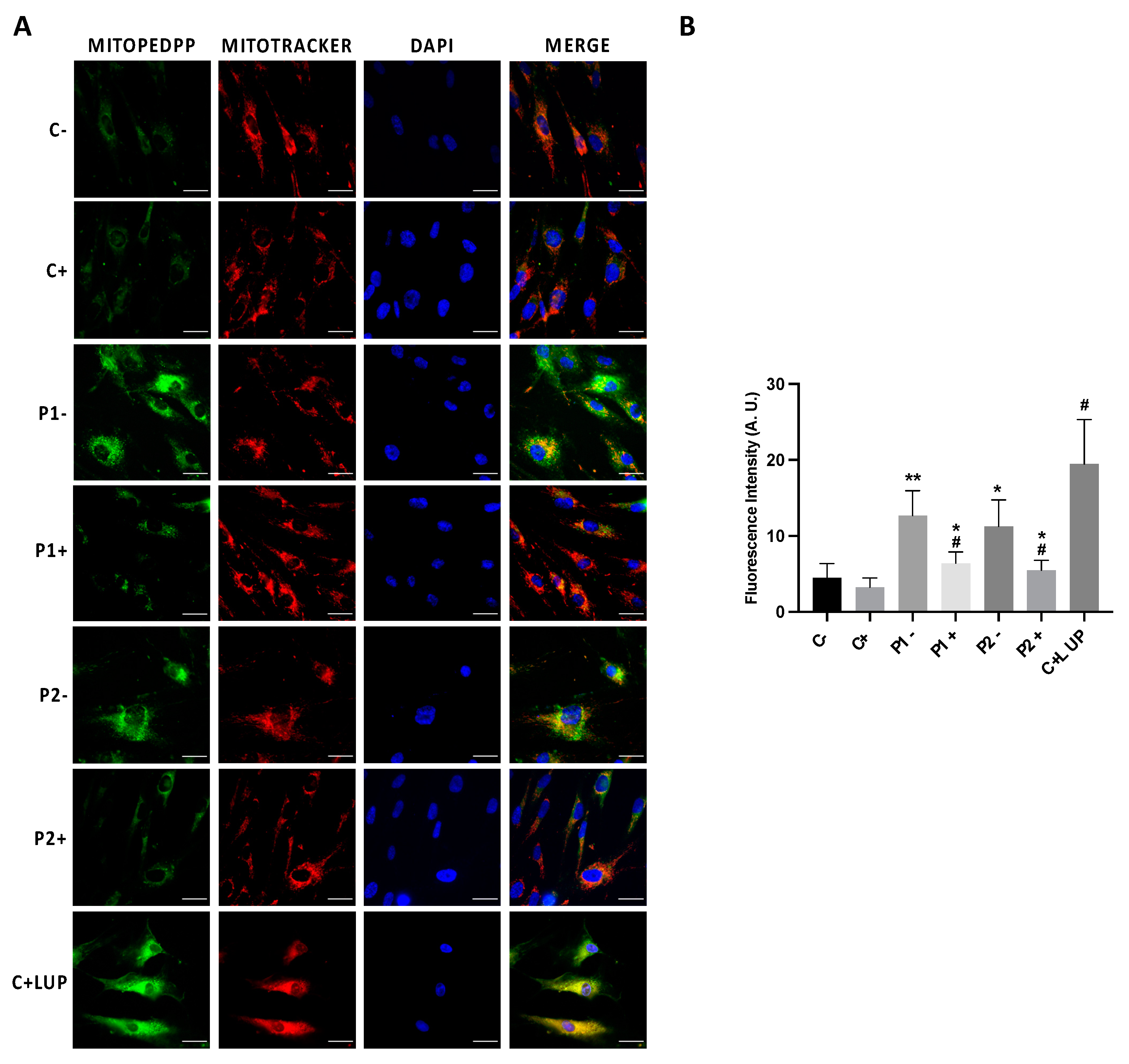
2.6. WDR45 Deficiency Impairs Mitochondrial Network and Function
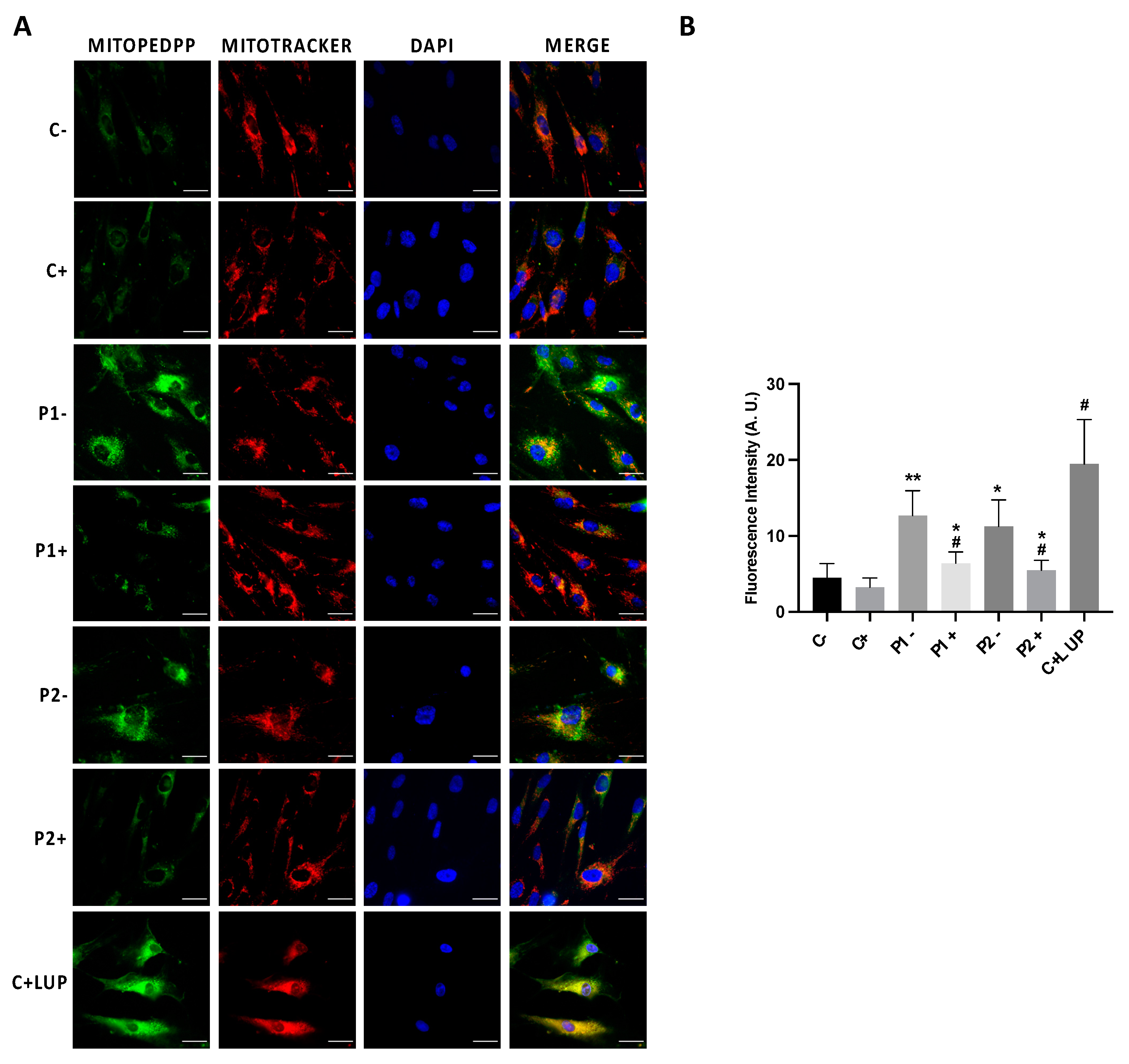
2.7. Mitochondrial Dysfunction Is Associated with Increased Mitochondrial Lipid Peroxidation in BPAN Cells
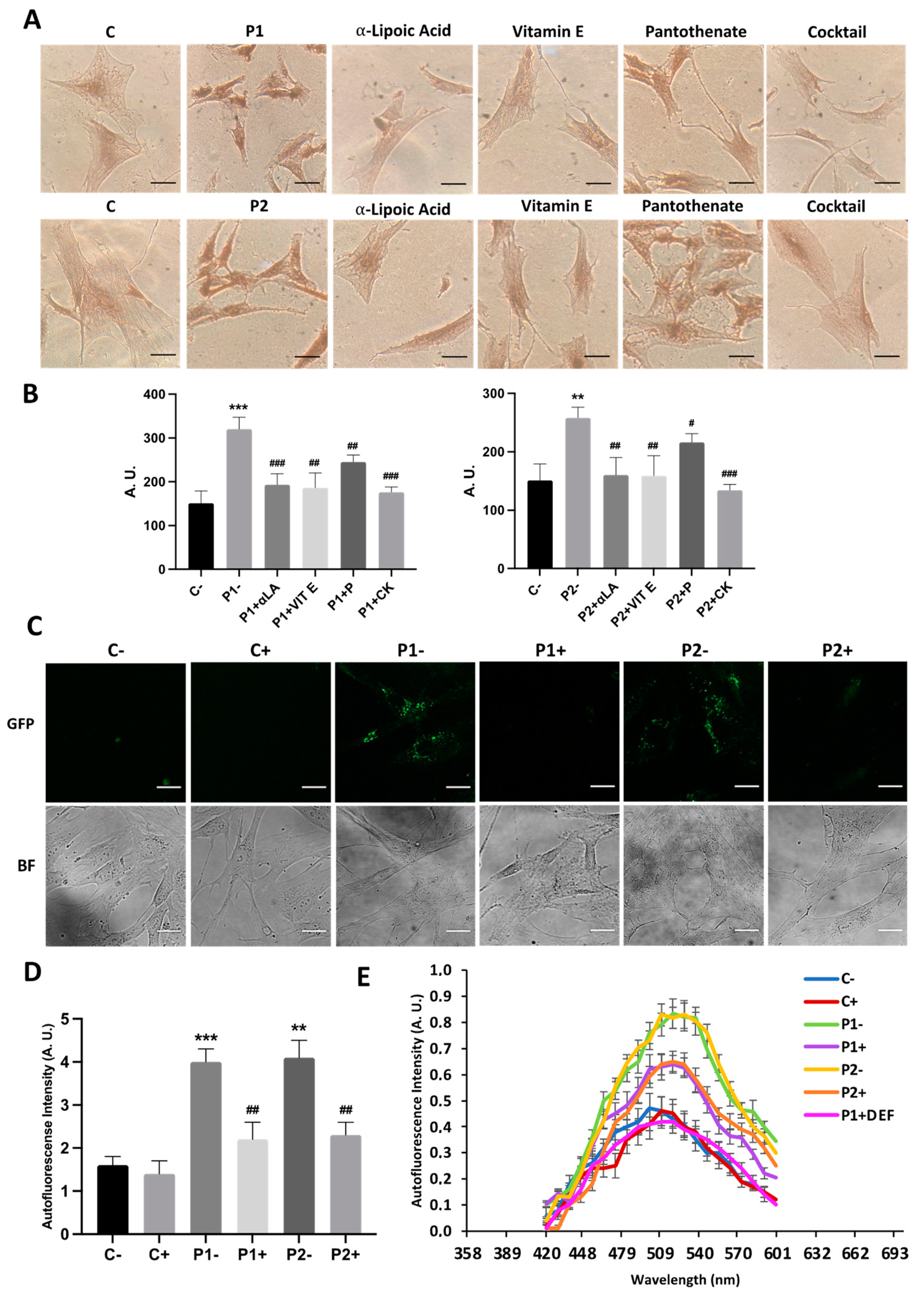
2.8. Antioxidants Prevent Iron Accumulation in BPAN Cells
2.9. Antioxidants Prevent the Accumulation of Lipofuscin-like Aggregates
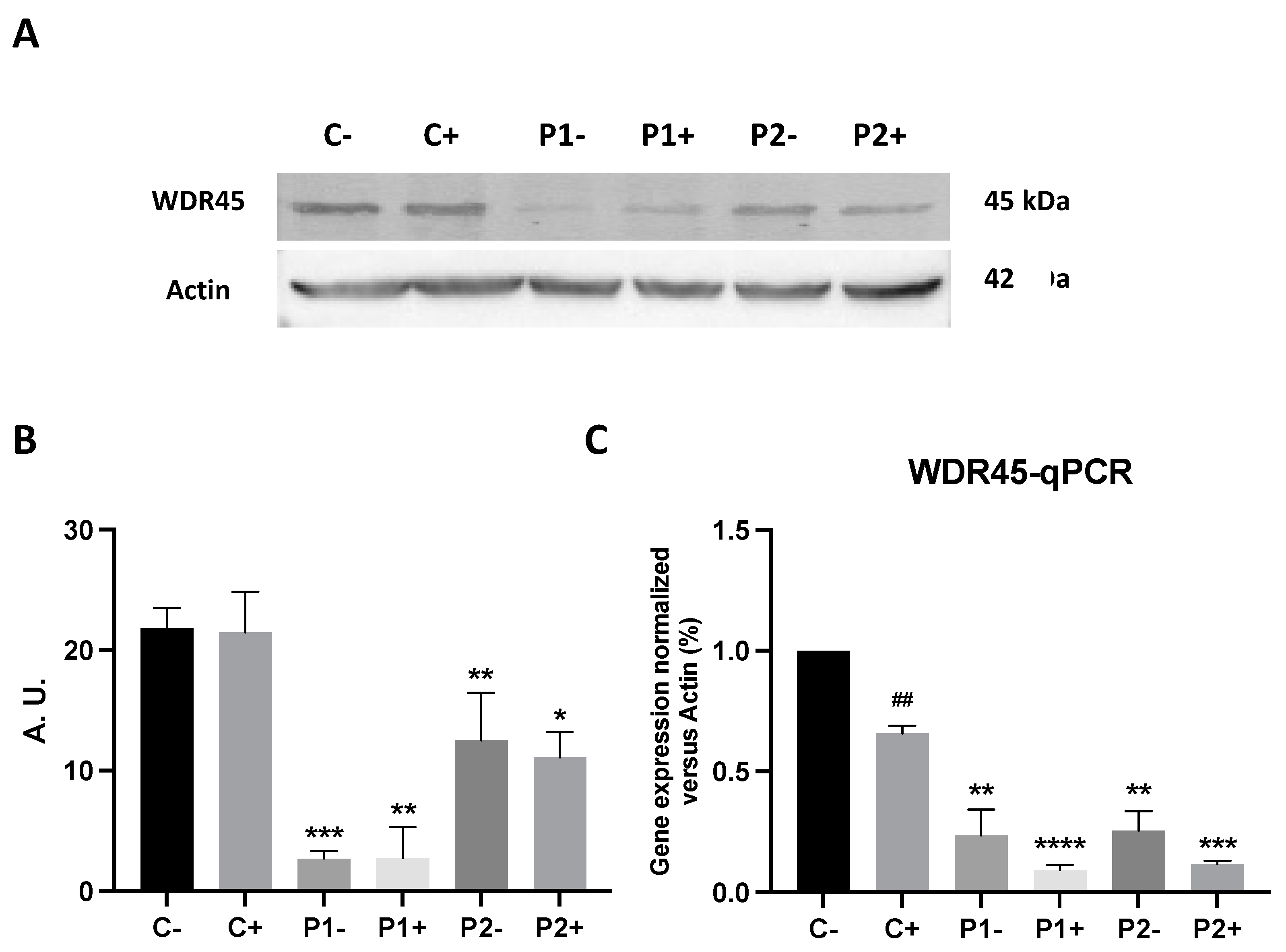
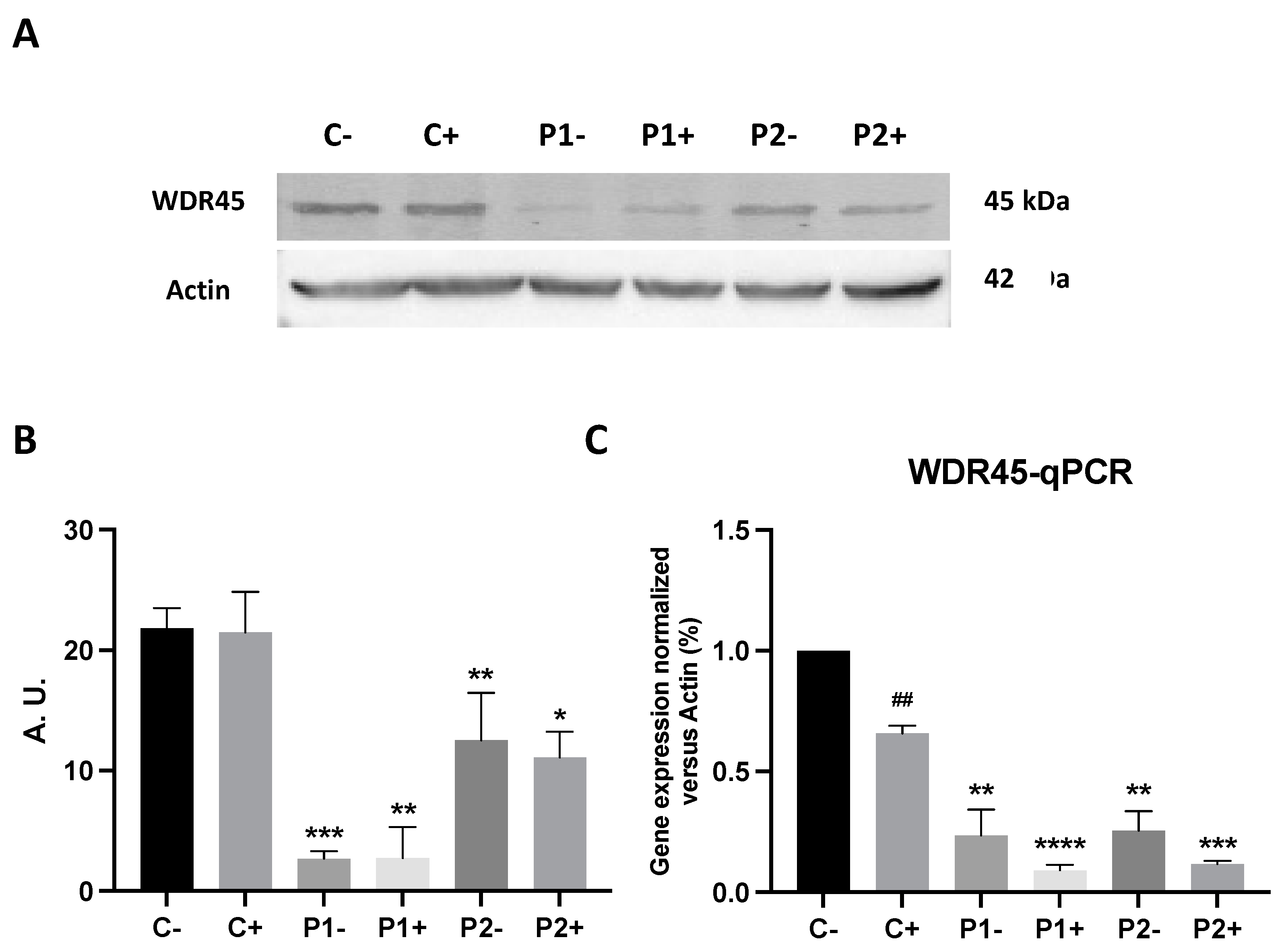
2.10. Antioxidants Do Not Correct WDR45 Transcripts or Protein Expression Levels in BPAN Fibroblasts
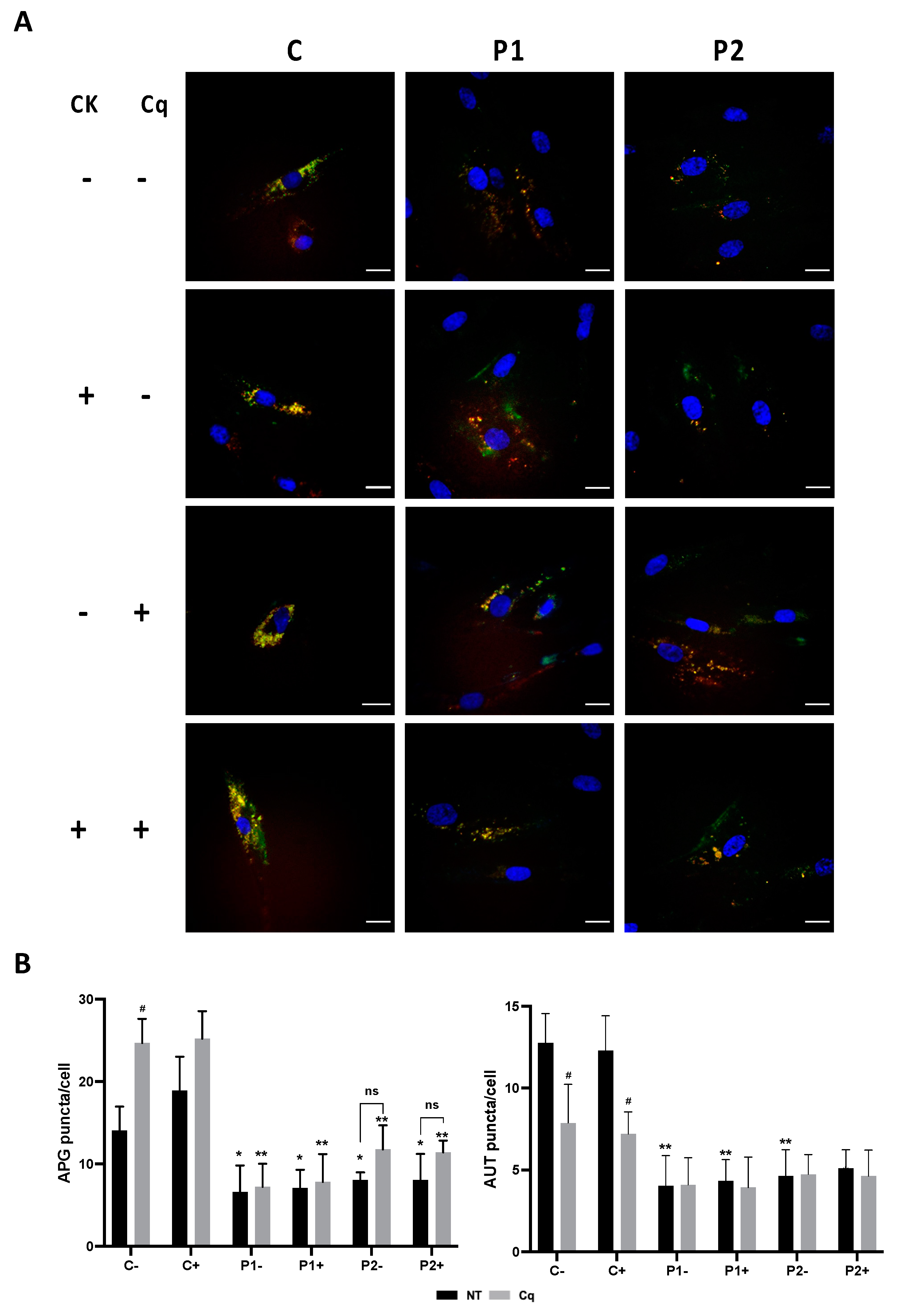
2.11. Antioxidants Do Not Correct Autophagic Failure in BPAN Fibroblasts
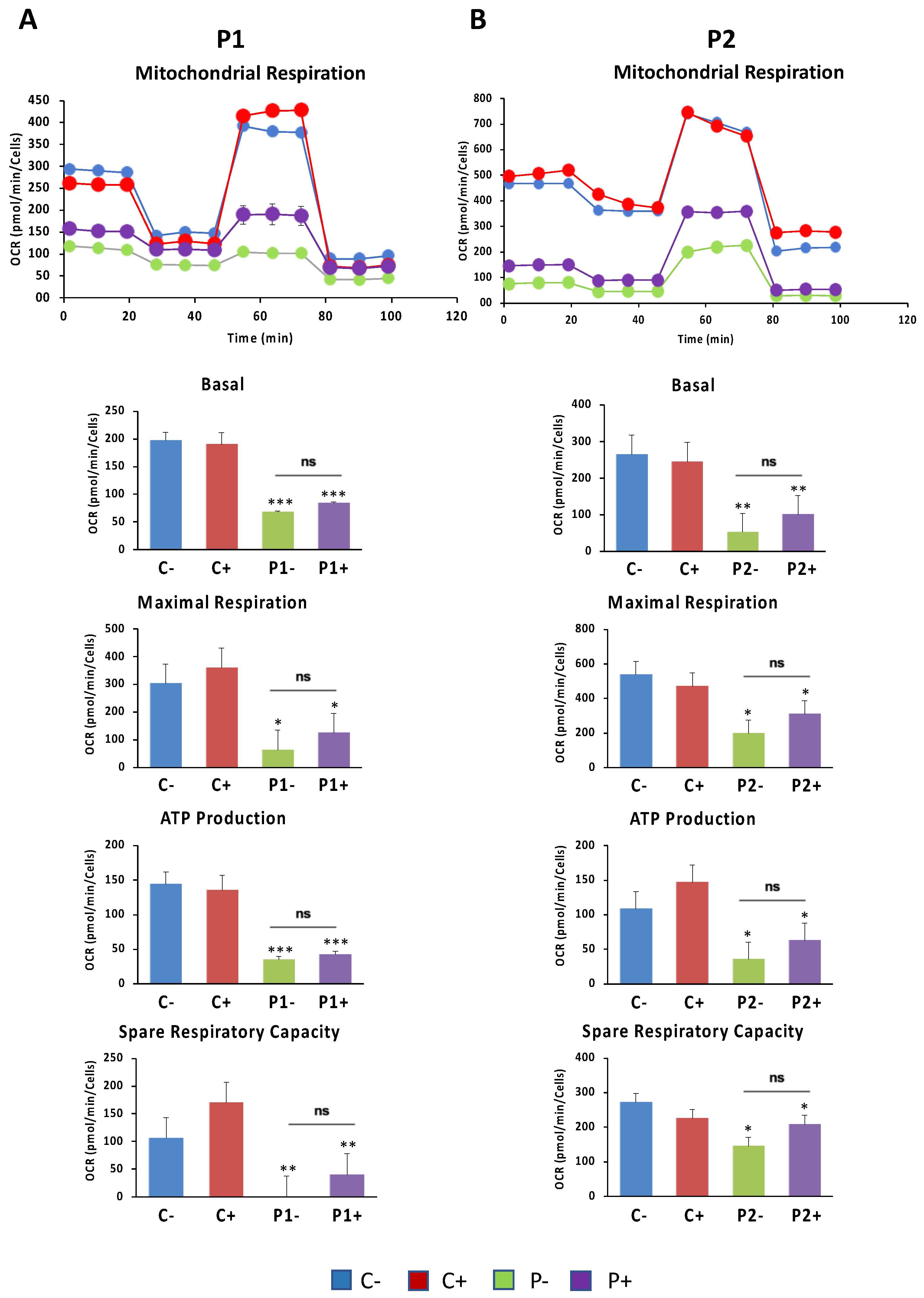
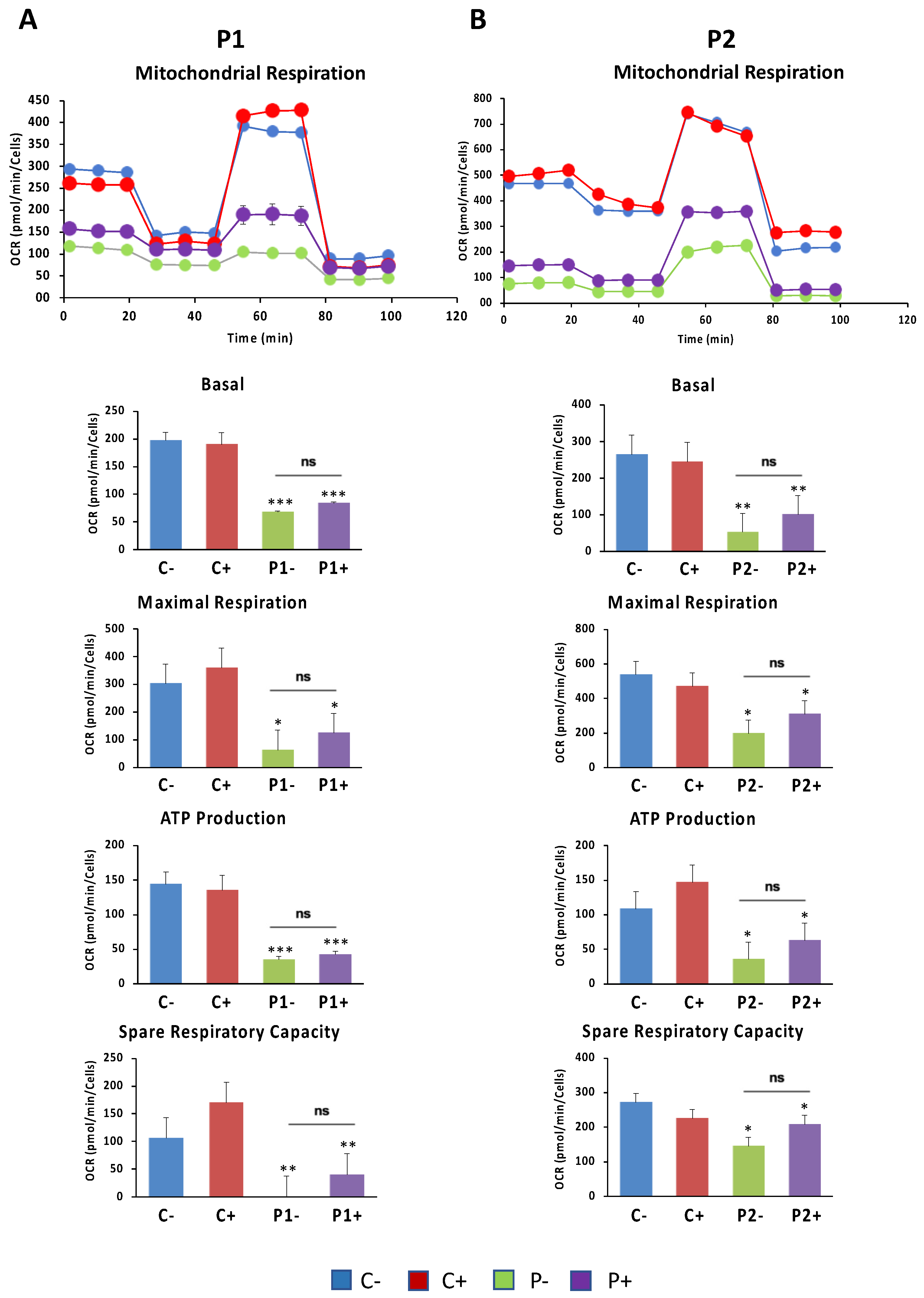
2.12. Antioxidants Do Not Correct Cell Bioenergetics in BPAN Fibroblasts
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Patient Cell Cultures
4.3. Cell Transfection with Human WDR45 Plasmid
4.4. Immunoblotting
4.5. Quantitative Real-Time PCR
4.6. Inmunofluorescence Microscopy
4.7. Lysotracker Staining
4.8. Tandem Sensor RFP-GFP-LC3B
4.9. Iron and Lipofuscin Accumulation
4.10. Iron Metabolism and Labile Iron Pool
4.11. TEM Analysis
4.12. Bioenergetic and Oxidative Stress Analysis
4.13. Mitotracker Staining: Analysis of Mitochondrial Network
4.14. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hayflick, S.J.; Kurian, M.A.; Hogarth, P. Neurodegeneration with brain iron accumulation. Handb. Clin. Neurol. 2018, 147, 293–305. [Google Scholar] [CrossRef] [PubMed]
- Adang, L.A.; Pizzino, A.; Malhotra, A.; Dubbs, H.; Williams, C.; Sherbini, O.; Anttonen, A.K.; Lesca, G.; Linnankivi, T.; Laurencin, C.; et al. Phenotypic and Imaging Spectrum Associated With WDR45. Pediatr. Neurol. 2020, 109, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Haack, T.B.; Hogarth, P.; Gregory, A.; Prokisch, H.; Hayflick, S.J. BPAN: The only X-linked dominant NBIA disorder. Int. Rev. Neurobiol. 2013, 110, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.L.; Gregory, A.; Kurian, M.A.; Bushlin, I.; Mochel, F.; Emrick, L.; Adang, L.; Group, B.G.C.A.; Hogarth, P.; Hayflick, S.J. Consensus clinical management guideline for beta-propeller protein-associated neurodegeneration. Dev. Med. Child Neurol. 2021, 63, 1402–1409. [Google Scholar] [CrossRef] [PubMed]
- Long, M.; Abdeen, N.; Geraghty, M.T.; Hogarth, P.; Hayflick, S.; Venkateswaran, S. Novel WDR45 Mutation and Pathognomonic BPAN Imaging in a Young Female With Mild Cognitive Delay. Pediatrics 2015, 136, e714–e717. [Google Scholar] [CrossRef]
- Abidi, A.; Mignon-Ravix, C.; Cacciagli, P.; Girard, N.; Milh, M.; Villard, L. Early-onset epileptic encephalopathy as the initial clinical presentation of WDR45 deletion in a male patient. Eur. J. Hum. Genet. 2016, 24, 615–618. [Google Scholar] [CrossRef]
- Saitsu, H.; Nishimura, T.; Muramatsu, K.; Kodera, H.; Kumada, S.; Sugai, K.; Kasai-Yoshida, E.; Sawaura, N.; Nishida, H.; Hoshino, A.; et al. De novo mutations in the autophagy gene WDR45 cause static encephalopathy of childhood with neurodegeneration in adulthood. Nat. Genet. 2013, 45, 445–449. [Google Scholar] [CrossRef]
- Lee, J.H.; Yun, J.Y.; Gregory, A.; Hogarth, P.; Hayflick, S.J. Brain MRI Pattern Recognition in Neurodegeneration with Brain Iron Accumulation. Front. Neurol. 2020, 11, 1024. [Google Scholar] [CrossRef]
- Stanga, D.; Zhao, Q.; Milev, M.P.; Saint-Dic, D.; Jimenez-Mallebrera, C.; Sacher, M. TRAPPC11 functions in autophagy by recruiting ATG2B-WIPI4/WDR45 to preautophagosomal membranes. Traffic 2019, 20, 325–345. [Google Scholar] [CrossRef]
- Wan, H.; Wang, Q.; Chen, X.; Zeng, Q.; Shao, Y.; Fang, H.; Liao, X.; Li, H.S.; Liu, M.G.; Xu, T.L.; et al. WDR45 contributes to neurodegeneration through regulation of ER homeostasis and neuronal death. Autophagy 2020, 16, 531–547. [Google Scholar] [CrossRef]
- Cong, Y.; So, V.; Tijssen, M.A.J.; Verbeek, D.S.; Reggiori, F.; Mauthe, M. WDR45, one gene associated with multiple neurodevelopmental disorders. Autophagy 2021, 17, 3908–3923. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Emr, S.D. Autophagy as a regulated pathway of cellular degradation. Science 2000, 290, 1717–1721. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Nam, S.O.; Kim, E.K.; Shin, J.H.; Oh, S.H.; Ryu, D.; Lee, H.E.; Mun, J.Y. Autophagic defects observed in fibroblasts from a patient with beta-propeller protein-associated neurodegeneration. Am. J. Med. Genet. Part A 2021, 185, 3866–3871. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.G.; Sun, L.; Miao, G.; Ji, C.; Zhao, H.; Sun, H.; Miao, L.; Yoshii, S.R.; Mizushima, N.; Wang, X.; et al. The autophagy gene Wdr45/Wipi4 regulates learning and memory function and axonal homeostasis. Autophagy 2015, 11, 881–890. [Google Scholar] [CrossRef]
- Ingrassia, R.; Memo, M.; Garavaglia, B. Ferrous Iron Up-regulation in Fibroblasts of Patients with Beta Propeller Protein-Associated Neurodegeneration (BPAN). Front. Genet. 2017, 8, 18. [Google Scholar] [CrossRef]
- Seibler, P.; Burbulla, L.F.; Dulovic, M.; Zittel, S.; Heine, J.; Schmidt, T.; Rudolph, F.; Westenberger, A.; Rakovic, A.; Munchau, A.; et al. Iron overload is accompanied by mitochondrial and lysosomal dysfunction in WDR45 mutant cells. Brain 2018, 141, 3052–3064. [Google Scholar] [CrossRef]
- Aring, L.; Choi, E.K.; Kopera, H.; Lanigan, T.; Iwase, S.; Klionsky, D.J.; Seo, Y.A. A neurodegeneration gene, WDR45, links impaired ferritinophagy to iron accumulation. J. Neurochem. 2022, 160, 356–375. [Google Scholar] [CrossRef]
- Mancias, J.D.; Wang, X.; Gygi, S.P.; Harper, J.W.; Kimmelman, A.C. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature 2014, 509, 105–109. [Google Scholar] [CrossRef]
- Diaw, S.H.; Ganos, C.; Zittel, S.; Plotze-Martin, K.; Kulikovskaja, L.; Vos, M.; Westenberger, A.; Rakovic, A.; Lohmann, K.; Dulovic-Mahlow, M. Mutant WDR45 Leads to Altered Ferritinophagy and Ferroptosis in beta-Propeller Protein-Associated Neurodegeneration. Int. J. Mol. Sci. 2022, 23, 9524. [Google Scholar] [CrossRef]
- Tsukida, K.; Muramatsu, S.I.; Osaka, H.; Yamagata, T.; Muramatsu, K. WDR45 variants cause ferrous iron loss due to impaired ferritinophagy associated with nuclear receptor coactivator 4 and WD repeat domain phosphoinositide interacting protein 4 reduction. Brain Commun. 2022, 4, fcac304. [Google Scholar] [CrossRef]
- Ganz, T. Systemic iron homeostasis. Physiol. Rev. 2013, 93, 1721–1741. [Google Scholar] [CrossRef]
- Salvador, G.A. Iron in neuronal function and dysfunction. Biofactors 2010, 36, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Uttara, B.; Singh, A.V.; Zamboni, P.; Mahajan, R.T. Oxidative stress and neurodegenerative diseases: A review of upstream and downstream antioxidant therapeutic options. Curr. Neuropharmacol. 2009, 7, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Villalon-Garcia, I.; Povea-Cabello, S.; Alvarez-Cordoba, M.; Talaveron-Rey, M.; Suarez-Rivero, J.M.; Suarez-Carrillo, A.; Munuera-Cabeza, M.; Reche-Lopez, D.; Cilleros-Holgado, P.; Pinero-Perez, R.; et al. Vicious cycle of lipid peroxidation and iron accumulation in neurodegeneration. Neural Regen. Res. 2023, 18, 1196–1202. [Google Scholar] [CrossRef] [PubMed]
- Cortes, C.J.; La Spada, A.R. TFEB dysregulation as a driver of autophagy dysfunction in neurodegenerative disease: Molecular mechanisms, cellular processes, and emerging therapeutic opportunities. Neurobiol. Dis. 2019, 122, 83–93. [Google Scholar] [CrossRef]
- Barth, S.; Glick, D.; Macleod, K.F. Autophagy: Assays and artifacts. J. Pathol. 2010, 221, 117–124. [Google Scholar] [CrossRef]
- Villanueva-Paz, M.; Povea-Cabello, S.; Villalon-Garcia, I.; Alvarez-Cordoba, M.; Suarez-Rivero, J.M.; Talaveron-Rey, M.; Jackson, S.; Falcon-Moya, R.; Rodriguez-Moreno, A.; Sanchez-Alcazar, J.A. Parkin-mediated mitophagy and autophagy flux disruption in cellular models of MERRF syndrome. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165726. [Google Scholar] [CrossRef]
- Alvarez-Cordoba, M.; Fernandez Khoury, A.; Villanueva-Paz, M.; Gomez-Navarro, C.; Villalon-Garcia, I.; Suarez-Rivero, J.M.; Povea-Cabello, S.; de la Mata, M.; Cotan, D.; Talaveron-Rey, M.; et al. Pantothenate Rescues Iron Accumulation in Pantothenate Kinase-Associated Neurodegeneration Depending on the Type of Mutation. Mol. Neurobiol. 2019, 56, 3638–3656. [Google Scholar] [CrossRef]
- Villalon-Garcia, I.; Alvarez-Cordoba, M.; Povea-Cabello, S.; Talaveron-Rey, M.; Villanueva-Paz, M.; Luzon-Hidalgo, R.; Suarez-Rivero, J.M.; Suarez-Carrillo, A.; Munuera-Cabeza, M.; Salas, J.J.; et al. Vitamin E prevents lipid peroxidation and iron accumulation in PLA2G6-Associated Neurodegeneration. Neurobiol. Dis. 2022, 165, 105649. [Google Scholar] [CrossRef]
- Bindewald-Wittich, A.; Han, M.; Schmitz-Valckenberg, S.; Snyder, S.R.; Giese, G.; Bille, J.F.; Holz, F.G. Two-photon-excited fluorescence imaging of human RPE cells with a femtosecond Ti:Sapphire laser. Investig. Ophthalmol. Vis. Sci. 2006, 47, 4553–4557. [Google Scholar] [CrossRef]
- Campanella, A.; Privitera, D.; Guaraldo, M.; Rovelli, E.; Barzaghi, C.; Garavaglia, B.; Santambrogio, P.; Cozzi, A.; Levi, S. Skin fibroblasts from pantothenate kinase-associated neurodegeneration patients show altered cellular oxidative status and have defective iron-handling properties. Hum. Mol. Genet. 2012, 21, 4049–4059. [Google Scholar] [CrossRef] [PubMed]
- Orellana, D.I.; Santambrogio, P.; Rubio, A.; Yekhlef, L.; Cancellieri, C.; Dusi, S.; Giannelli, S.G.; Venco, P.; Mazzara, P.G.; Cozzi, A.; et al. Coenzyme A corrects pathological defects in human neurons of PANK2-associated neurodegeneration. EMBO Mol. Med. 2016, 8, 1197–1211. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Cordoba, M.; Reche-Lopez, D.; Cilleros-Holgado, P.; Talaveron-Rey, M.; Villalon-Garcia, I.; Povea-Cabello, S.; Suarez-Rivero, J.M.; Suarez-Carrillo, A.; Munuera-Cabeza, M.; Pinero-Perez, R.; et al. Therapeutic approach with commercial supplements for pantothenate kinase-associated neurodegeneration with residual PANK2 expression levels. Orphanet J. Rare Dis. 2022, 17, 311. [Google Scholar] [CrossRef]
- Xiong, Q.; Li, X.; Li, W.; Chen, G.; Xiao, H.; Li, P.; Wu, C. WDR45 Mutation Impairs the Autophagic Degradation of Transferrin Receptor and Promotes Ferroptosis. Front. Mol. Biosci. 2021, 8, 645831. [Google Scholar] [CrossRef]
- Noda, M.; Ito, H.; Nagata, K.I. Physiological significance of WDR45, a responsible gene for beta-propeller protein associated neurodegeneration (BPAN), in brain development. Sci. Rep. 2021, 11, 22568. [Google Scholar] [CrossRef] [PubMed]
- Schonthal, A.H. Endoplasmic reticulum stress: Its role in disease and novel prospects for therapy. Scientifica 2012, 2012, 857516. [Google Scholar] [CrossRef] [PubMed]
- Kalia, L.V.; Lang, A.E. Parkinson’s disease. Lancet 2015, 386, 896–912. [Google Scholar] [CrossRef]
- Li, H.; Ham, A.; Ma, T.C.; Kuo, S.H.; Kanter, E.; Kim, D.; Ko, H.S.; Quan, Y.; Sardi, S.P.; Li, A.; et al. Mitochondrial dysfunction and mitophagy defect triggered by heterozygous GBA mutations. Autophagy 2019, 15, 113–130. [Google Scholar] [CrossRef]
- Urrutia, P.J.; Mena, N.P.; Nunez, M.T. The interplay between iron accumulation, mitochondrial dysfunction, and inflammation during the execution step of neurodegenerative disorders. Front. Pharmacol. 2014, 5, 38. [Google Scholar] [CrossRef]
- Hohn, A.; Grune, T. Lipofuscin: Formation, effects and role of macroautophagy. Redox Biol. 2013, 1, 140–144. [Google Scholar] [CrossRef]
- Lyamzaev, K.G.; Knorre, D.A.; Chernyak, B.V. Mitoptosis, Twenty Years after. Biochem. Biokhimiia 2020, 85, 1484–1498. [Google Scholar] [CrossRef]
- Tinari, A.; Garofalo, T.; Sorice, M.; Esposti, M.D.; Malorni, W. Mitoptosis: Different pathways for mitochondrial execution. Autophagy 2007, 3, 282–284. [Google Scholar] [CrossRef]
- Ilie, O.-D.; Ciobica, A.; Riga, S.; Dhunna, N.; McKenna, J.; Mavroudis, I.; Doroftei, B.; Ciobanu, A.-M.; Riga, D. Mini-Review on Lipofuscin and Aging: Focusing on the Molecular Interface, the Biological Recycling Mechanism, Oxidative Stress, and the Gut-Brain Axis Functionality. Medicina 2020, 56, 626. [Google Scholar] [CrossRef] [PubMed]
- Tonolli, P.N.; Martins, W.K.; Junqueira, H.C.; Silva, M.N.; Severino, D.; Santacruz-Perez, C.; Watanabe, I.; Baptista, M.S. Lipofuscin in keratinocytes: Production, properties, and consequences of the photosensitization with visible light. Free Radic. Biol. Med. 2020, 160, 277–292. [Google Scholar] [CrossRef] [PubMed]
- Kakimoto, Y.; Okada, C.; Kawabe, N.; Sasaki, A.; Tsukamoto, H.; Nagao, R.; Osawa, M. Myocardial lipofuscin accumulation in ageing and sudden cardiac death. Sci. Rep. 2019, 9, 3304. [Google Scholar] [CrossRef] [PubMed]
- Călin, E.F.; Popescu, S.I.P.; Cernat, C.C.C.; Patoni, C.; Popescu, M.-N.; Mușat, O. Lipofuscin: A key compound in ophthalmic practice. Rom. J. Ophthalmol. 2021, 65, 109–113. [Google Scholar] [CrossRef]
- Moreno-García, A.; Kun, A.; Calero, O.; Medina, M.; Calero, M. An Overview of the Role of Lipofuscin in Age-Related Neurodegeneration. Front. Neurosci. 2018, 12, 464. [Google Scholar] [CrossRef]
- Mijaljica, D.; Prescott, M.; Devenish, R.J. Mitophagy and Mitoptosis in Disease Processes. In Protein Misfolding and Cellular Stress in Disease and Aging: Concepts and Protocols (Methods and Protocols); Bross, P., Gregersen, N., Eds.; Humana Press: Totowa, NJ, USA, 2010; Volume 648. [Google Scholar]
- Brunk, U.T.; Terman, A. The mitochondrial-lysosomal axis theory of aging: Accumulation of damaged mitochondria as a result of imperfect autophagocytosis. Eur. J. Biochem. 2002, 269, 1996–2002. [Google Scholar] [CrossRef]
- Snyder, A.N. ; Crane., J.S. Histology, Lipofuscin; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Frolova, M.S.; Surin, A.M.; Braslavski, A.V.; Vekshin, N.L. Degradation of Mitochondria to Lipofuscin upon Heating and Illumination. Biofizika 2015, 60, 1125–1131. [Google Scholar] [CrossRef]
- Paul, B.T.; Manz, D.H.; Torti, F.M.; Torti, S.V. Mitochondria and Iron: Current Questions. Expert. Rev. Hematol. 2017, 10, 65–79. [Google Scholar] [CrossRef]
- Bindoli, A. Lipid peroxidation in mitochondria Free Radic. Biol. Med. 1988, 5, 247–261. [Google Scholar] [CrossRef]
- Arber, C.E.; Li, A.; Houlden, H.; Wray, S. Review: Insights into molecular mechanisms of disease in neurodegeneration with brain iron accumulation: Unifying theories. Neuropathol. Appl. Neurobiol. 2016, 42, 220–241. [Google Scholar] [CrossRef]
- Mena, N.P.; Urrutia, P.J.; Lourido, F.; Carrasco, C.M.; Nunez, M.T. Mitochondrial iron homeostasis and its dysfunctions in neurodegenerative disorders. Mitochondrion 2015, 21, 92–105. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.E.; Jung, M.K.; Noh, S.G.; Choi, H.B.; Chae, S.H.; Lee, J.H.; Mun, J.Y. Iron Accumulation and Changes in Cellular Organelles in WDR45 Mutant Fibroblasts. Int. J. Mol. Sci. 2021, 22, 11650. [Google Scholar] [CrossRef] [PubMed]
- Koksal, A.R.; Verne, G.N.; Zhou, Q. Endoplasmic reticulum stress in biological processing and disease. J. Investig. Med. 2021, 69, 309–315. [Google Scholar] [CrossRef]
- Moretti, R.; Peinkhofer, C. B Vitamins and Fatty Acids: What Do They Share with Small Vessel Disease-Related Dementia? Int. J. Mol. Sci. 2019, 20, 5797. [Google Scholar] [CrossRef]
- Kennedy, D.O. B Vitamins and the Brain: Mechanisms, Dose and Efficacy—A Review. Nutrients 2016, 8, 68. [Google Scholar] [CrossRef]
- Mikkelsen, K.; Apostolopoulos, V. B Vitamins and Ageing. Subcell. Biochem. 2018, 90, 451–470. [Google Scholar] [CrossRef]
- Patassini, S.; Begley, P.; Xu, J.; Church, S.J.; Kureishy, N.; Reid, S.J.; Waldvogel, H.J.; Faull, R.L.M.; Snell, R.G.; Unwin, R.D.; et al. Cerebral Vitamin B5 (D-Pantothenic Acid) Deficiency as a Potential Cause of Metabolic Perturbation and Neurodegeneration in Huntington’s Disease. Metabolites 2019, 9, 113. [Google Scholar] [CrossRef]
- Slyshenkov, V.S.; Dymkowska, D.; Wojtczak, L. Pantothenic acid and pantothenol increase biosynthesis of glutathione by boosting cell energetics. FEBS Lett. 2004, 569, 169–172. [Google Scholar] [CrossRef]
- Sm, S.; Hn, S.; Na, E.; As, H. Curative role of pantothenic acid in brain damage of gamma irradiated rats. Indian J. Clin. Biochem. 2018, 33, 314–321. [Google Scholar] [CrossRef]
- Wojtczak, L.; Slyshenkov, V.S. Protection by pantothenic acid against apoptosis and cell damage by oxygen free radicals—The role of glutathione. Biofactors 2003, 17, 61–73. [Google Scholar] [CrossRef] [PubMed]
- Lands, W.E.; Hart, P. Metabolism of Glycerolipids: Vi. Specificities of Acyl Coenzyme A: Phospholipid Acyltransferases. J. Biol. Chem. 1965, 240, 1905–1911. [Google Scholar] [CrossRef] [PubMed]
- Mustacich, D.J.; Bruno, R.S.; Traber, M.G. Vitamin E Vitam. Horm. 2007, 76, 1–21. [Google Scholar] [CrossRef]
- Ulatowski, L.M.; Manor, D. Vitamin E and neurodegeneration. Neurobiol. Dis. 2015, 84, 78–83. [Google Scholar] [CrossRef] [PubMed]
- Tibullo, D.; Li Volti, G.; Giallongo, C.; Grasso, S.; Tomassoni, D.; Anfuso, C.D.; Lupo, G.; Amenta, F.; Avola, R.; Bramanti, V. Biochemical and clinical relevance of alpha lipoic acid: Antioxidant and anti-inflammatory activity, molecular pathways and therapeutic potential. Inflamm. Res. 2017, 66, 947–959. [Google Scholar] [CrossRef]
- Smith, C.A.; de la Fuente, J.; Pelaz, B.; Furlani, E.P.; Mullin, M.; Berry, C.C. The effect of static magnetic fields and tat peptides on cellular and nuclear uptake of magnetic nanoparticles. Biomaterials 2010, 31, 4392–4400. [Google Scholar] [CrossRef]
- Khan, H.; Singh, T.G.; Dahiya, R.S.; Abdel-Daim, M.M. Alpha-Lipoic Acid, an Organosulfur Biomolecule a Novel Therapeutic Agent for Neurodegenerative Disorders: An Mechanistic Perspective. Neurochem. Res. 2022, 47, 1853–1864. [Google Scholar] [CrossRef]
- Abdelhalim, M.A.K.; Qaid, H.A.; Al-Mohy, Y.H.; Ghannam, M.M. The Protective Roles of Vitamin E and alpha-Lipoic Acid Against Nephrotoxicity, Lipid Peroxidation, and Inflammatory Damage Induced by Gold Nanoparticles. Int. J. Nanomed. 2020, 15, 729–734. [Google Scholar] [CrossRef]
- Androne, L.; Gavan, N.A.; Veresiu, I.A.; Orasan, R. In vivo effect of lipoic acid on lipid peroxidation in patients with diabetic neuropathy. Vivo 2000, 14, 327–330. [Google Scholar]
- Talaveron-Rey, M.; Alvarez-Cordoba, M.; Villalon-Garcia, I.; Povea-Cabello, S.; Suarez-Rivero, J.M.; Gomez-Fernandez, D.; Romero-Gonzalez, A.; Suarez-Carrillo, A.; Munuera-Cabeza, M.; Cilleros-Holgado, P.; et al. Alpha-lipoic acid supplementation corrects pathological alterations in cellular models of pantothenate kinase-associated neurodegeneration with residual PANK2 expression levels. Orphanet J. Rare Dis. 2023, 18, 80. [Google Scholar] [CrossRef] [PubMed]
- Hayflick, S.J.; Kruer, M.C.; Gregory, A.; Haack, T.B.; Kurian, M.A.; Houlden, H.H.; Anderson, J.; Boddaert, N.; Sanford, L.; Harik, S.I.; et al. Beta-Propeller protein-associated neurodegeneration: A new X-linked dominant disorder with brain iron accumulation. Brain 2013, 136, 1708–1717. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Rubio, D.; Hinarejos, I.; Sancho, P.; Gorria-Redondo, N.; Bernado-Fonz, R.; Tello, C.; Marco-Marin, C.; Marti-Carrera, I.; Martinez-Gonzalez, M.J.; Garcia-Ribes, A.; et al. Mutations, Genes, and Phenotypes Related to Movement Disorders and Ataxias. Int. J. Mol. Sci. 2022, 23, 11847. [Google Scholar] [CrossRef]
- Haack, T.B.; Hogarth, P.; Kruer, M.C.; Gregory, A.; Wieland, T.; Schwarzmayr, T.; Graf, E.; Sanford, L.; Meyer, E.; Kara, E.; et al. Exome sequencing reveals de novo WDR45 mutations causing a phenotypically distinct, X-linked dominant form of NBIA. Am. J. Hum. Genet. 2012, 91, 1144–1149. [Google Scholar] [CrossRef] [PubMed]
- Georgakopoulou, E.A.; Tsimaratou, K.; Evangelou, K.; Fernandez Marcos, P.J.; Zoumpourlis, V.; Trougakos, I.P.; Kletsas, D.; Bartek, J.; Serrano, M.; Gorgoulis, V.G. Specific lipofuscin staining as a novel biomarker to detect replicative and stress-induced senescence. A method applicable in cryo-preserved and archival tissues. Aging 2013, 5, 37–50. [Google Scholar] [CrossRef] [PubMed]
- Tarohda, T.; Ishida, Y.; Kawai, K.; Yamamoto, M.; Amano, R. Regional distributions of manganese, iron, copper, and zinc in the brains of 6-hydroxydopamine-induced parkinsonian rats. Anal. Bioanal. Chem. 2005, 383, 224–234. [Google Scholar] [CrossRef] [PubMed]
- Le Boedec, K. Sensitivity and specificity of normality tests and consequences on reference interval accuracy at small sample size: A computer-simulation study. Vet. Clin. Pathol. 2016, 45, 648–656. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suárez-Carrillo, A.; Álvarez-Córdoba, M.; Romero-González, A.; Talaverón-Rey, M.; Povea-Cabello, S.; Cilleros-Holgado, P.; Piñero-Pérez, R.; Reche-López, D.; Gómez-Fernández, D.; Romero-Domínguez, J.M.; et al. Antioxidants Prevent Iron Accumulation and Lipid Peroxidation, but Do Not Correct Autophagy Dysfunction or Mitochondrial Bioenergetics in Cellular Models of BPAN. Int. J. Mol. Sci. 2023, 24, 14576. https://doi.org/10.3390/ijms241914576
Suárez-Carrillo A, Álvarez-Córdoba M, Romero-González A, Talaverón-Rey M, Povea-Cabello S, Cilleros-Holgado P, Piñero-Pérez R, Reche-López D, Gómez-Fernández D, Romero-Domínguez JM, et al. Antioxidants Prevent Iron Accumulation and Lipid Peroxidation, but Do Not Correct Autophagy Dysfunction or Mitochondrial Bioenergetics in Cellular Models of BPAN. International Journal of Molecular Sciences. 2023; 24(19):14576. https://doi.org/10.3390/ijms241914576
Chicago/Turabian StyleSuárez-Carrillo, Alejandra, Mónica Álvarez-Córdoba, Ana Romero-González, Marta Talaverón-Rey, Suleva Povea-Cabello, Paula Cilleros-Holgado, Rocío Piñero-Pérez, Diana Reche-López, David Gómez-Fernández, José Manuel Romero-Domínguez, and et al. 2023. "Antioxidants Prevent Iron Accumulation and Lipid Peroxidation, but Do Not Correct Autophagy Dysfunction or Mitochondrial Bioenergetics in Cellular Models of BPAN" International Journal of Molecular Sciences 24, no. 19: 14576. https://doi.org/10.3390/ijms241914576
APA StyleSuárez-Carrillo, A., Álvarez-Córdoba, M., Romero-González, A., Talaverón-Rey, M., Povea-Cabello, S., Cilleros-Holgado, P., Piñero-Pérez, R., Reche-López, D., Gómez-Fernández, D., Romero-Domínguez, J. M., Munuera-Cabeza, M., Díaz, A., González-Granero, S., García-Verdugo, J. M., & Sánchez-Alcázar, J. A. (2023). Antioxidants Prevent Iron Accumulation and Lipid Peroxidation, but Do Not Correct Autophagy Dysfunction or Mitochondrial Bioenergetics in Cellular Models of BPAN. International Journal of Molecular Sciences, 24(19), 14576. https://doi.org/10.3390/ijms241914576