
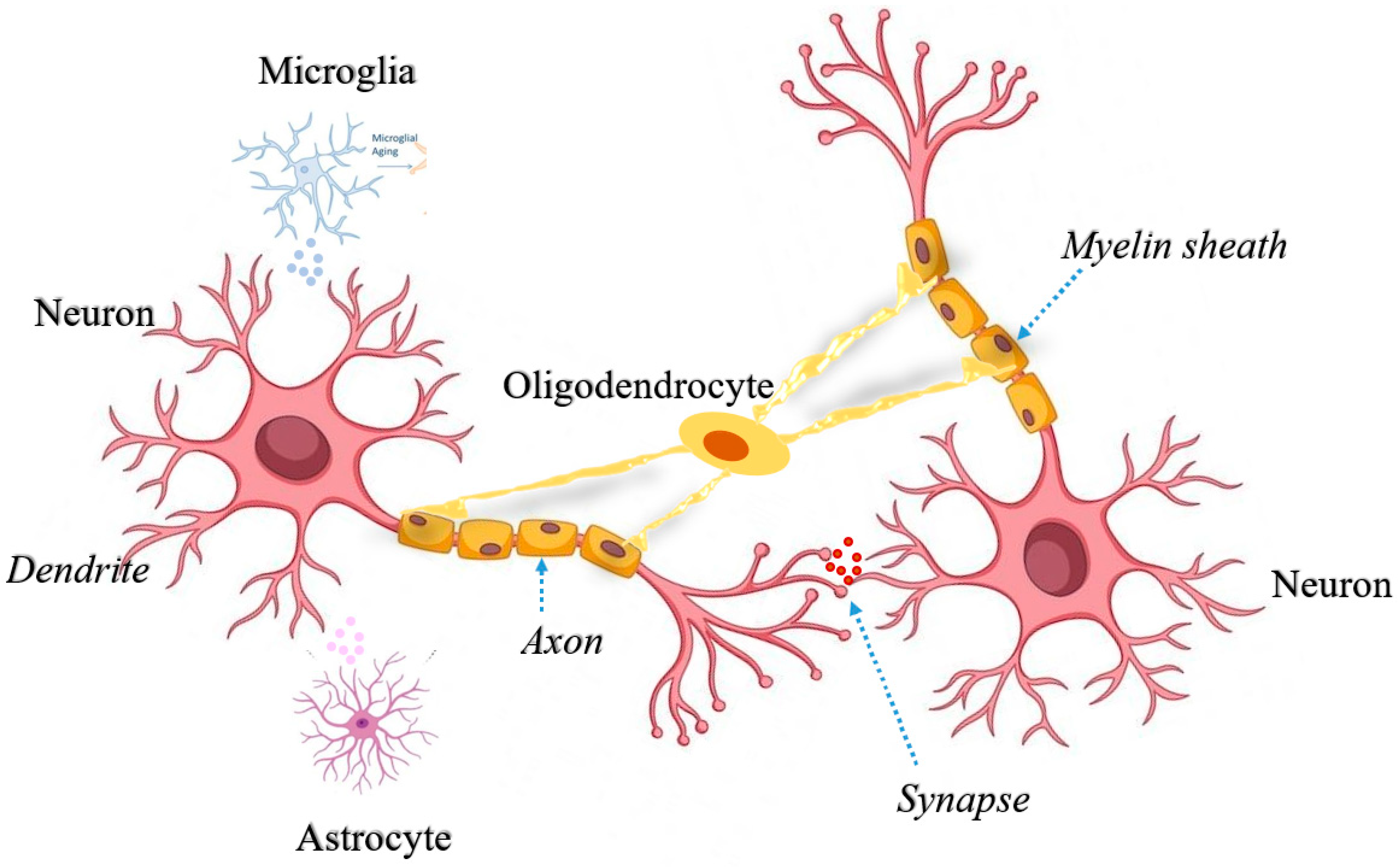
Cell-Type-Specific Mitochondrial Quality Control in the Brain: A Plausible Mechanism of Neurodegeneration


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Mitochondrial Quality Control in Neurons: A Promising Mechanism of Neurodegeneration
2.1. Mitochondrial Dynamics in Neurons
2.2. Mitochondrial Bioenergetics
2.3. Intracellular Translocation of Mitochondria in Neurons
2.4. Mitochondrial Dysfunction and Neurodegeneration
2.4.1. Parkinson’s Disease
2.4.2. Frontotemporal Dementia and Alzheimer’s Disease
2.4.3. Amyotrophic Lateral Sclerosis
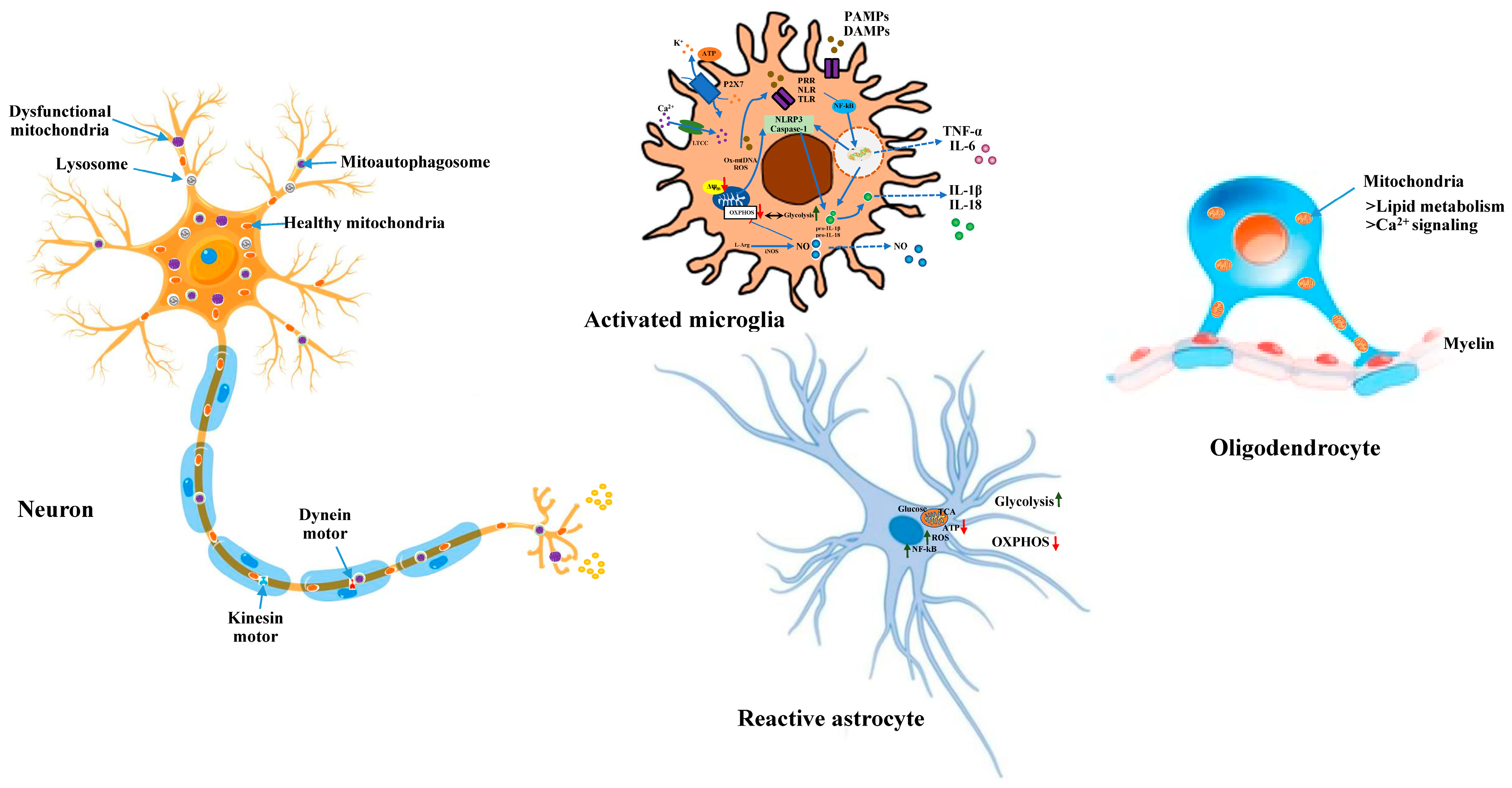
3. Astrocytic Mitochondria and Their Contribution in Neurodegeneration
4. Microglial Mitochondria and Their Significance in Neurodegeneration
5. Significance of Oligodendrocytic Mitochondria in Axonal Dysfunction and Neurodegeneration
6. Conclusions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Misgeld, T.; Schwarz, T.L. Mitostasis in Neurons: Maintaining Mitochondria in an Extended Cellular Architecture. Neuron 2017, 96, 651–666. [Google Scholar] [CrossRef]
- Devine, M.J.; Kittler, J.T. Mitochondria at the neuronal presynapse in health and disease. Nat. Rev. Neurosci. 2018, 19, 63–80. [Google Scholar] [CrossRef]
- Lin, M.Y.; Cheng, X.T.; Tammineni, P.; Xie, Y.; Zhou, B.; Cai, Q.; Sheng, Z.H. Releasing Syntaphilin Removes Stressed Mitochondria from Axons Independent of Mitophagy under Pathophysiological Conditions. Neuron 2017, 94, 595–610. [Google Scholar] [CrossRef]
- Hoffmann, L.; Rust, M.B.; Culmsee, C. Actin(g) on Mitochondria—A Role for cofilin1 in Neuronal Cell Death Pathways. Biol. Chem. 2019, 400, 1089–1097. [Google Scholar] [CrossRef]
- von Bernhardi, R.; Eugenín-von Bernhardi, J.; Flores, B.; Eugenín León, J. Glial Cells and Integrity of the Nervous System. Adv. Exp. Med. Biol. 2016, 949, 1–24. [Google Scholar]
- MacVicar, B.A.; Newman, E.A. Astrocyte Regulation of Blood Flow in the Brain. Cold Spring Harb. Perspect. Biol. 2015, 7, a020388. [Google Scholar] [CrossRef]
- Verkhratsky, A.; Steardo, L.; Peng, L.; Parpura, V. Astroglia, Glutamatergic Transmission and Psychiatric Diseases. Adv. Neurobiol. 2016, 13, 307–326. [Google Scholar]
- Jeon, Y.-M.; Kwon, Y.; Jo, M.; Lee, S.; Kim, S.; Kim, H.-J. The Role of Glial Mitochondria in α-Synuclein Toxicity. Front. Cell Dev. Biol. 2020, 8, 548283. [Google Scholar] [CrossRef]
- Sirabella, R.; Sisalli, M.J.; Costa, G.; Omura, K.; Ianniello, G.; Pinna, A.; Morelli, M.; Di Renzo, G.M.; Annunziato, L.; Scorziello, A. NCX1 and NCX3 as Potential Factors Contributing to Neurodegeneration and Neuroinflammation in the A53T Transgenic Mouse Model of Parkinson’s Disease. Cell Death Dis. 2018, 9, 725. [Google Scholar] [CrossRef]
- Nunnari, J.; Suomalainen, A. Mitochondria: In Sickness and in Health. Cell 2012, 148, 1145–1159. [Google Scholar] [CrossRef]
- Restelli, L.M.; Oettinghaus, B.; Halliday, M.; Agca, C.; Licci, M.; Sironi, L.; Savoia, C.; Hench, J.; Tolnay, M.; Neutzner, A.; et al. Neuronal Mitochondrial Dysfunction Activates the Integrated Stress Response to Induce Fibroblast Growth Factor 21. Cell Rep. 2018, 24, 1407–1414. [Google Scholar] [CrossRef] [PubMed]
- Oettinghaus, B.; Schulz, J.M.; Restelli, L.M.; Licci, M.; Savoia, C.; Schmidt, A.; Schmitt, K.; Grimm, A.; Morè, L.; Hench, J.; et al. Synaptic Dysfunction, Memory Deficits and Hippocampal Atrophy due to Ablation of Mitochondrial Fission in Adult Forebrain Neurons. Cell Death Differ. 2016, 23, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Puri, R.; Cheng, X.-T.; Lin, M.-Y.; Huang, N.; Sheng, Z.-H. Mul1 Restrains Parkin-Mediated Mitophagy in Mature Neurons by Maintaining ER-Mitochondrial Contacts. Nat. Commun. 2019, 10, 3645. [Google Scholar] [CrossRef]
- Han, S.; Nandy, P.; Austria, Q.; Siedlak, S.L.; Torres, S.; Fujioka, H.; Wang, W.; Zhu, X. Mfn2 Ablation in the Adult Mouse Hippocampus and Cortex Causes Neuronal Death. Cells 2020, 9, 116. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zhang, F.; Li, L.; Tang, F.; Siedlak, S.L.; Fujioka, H.; Liu, Y.; Su, B.; Pi, Y.; Wang, X. MFN2 Couples Glutamate Excitotoxicity and Mitochondrial Dysfunction in Motor Neurons*. J. Biol. Chem. 2015, 290, 168–182. [Google Scholar] [CrossRef]
- Pirooznia, S.K.; Yuan, C.; Khan, M.R.; Karuppagounder, S.S.; Wang, L.; Xiong, Y.; Kang, S.U.; Lee, Y.; Dawson, V.L.; Dawson, T.M. PARIS Induced Defects in Mitochondrial Biogenesis Drive Dopamine Neuron Loss under Conditions of Parkin or PINK1 Deficiency. Mol. Neurodegener. 2020, 15, 17. [Google Scholar] [CrossRef]
- Kumar, H.; Lim, H.W.; More, S.V.; Kim, B.W.; Koppula, S.; Kim, I.S.; Choi, D.K. The role of free radicals in the aging brain and Parkinson’s Disease: Convergence and parallelism. Int. J. Mol. Sci. 2012, 13, 10478–10504. [Google Scholar] [CrossRef]
- Dumont, M.; Ho, D.J.; Calingasan, N.Y.; Xu, H.; Gibson, G.; Beal, M.F. Mitochondrial dihydrolipoyl succinyltransferase deficiency accelerates amyloid pathology and memory deficit in a transgenic mouse model of amyloid deposition. Free Radic. Biol. Med. 2009, 47, 1019–1027. [Google Scholar] [CrossRef]
- Shi, Q.; Xu, H.; Yu, H.; Zhang, N.; Ye, Y.; Estevez, A.G.; Deng, H.; Gibson, G.E. Inactivation and reactivation of the mitochondrial α-ketoglutarate dehydrogenase complex. J. Biol. Chem. 2011, 286, 17640–17648. [Google Scholar] [CrossRef]
- Ferri, A.; Cozzolino, M.; Crosio, C.; Nencini, M.; Casciati, A.; Gralla, E.B.; Rotilio, G.; Valentine, J.S.; Carrì, M.T. Familial ALS-superoxide dismutases associate with mitochondria and shift their redox potentials. Proc. Natl. Acad. Sci. USA 2006, 103, 13860–13865. [Google Scholar] [CrossRef]
- Obeso, J.A.; Rodriguez-Oroz, M.C.; Goetz, C.G.; Marin, C.; Kordower, J.H.; Rodriguez, M.; Hirsch, E.C.; Farrer, M.; Schapira, A.H.; Halliday, G. Missing pieces in the Parkinson’s disease puzzle. Nat. Med. 2010, 16, 653–661. [Google Scholar] [CrossRef]
- Quintanilla, R.A.; Tapia-Monsalves, C.; Vergara, E.H.; Pérez, M.J.; Aranguiz, A. Truncated Tau Induces Mitochondrial Transport Failure Through the Impairment of TRAK2 Protein and Bioenergetics Decline in Neuronal Cells. Front. Cell. Neurosci. 2020, 14, 175. [Google Scholar] [CrossRef] [PubMed]
- Joshi, D.C.; Zhang, C.-L.; Mathur, D.; Li, A.; Kaushik, G.; Sheng, Z.-H.; Chiu, S.Y. Tripartite Crosstalk between Cytokine IL-1β, NMDA-R and Misplaced Mitochondrial Anchor in Neuronal Dendrites Is a Novel Pathway for Neurodegeneration in Inflammatory Diseases. J. Neurosci. 2022, 42, 7318–7329. [Google Scholar] [CrossRef] [PubMed]
- Stykel, M.G.; Humphries, K.; Kirby, M.P.; Czaniecki, C.; Wang, T.; Ryan, T.; Bamm, V.; Ryan, S.D. Nitration of Microtubules Blocks Axonal Mitochondrial Transport in a Human Pluripotent Stem Cell Model of Parkinson’s Disease. FASEB J. 2018, 32, 5350–5364. [Google Scholar] [CrossRef] [PubMed]
- Esteves, A.R.; Arduíno, D.M.; Silva, D.F.; Viana, S.D.; Pereira, F.C.; Cardoso, S.M. Mitochondrial Metabolism Regulates Microtubule Acetylome and Autophagy Trough Sirtuin-2: Impact for Parkinson’s Disease. Mol. Neurobiol. 2018, 55, 1440–1462. [Google Scholar] [CrossRef] [PubMed]
- Barodia, S.K.; Creed, R.B.; Goldberg, M.S. Parkin and PINK1 Functions in Oxidative Stress and Neurodegeneration. Brain Res. Bull. 2017, 133, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Wang, X.; Fujioka, H.; Hoppel, C.; Whone, A.L.; Caldwell, M.A.; Cullen, P.J.; Liu, J.; Zhu, X. Parkinson’s Disease-Associated Mutant VPS35 Causes Mitochondrial Dysfunction by Recycling DLP1 Complexes. Nat. Med. 2016, 22, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, C.-H.; Shaltouki, A.; Gonzalez, A.E.; da Cruz, A.B.; Burbulla, L.F.; Lawrence, E.S.; Schüle, B.; Krainc, D.; Palmer, T.D.; Wang, X. Functional Impairment in Miro Degradation and Mitophagy Is a Shared Feature in Familial and Sporadic Parkinson’s Disease. Cell Stem Cell 2016, 19, 709–724. [Google Scholar] [CrossRef] [PubMed]
- Shaltouki, A.; Hsieh, C.-H.; Kim, M.J.; Wang, X. Alpha-Synuclein Delays Mitophagy and Targeting Miro Rescues Neuron Loss in Parkinson’s Models. Acta Neuropathol. 2018, 136, 607–620. [Google Scholar] [CrossRef]
- Almikhlafi, M.A.; Stauch, K.L.; Villeneuve, L.M.; Purnell, P.R.; Lamberty, B.G.; Fox, H.S. Deletion of DJ-1 in Rats Affects Protein Abundance and Mitochondrial Function at the Synapse. Sci. Rep. 2020, 10, 13719. [Google Scholar] [CrossRef]
- Esteras, N.; Rohrer, J.D.; Hardy, J.; Wray, S.; Abramov, A.Y. Mitochondrial Hyperpolarization in iPSC-Derived Neurons from Patients of FTDP-17 with 10 + 16 MAPT Mutation Leads to Oxidative Stress and Neurodegeneration. Redox Biol. 2017, 12, 410–422. [Google Scholar] [CrossRef] [PubMed]
- Boczonadi, V.; Horvath, R. Amyloid-β in Mitochondrial Disease: Mutation in a Human Metallopeptidase Links Amyloidotic Neurodegeneration with Mitochondrial Processing. EMBO Mol. Med. 2016, 8, 173–175. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Kim, Y.; Liu, T.; Hwang, Y.J.; Hyeon, S.J.; Im, H.; Lee, K.; Alvarez, V.E.; McKee, A.C.; Um, S.-J.; et al. SIRT3 Deregulation Is Linked to Mitochondrial Dysfunction in Alzheimer’s Disease. Aging Cell 2018, 17, e12679. [Google Scholar] [CrossRef]
- Cheng, A.; Wang, J.; Ghena, N.; Zhao, Q.; Perone, I.; King, T.M.; Veech, R.L.; Gorospe, M.; Wan, R.; Mattson, M.P. SIRT3 Haploinsufficiency Aggravates Loss of GABAergic Interneurons and Neuronal Network Hyperexcitability in an Alzheimer’s Disease Model. J. Neurosci. 2020, 40, 694–709. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Kim, S.; Choi, J.E.; Han, D.; Koh, S.M.; Kim, H.-S.; Kaang, B.-K. Decreased Neuron Number and Synaptic Plasticity in SIRT3-Knockout Mice with Poor Remote Memory. Neurochem. Res. 2019, 44, 676–682. [Google Scholar] [CrossRef]
- Orr, A.L.; Kim, C.; Jimenez-Morales, D.; Newton, B.W.; Johnson, J.R.; Krogan, N.J.; Swaney, D.L.; Mahley, R.W. Neuronal Apolipoprotein E4 Expression Results in Proteome-Wide Alterations and Compromises Bioenergetic Capacity by Disrupting Mitochondrial Function. J. Alzheimers. Dis. 2019, 68, 991–1011. [Google Scholar] [CrossRef]
- Salvatori, I.; Ferri, A.; Scaricamazza, S.; Giovannelli, I.; Serrano, A.; Rossi, S.; D’Ambrosi, N.; Cozzolino, M.; Di Giulio, A.; Moreno, S.; et al. Differential Toxicity of TAR DNA-binding Protein 43 Isoforms Depends on Their Submitochondrial Localization in Neuronal Cells. J. Neurochem. 2018, 146, 585–597. [Google Scholar] [CrossRef]
- Ravera, S.; Torazza, C.; Bonifacino, T.; Provenzano, F.; Rebosio, C.; Milanese, M.; Usai, C.; Panfoli, I.; Bonanno, G. Altered Glucose Catabolism in the Presynaptic and Perisynaptic Compartments of SOD1G93A mouse Spinal Cord and Motor Cortex Indicates That Mitochondria Are the Site of Bioenergetic Imbalance in ALS. J. Neurochem. 2019, 151, 336–350. [Google Scholar] [CrossRef]
- Mehta, A.R.; Gregory, J.M.; Dando, O.; Carter, R.N.; Burr, K.; Nanda, J.; Story, D.; McDade, K.; Smith, C.; Morton, N.M.; et al. Mitochondrial Bioenergetic Deficits in C9orf72 Amyotrophic Lateral Sclerosis Motor Neurons Cause Dysfunctional Axonal Homeostasis. Acta Neuropathol. 2021, 141, 257–279. [Google Scholar] [CrossRef]
- Wang, T.; Liu, H.; Itoh, K.; Oh, S.; Zhao, L.; Murata, D.; Sesaki, H.; Hartung, T.; Na, C.H.; Wang, J. C9orf72 Regulates Energy Homeostasis by Stabilizing Mitochondrial Complex I Assembly. Cell Metab. 2021, 33, 531–546.e9. [Google Scholar] [CrossRef]
- Weil, R.; Laplantine, E.; Curic, S.; Génin, P. Role of Optineurin in the Mitochondrial Dysfunction: Potential Implications in Neurodegenerative Diseases and Cancer. Front. Immunol. 2018, 9, 1243. [Google Scholar] [CrossRef] [PubMed]
- Fang, E.F.; Hou, Y.; Palikaras, K.; Adriaanse, B.A.; Kerr, J.S.; Yang, B.; Lautrup, S.; Hasan-Olive, M.M.; Caponio, D.; Dan, X.; et al. Mitophagy inhibits amyloid-β and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat. Neurosci. 2019, 22, 401–412. [Google Scholar] [CrossRef] [PubMed]
- Mulica, P.; Grünewald, A.; Pereira, S.L. Astrocyte-Neuron Metabolic Crosstalk in Neurodegeneration: A Mitochondrial Perspective. Front. Endocrinol. 2021, 12, 668517. [Google Scholar] [CrossRef] [PubMed]
- Casaril, A.M.; Katsalifis, A.; Schmidt, R.M.; Bas-Orth, C. Activated Glia Cells Cause Bioenergetic Impairment of Neurons That Can Be Rescued by Knock-down of the Mitochondrial Calcium Uniporter. Biochem. Biophys. Res. Commun. 2022, 608, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, A.; Wu, P.-H.; Hughes, E.G.; Fukaya, M.; Tischfield, M.A.; Langseth, A.J.; Wirtz, D.; Bergles, D.E. Transient Opening of the Mitochondrial Permeability Transition Pore Induces Microdomain Calcium Transients in Astrocyte Processes. Neuron 2017, 93, 587–605.e7. [Google Scholar] [CrossRef]
- Logan, S.; Pharaoh, G.A.; Marlin, M.C.; Masser, D.R.; Matsuzaki, S.; Wronowski, B.; Yeganeh, A.; Parks, E.E.; Premkumar, P.; Farley, J.A.; et al. Insulin-like Growth Factor Receptor Signaling Regulates Working Memory, Mitochondrial Metabolism, and Amyloid-β Uptake in Astrocytes. Mol. Metab. 2018, 9, 141–155. [Google Scholar] [CrossRef]
- Murru, S.; Hess, S.; Barth, E.; Almajan, E.R.; Schatton, D.; Hermans, S.; Brodesser, S.; Langer, T.; Kloppenburg, P.; Rugarli, E.I. Astrocyte-Specific Deletion of the Mitochondrial M-AAA Protease Reveals Glial Contribution to Neurodegeneration. Glia 2019, 67, 1526–1541. [Google Scholar] [CrossRef]
- Aldana, B.I.; Zhang, Y.; Jensen, P.; Chandrasekaran, A.; Christensen, S.K.; Nielsen, T.T.; Nielsen, J.; Hyttel, P.; Larsen, M.R.; Waagepetersen, H.; et al. Glutamate-Glutamine Homeostasis Is Perturbed in Neurons and Astrocytes Derived from Patient iPSC Models of Frontotemporal Dementia. Mol. Brain 2020, 13, 125. [Google Scholar] [CrossRef]
- Chandrasekaran, A.; Dittlau, K.S.; Corsi, G.I.; Haukedal, H.; Doncheva, N.T.; Ramakrishna, S.; Ambardar, S.; Salcedo, C.; Schmidt, S.I.; Zhang, Y.; et al. Astrocytic Reactivity Triggered by Defective Autophagy and Metabolic Failure Causes Neurotoxicity in Frontotemporal Dementia Type 3. Stem Cell Rep. 2021, 16, 2736–2751. [Google Scholar] [CrossRef]
- Barbosa, M.; Gomes, C.; Sequeira, C.; Gonçalves-Ribeiro, J.; Pina, C.C.; Carvalho, L.A.; Moreira, R.; Vaz, S.H.; Vaz, A.R.; Brites, D. Recovery of Depleted miR-146a in ALS Cortical Astrocytes Reverts Cell Aberrancies and Prevents Paracrine Pathogenicity on Microglia and Motor Neurons. Front. Cell Dev. Biol. 2021, 9, 634355. [Google Scholar] [CrossRef]
- Hall, C.E.; Yao, Z.; Choi, M.; Tyzack, G.E.; Serio, A.; Luisier, R.; Harley, J.; Preza, E.; Arber, C.; Crisp, S.J.; et al. Progressive Motor Neuron Pathology and the Role of Astrocytes in a Human Stem Cell Model of VCP-Related ALS. Cell Rep. 2017, 19, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Saba, J.; Couselo, F.L.; Turati, J.; Carniglia, L.; Durand, D.; de Laurentiis, A.; Lasaga, M.; Caruso, C. Astrocytes from Cortex and Striatum Show Differential Responses to Mitochondrial Toxin and BDNF: Implications for Protection of Striatal Neurons Expressing Mutant Huntingtin. J. Neuroinflamm. 2020, 17, 290. [Google Scholar] [CrossRef]
- Lampinen, R.; Belaya, I.; Saveleva, L.; Liddell, J.R.; Rait, D.; Huuskonen, M.T.; Giniatullina, R.; Sorvari, A.; Soppela, L.; Mikhailov, N.; et al. Neuron-Astrocyte Transmitophagy Is Altered in Alzheimer’s Disease. Neurobiol. Dis. 2022, 170, 105753. [Google Scholar] [CrossRef] [PubMed]
- Loría, F.; Díaz-Nido, J. Frataxin Knockdown in Human Astrocytes Triggers Cell Death and the Release of Factors That Cause Neuronal Toxicity. Neurobiol. Dis. 2015, 76, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Ignatenko, O.; Malinen, S.; Rybas, S.; Vihinen, H.; Nikkanen, J.; Kononov, A.; Jokitalo, E.S.; Ince-Dunn, G.; Suomalainen, A. Mitochondrial Dysfunction Compromises Ciliary Homeostasis in Astrocytes. J. Cell Biol. 2023, 222, e202203019. [Google Scholar] [CrossRef]
- Rawat, P.; Teodorof-Diedrich, C.; Spector, S.A. Human Immunodeficiency Virus Type-1 Single-stranded RNA Activates the NLRP3 Inflammasome and Impairs Autophagic Clearance of Damaged Mitochondria in Human Microglia. Glia 2019, 67, 802–824. [Google Scholar] [CrossRef]
- von Bernhardi, R.; von Bernhardi, R.; Bernhardi, L.E.; Eugenín, J. Microglial Cell Dysregulation in Brain Aging and Neurodegeneration. Front. Aging Neurosci. 2015, 7, 124. [Google Scholar] [CrossRef]
- Simpson, D.S.A.; Oliver, P.L. ROS Generation in Microglia: Understanding Oxidative Stress and Inflammation in Neurodegenerative Disease. Antioxidants 2020, 9, 743. [Google Scholar] [CrossRef]
- Bajwa, E.; Pointer, C.B.; Klegeris, A. The Role of Mitochondrial Damage-Associated Molecular Patterns in Chronic Neuroinflammation. Mediat. Inflamm. 2019, 2019, 4050796. [Google Scholar] [CrossRef]
- Sarkar, S.; Malovic, E.; Harishchandra, D.S.; Ghaisas, S.; Panicker, N.; Charli, A.; Palanisamy, B.N.; Rokad, D.; Jin, H.; Anantharam, V.; et al. Mitochondrial Impairment in Microglia Amplifies NLRP3 Inflammasome Proinflammatory Signaling in Cell Culture and Animal Models of Parkinson’s Disease. Npj Park. Dis. 2017, 3, 30. [Google Scholar] [CrossRef]
- Joshi, A.U.; Minhas, P.S.; Liddelow, S.A.; Haileselassie, B.; Andreasson, K.I.; Dorn, G.W.; Mochly-Rosen, D. Fragmented Mitochondria Released from Microglia Trigger A1 Astrocytic Response and Propagate Inflammatory Neurodegeneration. Nat. Neurosci. 2019, 22, 1635–1648. [Google Scholar] [CrossRef]
- Jassim, A.H.; Inman, D.M.; Mitchell, C.H. Crosstalk between Dysfunctional Mitochondria and Inflammation in Glaucomatous Neurodegeneration. Front. Pharmacol. 2021, 12, 699623. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, G.M.E.P.; Lawrence, G.M.E.; Holley, C.L.; Schroder, K. Parkinson’s Disease: Connecting Mitochondria to Inflammasomes. Trends Immunol. 2022, 43, 877–885. [Google Scholar] [CrossRef] [PubMed]
- Chausse, B.; Lewen, A.; Poschet, G.; Kann, O. Selective Inhibition of Mitochondrial Respiratory Complexes Controls the Transition of Microglia into a Neurotoxic Phenotype in Situ. Brain Behav. Immun. 2020, 88, 802–814. [Google Scholar] [CrossRef]
- Simone, R.D.; De Simone, R.; Ajmone-Cat, M.A.; Pandolfi, M.; Bernardo, A.; De Nuccio, C.; Minghetti, L.; Visentin, S. The Mitochondrial Uncoupling Protein-2 Is a Master Regulator of Both M1 and M2 Microglial Responses. J. Neurochem. 2015, 135, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Morrison, B.M.; Li, Y.; Lengacher, S.; Farah, M.H.; Hoffman, P.N.; Liu, Y.; Tsingalia, A.; Jin, L.; Zhang, P.W.; et al. Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature 2012, 487, 443–448. [Google Scholar] [CrossRef]
- Fünfschilling, U.; Supplie, L.M.; Mahad, D.; Boretius, S.; Saab, A.S.; Edgar, J.; Brinkmann, B.G.; Kassmann, C.M.; Tzvetanova, I.D.; Möbius, W.; et al. Glycolytic oligodendrocytes maintain myelin and long-term axonal integrity. Nature 2012, 485, 517–521. [Google Scholar] [CrossRef] [PubMed]
- Patergnani, S.; Fossati, V.; Bonora, M.; Giorgi, C.; Marchi, S.; Missiroli, S.; Rusielewicz, T.; Wieckowski, M.R.; Pinton, P. Mitochondria in Multiple Sclerosis: Molecular Mechanisms of Pathogenesis. Int. Rev. Cell Mol. Biol. 2017, 328, 49–103. [Google Scholar]
- Fetisova, E.K.; Muntyan, M.S.; Lyamzaev, K.G.; Chernyak, B.V. Therapeutic Effect of the Mitochondria-Targeted Antioxidant SkQ1 on the Culture Model of Multiple Sclerosis. Oxidative Med. Cell. Longev. 2019, 2019, 2082561. [Google Scholar] [CrossRef]
- Steudler, J.; Ecott, T.; Ivan, D.C.; Bouillet, E.; Walthert, S.; Berve, K.; Dick, T.P.; Engelhardt, B.; Locatelli, G. Autoimmune Neuroinflammation Triggers Mitochondrial Oxidation in Oligodendrocytes. Glia 2022, 70, 2045–2061. [Google Scholar] [CrossRef]
- Valentino, R.R.; Heckman, M.G.; Johnson, P.W.; Soto-Beasley, A.I.; Walton, R.L.; Koga, S.; Uitti, R.J.; Wszolek, Z.K.; Dickson, D.W.; Ross, O.A. Association of Mitochondrial Genomic Background with Risk of Multiple System Atrophy. Park. Relat. Disord. 2020, 81, 200–204. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Kidd, G.J.; Ohno, N.; Perkins, G.A.; Ellisman, M.H.; Bastian, C.; Brunet, S.; Baltan, S.; Trapp, B.D. Proteolipid Protein–deficient Myelin Promotes Axonal Mitochondrial Dysfunction via Altered Metabolic Coupling. J. Cell Biol. 2016, 215, 531–542. [Google Scholar] [CrossRef] [PubMed]
- Madsen, P.M.; Pinto, M.; Patel, S.; McCarthy, S.; Gao, H.; Taherian, M.; Karmally, S.; Pereira, C.V.; Dvoriantchikova, G.; Ivanov, D.; et al. Mitochondrial DNA Double-Strand Breaks in Oligodendrocytes Cause Demyelination, Axonal Injury, and CNS Inflammation. J. Neurosci. 2017, 37, 10185–10199. [Google Scholar] [CrossRef] [PubMed]
- Paschon, V.; Morena, B.C.; Correia, F.F.; Beltrame, G.R.; Dos Santos, G.B.; Cristante, A.F.; Kihara, A.H. VDAC1 Is Essential for Neurite Maintenance and the Inhibition of Its Oligomerization Protects Spinal Cord from Demyelination and Facilitates Locomotor Function Recovery after Spinal Cord Injury. Sci. Rep. 2019, 9, 14063. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, A.; Quintela-López, T.; Sánchez-Gómez, M.V.; Gaminde-Blasco, A.; Alberdi, E.; Matute, C. Mitochondrial Division Inhibitor 1 Disrupts Oligodendrocyte Ca Homeostasis and Mitochondrial Function. Glia 2020, 68, 1743–1756. [Google Scholar] [CrossRef] [PubMed]
- Ineichen, B.V.; Zhu, K.; Carlström, K.E. Axonal Mitochondria Adjust in Size Depending on G-Ratio of Surrounding Myelin during Homeostasis and Advanced Remyelination. J. Neurosci. Res. 2021, 99, 793–805. [Google Scholar] [CrossRef]
- Rosato-Siri, M.V.; Adami, P.V.M.; Guitart, M.E.; Verstraeten, S.; Morelli, L.; Correale, J.; Pasquini, J.M. Glial Cell Metabolic Profile upon Iron Deficiency: Oligodendroglial and Astroglial Casualties of Bioenergetic Adjustments. Mol. Neurobiol. 2023, 60, 1949–1963. [Google Scholar] [CrossRef]
- Bhaskaran, S.; Kumar, G.; Thadathil, N.; Piekarz, K.M.; Mohammed, S.; Lopez, S.D.; Qaisar, R.; Walton, D.; Brown, J.L.; Murphy, A.; et al. Neuronal Deletion of MnSOD in Mice Leads to Demyelination, Inflammation and Progressive Paralysis That Mimics Phenotypes Associated with Progressive Multiple Sclerosis. Redox Biol. 2023, 59, 102550. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ragupathy, H.; Vukku, M.; Barodia, S.K. Cell-Type-Specific Mitochondrial Quality Control in the Brain: A Plausible Mechanism of Neurodegeneration. Int. J. Mol. Sci. 2023, 24, 14421. https://doi.org/10.3390/ijms241914421
Ragupathy H, Vukku M, Barodia SK. Cell-Type-Specific Mitochondrial Quality Control in the Brain: A Plausible Mechanism of Neurodegeneration. International Journal of Molecular Sciences. 2023; 24(19):14421. https://doi.org/10.3390/ijms241914421
Chicago/Turabian StyleRagupathy, Hariprasath, Manasvi Vukku, and Sandeep Kumar Barodia. 2023. "Cell-Type-Specific Mitochondrial Quality Control in the Brain: A Plausible Mechanism of Neurodegeneration" International Journal of Molecular Sciences 24, no. 19: 14421. https://doi.org/10.3390/ijms241914421
APA StyleRagupathy, H., Vukku, M., & Barodia, S. K. (2023). Cell-Type-Specific Mitochondrial Quality Control in the Brain: A Plausible Mechanism of Neurodegeneration. International Journal of Molecular Sciences, 24(19), 14421. https://doi.org/10.3390/ijms241914421