

scTIGER: A Deep-Learning Method for Inferring Gene Regulatory Networks from Case versus Control scRNA-seq Datasets




Abstract
1. Introduction
2. Results
2.1. scTIGER Pipeline
2.2. Comparing scTIGER Performance with Other Methods
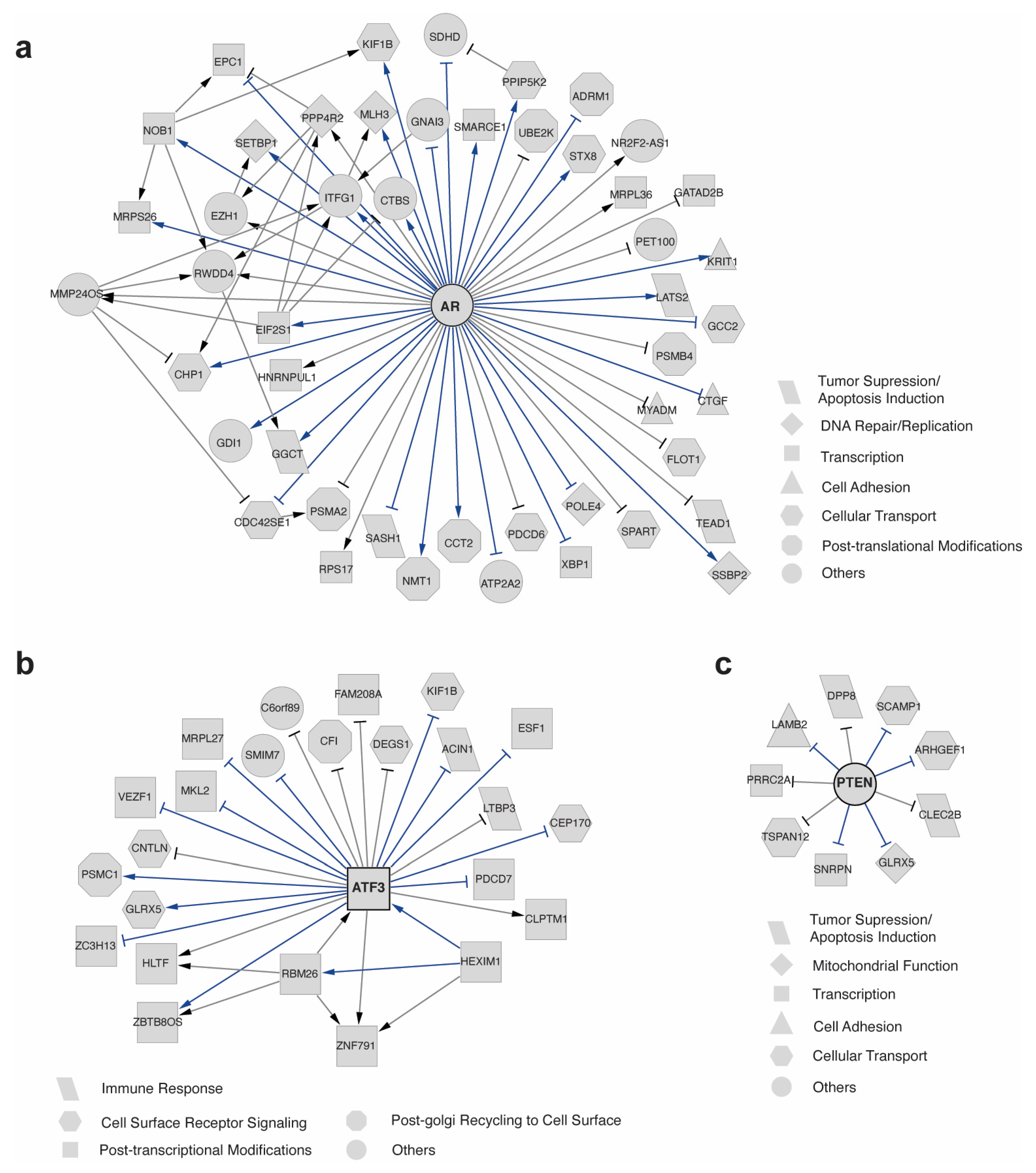
2.3. scTIGER Detected Potential AR Regulatory Network in Prostatic Cancerous vs. Normal Cells
2.4. scTIGER Detected Potential AR Regulatory Network in Different Cell Types in Prostatic Environments
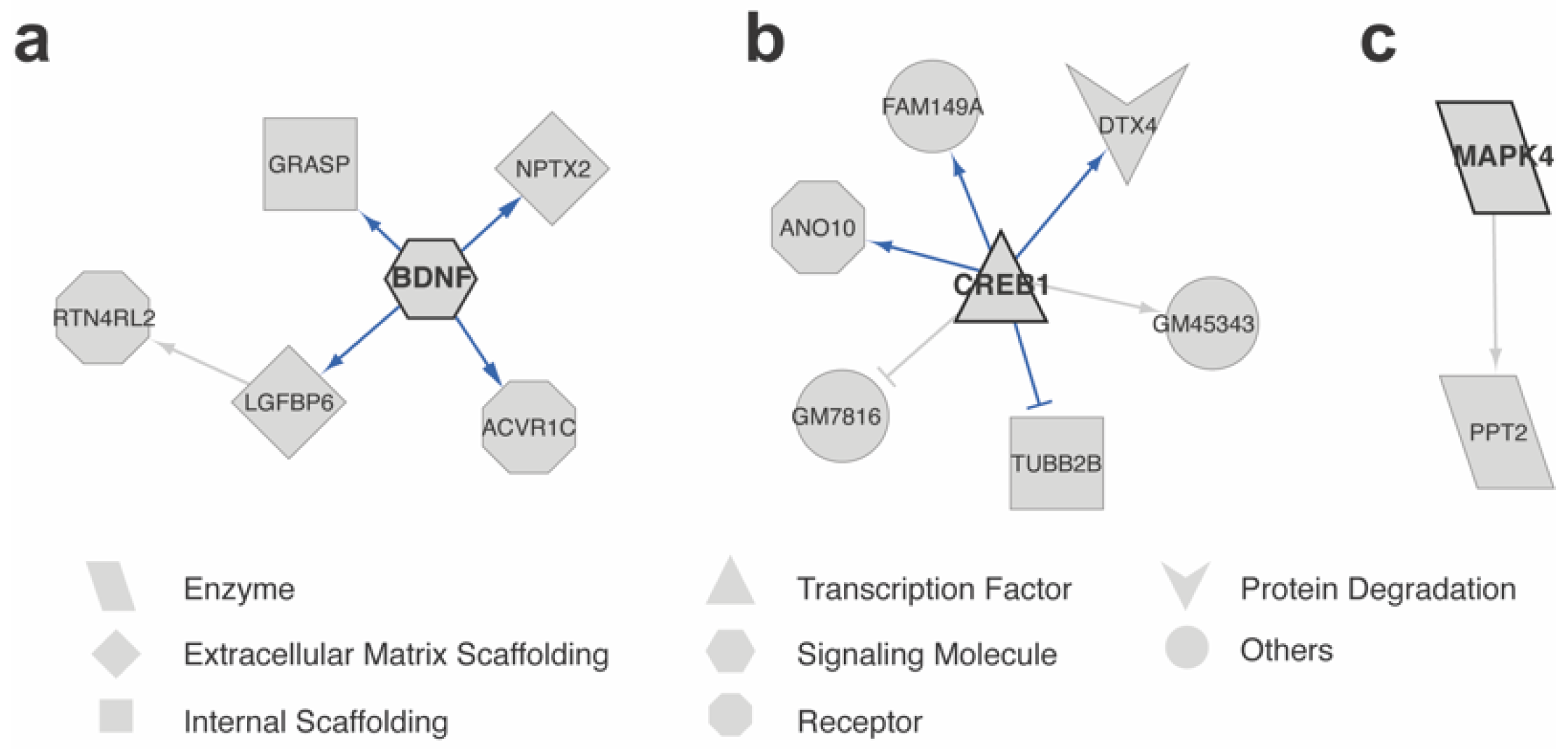
2.5. scTIGER Detected Potential Regulatory Networks in Neurons with and without Memories
2.6. Robustness against Dropouts and Cell Orders
3. Discussion
4. Materials and Methods
4.1. scTIGER Overview and Design
4.2. Data Preparation and Pseudotiming
4.3. Detecting Causal Interactions
4.4. Permutation Testing
4.5. Benchmarking against Existing Methods
4.6. Validations on Real scRNA-seq Datasets
4.7. Testing Robustness against Dropout Noise
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mereu, E.; Lafzi, A.; Moutinho, C.; Ziegenhain, C.; McCarthy, D.J.; Álvarez-Varela, A.; Batlle, E.; Sagar; Grün, D.; Lau, J.K.; et al. Benchmarking single-cell RNA-sequencing protocols for cell atlas projects. Nat. Biotechnol. 2020, 38, 747–755. [Google Scholar] [CrossRef]
- Choi, Y.H.; Kim, J.K. Dissecting Cellular Heterogeneity Using Single-Cell RNA Sequencing. Mol. Cells 2019, 42, 189–199. [Google Scholar] [CrossRef]
- Junaid, M.; Lee, A.; Kim, J.; Park, T.J.; Bin Lim, S. Transcriptional Heterogeneity of Cellular Senescence in Cancer. Mol. Cells 2022, 45, 610–619. [Google Scholar] [CrossRef]
- Haniffa, M.; Taylor, D.; Linnarsson, S.; Aronow, B.J.; Bader, G.D.; Barker, R.A.; Camara, P.G.; Camp, J.G.; Chédotal, A.; Copp, A.; et al. A roadmap for the Human Developmental Cell Atlas. Nature 2021, 597, 196–205. [Google Scholar] [CrossRef]
- Nguyen, H.; Tran, D.; Tran, B.; Pehlivan, B.; Nguyen, T. A comprehensive survey of regulatory network inference methods using single cell RNA sequencing data. Brief. Bioinform. 2021, 22, bbaa190. [Google Scholar] [CrossRef]
- Mercatelli, D.; Scalambra, L.; Triboli, L.; Ray, F.; Giorgi, F.M. Gene regulatory network inference resources: A practical overview. Biochim. Et Biophys. Acta Gene Regul. Mech. 2020, 1863, 194430. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.; Thieffry, D.; Cantini, L. Evaluating the Reproducibility of Single-Cell Gene Regulatory Network Inference Algorithms. Front. Genet. 2021, 12, 617282. [Google Scholar] [CrossRef] [PubMed]
- Erbe, R.; Gore, J.; Gemmill, K.; Gaykalova, D.A.; Fertig, E.J. The use of machine learning to discover regulatory networks controlling biological systems. Mol. Cell 2022, 82, 260–273. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Cui, M.; Shah, A.M.; Tan, W.; Liu, N.; Bassel-Duby, R.; Olson, E.N. Cell-Type-Specific Gene Regulatory Networks Underlying Murine Neonatal Heart Regeneration at Single-Cell Resolution. Cell Rep. 2020, 33, 108472. [Google Scholar] [CrossRef] [PubMed]
- Rogers, J.D.; Aguado, B.A.; Watts, K.M.; Anseth, K.S.; Richardson, W.J. Network modeling predicts personalized gene expression and drug responses in valve myofibroblasts cultured with patient sera. Proc. Natl. Acad. Sci. USA 2022, 119, e2117323119. [Google Scholar] [CrossRef]
- Singh, A.J.; Ramsey, S.A.; Filtz, T.M.; Kioussi, C. Differential gene regulatory networks in development and disease. Cell. Mol. Life Sci. 2017, 75, 1013–1025. [Google Scholar] [CrossRef] [PubMed]
- Jovic, D.; Liang, X.; Zeng, H.; Lin, L.; Xu, F.; Luo, Y. Single-cell RNA sequencing technologies and applications: A brief overview. Clin. Transl. Med. 2022, 12, e694. [Google Scholar] [CrossRef]
- Aibar, S.; González-Blas, C.B.; Moerman, T.; Huynh-Thu, V.A.; Imrichova, H.; Hulselmans, G.; Rambow, F.; Marine, J.-C.; Geurts, P.; Aerts, J.; et al. SCENIC: Single-cell regulatory network inference and clustering. Nat. Methods 2017, 14, 1083–1086. [Google Scholar] [CrossRef]
- Matsumoto, H.; Kiryu, H.; Furusawa, C.; Ko, M.S.H.; Ko, S.B.H.; Gouda, N.; Hayashi, T.; Nikaido, I. SCODE: An efficient regulatory network inference algorithm from single-cell RNA-Seq during differentiation. Bioinformatics 2017, 33, 2314–2321. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, H.; Kiryu, H. SCOUP: A probabilistic model based on the Ornstein-Uhlenbeck process to analyze single-cell expression data during differentiation. BMC Bioinform. 2016, 17, 232. [Google Scholar] [CrossRef]
- Chan, T.E.; Stumpf, M.P.; Babtie, A.C. Gene Regulatory Network Inference from Single-Cell Data Using Multivariate Information Measures. Cell Syst. 2017, 5, 251–267.e3. [Google Scholar] [CrossRef] [PubMed]
- Huynh-Thu, V.A.; Irrthum, A.; Wehenkel, L.; Geurts, P. Inferring Regulatory Networks from Expression Data Using Tree-Based Methods. PLoS ONE 2010, 5, e12776. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Wu, A.P.; Peng, J.; Berger, B.; Cho, H. Bayesian information sharing enhances detection of regulatory associations in rare cell types. Bioinformatics 2021, 37, i349–i357. [Google Scholar] [CrossRef]
- Hu, X.; Hu, Y.; Wu, F.; Leung, R.W.T.; Qin, J. Integration of single-cell multi-omics for gene regulatory network inference. Comput. Struct. Biotechnol. J. 2020, 18, 1925–1938. [Google Scholar] [CrossRef]
- Su, K.; Katebi, A.; Kohar, V.; Clauss, B.; Gordin, D.; Qin, Z.S.; Karuturi, R.K.M.; Li, S.; Lu, M. NetAct: A computational platform to construct core transcription factor regulatory networks using gene activity. Genome Biol. 2022, 23, 270. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Cheong, C.; Lan, L.; Zhou, X.; Liu, J.; Lyu, A.; Cheung, W.K.; Zhang, L. DeepDRIM: A deep neural network to reconstruct cell-type-specific gene regulatory network using single-cell RNA-seq data. Brief. Bioinform. 2021, 22, bbab325. [Google Scholar] [CrossRef] [PubMed]
- Zarayeneh, N.; Ko, E.; Oh, J.H.; Suh, S.; Liu, C.; Gao, J.; Kim, D.; Kang, M. Integration of multi-omics data for integrative gene regulatory network inference. Int. J. Data Min. Bioinform. 2017, 18, 223–239. [Google Scholar] [CrossRef]
- Wolf, F.A.; Hamey, F.K.; Plass, M.; Solana, J.; Dahlin, J.S.; Göttgens, B.; Rajewsky, N.; Simon, L.; Theis, F.J. PAGA: Graph abstraction reconciles clustering with trajectory inference through a topology preserving map of single cells. Genome Biol. 2019, 20, 59. [Google Scholar] [CrossRef]
- Nauta, M.; Bucur, D.; Seifert, C. Causal Discovery with Attention-Based Convolutional Neural Networks. Mach. Learn. Knowl. Extr. 2019, 1, 312–340. [Google Scholar] [CrossRef]
- Gao, N.P.; Ud-Dean, S.M.M.; Gandrillon, O.; Gunawan, R. SINCERITIES: Inferring gene regulatory networks from time-stamped single cell transcriptional expression profiles. Bioinformatics 2018, 34, 258–266. [Google Scholar] [CrossRef]
- Moerman, T.; Santos, S.A.; González-Blas, C.B.; Simm, J.; Moreau, Y.; Aerts, J.; Aerts, S. GRNBoost2 and Arboreto: Efficient and scalable inference of gene regulatory networks. Bioinformatics 2019, 35, 2159–2161. [Google Scholar] [CrossRef] [PubMed]
- Gan, Y.; Hu, X.; Zou, G.; Yan, C.; Xu, G. Inferring Gene Regulatory Networks From Single-Cell Transcriptomic Data Using Bidirectional RNN. Front. Oncol. 2022, 12, 899825. [Google Scholar] [CrossRef]
- Pratapa, A.; Jalihal, A.P.; Law, J.N.; Bharadwaj, A.; Murali, T.M. Benchmarking algorithms for gene regulatory network inference from single-cell transcriptomic data. Nat. Methods 2020, 17, 147–154. [Google Scholar] [CrossRef]
- Heidegger, I.; Fotakis, G.; Offermann, A.; Goveia, J.; Daum, S.; Salcher, S.; Noureen, A.; Timmer-Bosscha, H.; Schäfer, G.; Walenkamp, A.; et al. Comprehensive characterization of the prostate tumor microenvironment identifies CXCR4/CXCL12 crosstalk as a novel antiangiogenic therapeutic target in prostate cancer. Mol. Cancer 2022, 21, 132. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Su, Y.; Zhang, Y.; Han, B.; Liu, H.; Wang, X. Endothelial Cells Promote Docetaxel Resistance of Prostate Cancer Cells by Inducing ERG Expression and Activating Akt/mTOR Signaling Pathway. Front. Oncol. 2020, 10, 584505. [Google Scholar] [CrossRef]
- Zhao, R.; Bei, X.; Yang, B.; Wang, X.; Jiang, C.; Shi, F.; Wang, X.; Zhu, Y.; Jing, Y.; Han, B.; et al. Endothelial cells promote metastasis of prostate cancer by enhancing autophagy. J. Exp. Clin. Cancer Res. 2018, 37, 221. [Google Scholar] [CrossRef] [PubMed]
- La Manna, F.; Karkampouna, S.; Zoni, E.; De Menna, M.; Hensel, J.; Thalmann, G.N.; Kruithof-de Julio, M. Metastases in Prostate Cancer. Cold Spring Harb. Perspect. Med. 2019, 9, a033688. [Google Scholar] [CrossRef] [PubMed]
- Clarke, N.W.; Hart, C.A.; Brown, M.D. Molecular mechanisms of metastasis in prostate cancer. Asian J. Androl. 2008, 11, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Guo, P.; He, T.; Powell, C.A. Role of endothelial cells in tumor microenvironment. Clin. Transl. Med. 2021, 11, e450. [Google Scholar] [CrossRef]
- Harryman, W.L.; Marr, K.D.; Hernandez-Cortes, D.; Nagle, R.B.; Garcia, J.G.N.; Cress, A.E. Cohesive cancer invasion of the biophysical barrier of smooth muscle. Cancer Metastasis Rev. 2021, 40, 205–219. [Google Scholar] [CrossRef]
- Pederzoli, F.; Raffo, M.; Pakula, H.; Ravera, F.; Nuzzo, P.V.; Loda, M. Stromal cells in prostate cancer pathobiology: Friends or foes? Br. J. Cancer 2023, 128, 930–939. [Google Scholar] [CrossRef]
- O’Leary, N.A.; Wright, M.W.; Brister, J.R.; Ciufo, S.; Haddad, D.; McVeigh, R.; Rajput, B.; Robbertse, B.; Smith-White, B.; Ako-Adjei, D.; et al. Reference sequence (RefSeq) database at NCBI: Current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016, 44, D733–D745. [Google Scholar] [CrossRef]
- Pisano, C.; Tucci, M.; Di Stefano, R.F.; Turco, F.; Scagliotti, G.V.; Di Maio, M.; Buttigliero, C. Interactions between androgen receptor signaling and other molecular pathways in prostate cancer progression: Current and future clinical implications. Crit. Rev. Oncol. 2021, 157, 103185. [Google Scholar] [CrossRef]
- Chaturvedi, A.P.; Dehm, S.M. Androgen Receptor Dependence. In Prostate Cancer: Cellular and Genetic Mechanisms of Disease Development and Progression; Dehm, S.M., Tindall, D.J., Eds.; Springer International Publishing: Cham, Switzerland, 2019; pp. 333–350. [Google Scholar]
- Datta, D.; Aftabuddin; Gupta, D.K.; Raha, S.; Sen, P. Human Prostate Cancer Hallmarks Map. Sci. Rep. 2016, 6, 30691. [Google Scholar] [CrossRef]
- Ardura, J.A.; Álvarez-Carrión, L.; Gutiérrez-Rojas, I.; Alonso, V. Role of Calcium Signaling in Prostate Cancer Progression: Effects on Cancer Hallmarks and Bone Metastatic Mechanisms. Cancers 2020, 12, 1071. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Xu, M.; Li, Z.; Shi, M.; Zhou, X.; Jiang, X.; Bryant, J.; Balk, S.; Ma, J.; Isaacs, W.; et al. Calcium and CaSR/IP3R in prostate cancer development. Cell Biosci. 2018, 8, 16. [Google Scholar] [CrossRef]
- Henrich, S.E.; McMahon, K.M.; Plebanek, M.P.; Calvert, A.E.; Feliciano, T.J.; Parrish, S.; Tavora, F.; Mega, A.; De Souza, A.; Carneiro, B.A.; et al. Prostate cancer extracellular vesicles mediate intercellular communication with bone marrow cells and promote metastasis in a cholesterol-dependent manner. J. Extracell. Vesicles 2020, 10, e12042. [Google Scholar] [CrossRef] [PubMed]
- Lelong, E.I.J.; Khelifi, G.; Adjibade, P.; Joncas, F.-H.; St-Sauveur, V.G.; Paquette, V.; Gris, T.; Zoubeidi, A.; Audet-Walsh, E.; Lambert, J.-P.; et al. Prostate cancer resistance leads to a global deregulation of translation factors and unconventional translation. NAR Cancer 2022, 4, zcac034. [Google Scholar] [CrossRef] [PubMed]
- Hernández, G.; Ramírez, J.L.; Pedroza-Torres, A.; Herrera, L.A.; Jiménez-Ríos, M.A. The Secret Life of Translation Initiation in Prostate Cancer. Front. Genet. 2019, 10, 14. [Google Scholar] [CrossRef]
- Selmi, T.; Hussain, S.; Dietmann, S.; Heiß, M.; Borland, K.; Flad, S.; Carter, J.M.; Dennison, R.; Huang, Y.L.; Kellner, S.; et al. Sequence- and structure-specific cytosine-5 mRNA methylation by NSUN6. Nucleic Acids Res. 2021, 49, 1006–1022. [Google Scholar] [CrossRef]
- Somasekharan, S.P.; Saxena, N.; Zhang, F.; Beraldi, E.; Ni Huang, J.; Gentle, C.; Fazli, L.; Thi, M.; Sorensen, P.H.; Gleave, M. Regulation of AR mRNA translation in response to acute AR pathway inhibition. Nucleic Acids Res. 2022, 50, 1069–1091. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, D.; Qu, F.; Hong, Y.; Cao, J.; Pan, X.; Li, L.; Huang, Y.; Huang, H.; Yin, L.; et al. Knockdown of NOB1 expression inhibits the malignant transformation of human prostate cancer cells. Mol. Cell. Biochem. 2014, 396, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Schulz, W.A.; Ingenwerth, M.; Djuidje, C.E.; Hader, C.; Rahnenführer, J.; Engers, R. Changes in cortical cytoskeletal and extracellular matrix gene expression in prostate cancer are related to oncogenic ERG deregulation. BMC Cancer 2010, 10, 505–509. [Google Scholar] [CrossRef]
- Haffner, M.C.; Esopi, D.M.; Chaux, A.; Gürel, M.; Ghosh, S.; Vaghasia, A.M.; Tsai, H.; Kim, K.; Castagna, N.; Lam, H.; et al. AIM1 is an actin-binding protein that suppresses cell migration and micrometastatic dissemination. Nat. Commun. 2017, 8, 142. [Google Scholar] [CrossRef]
- Donald, C.D.; Cooper, C.R.; Harris-Hooker, S.; Emmett, N.; Scanlon, M.; Cooke, D.B. Cytoskeletal organization and cell motility correlates with metastatic potential and state of differentiation in prostate cancer. Cell. Mol. Biol. 2001, 47, 1033–1038. [Google Scholar]
- Mei, Y.; Li, K.; Zhang, Z.; Li, M.; Yang, H.; Wang, H.; Huang, X.; Li, X.; Shi, S.; Yang, H. miR-33b-3p Acts as a Tumor Suppressor by Targeting DOCK4 in Prostate Cancer. Front. Oncol. 2021, 11, 740452. [Google Scholar] [CrossRef]
- Lachmann, A.; Xu, H.; Krishnan, J.; Berger, S.I.; Mazloom, A.R.; Ma’ayan, A. ChEA: Transcription factor regulation inferred from integrating genome-wide ChIP-X experiments. Bioinformatics 2010, 26, 2438–2444. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, B.D.; Hodakoski, C.; Barrows, D.; Mense, S.M.; Parsons, R.E. PTEN function: The long and the short of it. Trends Biochem. Sci. 2014, 39, 183–190. [Google Scholar] [CrossRef]
- Shen, S.M.; Ji, Y.; Zhang, C.; Dong, S.S.; Yang, S.; Xiong, Z.; Ge, M.K.; Yu, Y.; Xia, L.; Guo, M.; et al. Nuclear PTEN safeguards pre-mRNA splicing to link Golgi apparatus for its tumor suppressive role. Nat. Commun. 2018, 9, 2392. [Google Scholar] [CrossRef]
- Li, D.; Stovall, D.B.; Wang, W.; Sui, G. Advances of Zinc Signaling Studies in Prostate Cancer. Int. J. Mol. Sci. 2020, 21, 667. [Google Scholar] [CrossRef] [PubMed]
- Prasad, R.R.; Raina, K.; Mishra, N.; Tomar, M.S.; Kumar, R.; Palmer, A.E.; Maroni, P.; Agarwal, R. Stage-specific differential expression of zinc transporter SLC30A and SLC39A family proteins during prostate tumorigenesis. Mol. Carcinog. 2022, 61, 454–471. [Google Scholar] [CrossRef] [PubMed]
- Costello, L.C.; Feng, P.; Milon, B.; Tan, M.; Franklin, R.B. Role of zinc in the pathogenesis and treatment of prostate cancer: Critical issues to resolve. Prostate Cancer Prostatic Dis. 2004, 7, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Yin, L.; Li, W.; Xu, A.; Shi, H.; Wang, K.; Yang, H.; Wang, R.; Peng, B. SH3BGRL2 inhibits growth and metastasis in clear cell renal cell carcinoma via activating hippo/TEAD1-Twist1 pathway. Ebiomedicine 2020, 51, 102596. [Google Scholar] [CrossRef]
- Abdulfatah, E.; Reichert, Z.R.; Davenport, M.S.; Chinnaiyan, A.M.; Dadhania, V.; Wang, X.; Mannan, R.; Kunju, L.P.; Hollenbeck, B.K.; Montgomery, J.S.; et al. De novo neuroendocrine transdifferentiation in primary prostate cancer–a phenotype associated with advanced clinico-pathologic features and aggressive outcome. Med. Oncol. 2021, 38, 26. [Google Scholar] [CrossRef]
- Merkens, L.; Sailer, V.; Lessel, D.; Janzen, E.; Greimeier, S.; Kirfel, J.; Perner, S.; Pantel, K.; Werner, S.; von Amsberg, G. Aggressive variants of prostate cancer: Underlying mechanisms of neuroendocrine transdifferentiation. J. Exp. Clin. Cancer Res. 2022, 41, 46. [Google Scholar] [CrossRef]
- Liu, X.; Li, W.; Puzanov, I.; Goodrich, D.W.; Chatta, G.; Tang, D.G. Prostate cancer as a dedifferentiated organ: Androgen receptor, cancer stem cells, and cancer stemness. Essays Biochem. 2022, 66, 291–303. [Google Scholar] [CrossRef]
- Zhang, S.; Xie, L.; Cui, Y.; Carone, B.R.; Chen, Y. Detecting Fear-Memory-Related Genes from Neuronal scRNA-seq Data by Diverse Distributions and Bhattacharyya Distance. Biomolecules 2022, 12, 1130. [Google Scholar] [CrossRef]
- Miranda, M.; Morici, J.F.; Zanoni, M.B.; Bekinschtein, P. Brain-Derived Neurotrophic Factor: A Key Molecule for Memory in the Healthy and the Pathological Brain. Front. Cell. Neurosci. 2019, 13, 363. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, M.C.; Radiske, A.; Cammarota, M. On the Involvement of BDNF Signaling in Memory Reconsolidation. Front. Cell. Neurosci. 2019, 13, 383. [Google Scholar] [CrossRef] [PubMed]
- Lakhina, V.; Arey, R.N.; Kaletsky, R.; Kauffman, A.; Stein, G.; Keyes, W.; Xu, D.; Murphy, C.T. Genome-wide Functional Analysis of CREB/Long-Term Memory-Dependent Transcription Reveals Distinct Basal and Memory Gene Expression Programs. Neuron 2015, 85, 330–345. [Google Scholar] [CrossRef] [PubMed]
- Bourtchuladze, R.; Frenguelli, B.; Blendy, J.; Cioffi, D.; Schutz, G.; Silva, A.J. Deficient long-term memory in mice with a targeted mutation of the cAMP-responsive element-binding protein. Cell 1994, 79, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Matos, M.R.; Visser, E.; Kramvis, I.; van der Loo, R.J.; Gebuis, T.; Zalm, R.; Rao-Ruiz, P.; Mansvelder, H.D.; Smit, A.B.; Oever, M.C.v.D. Memory strength gates the involvement of a CREB-dependent cortical fear engram in remote memory. Nat. Commun. 2019, 10, 2315. [Google Scholar] [CrossRef]
- Honda, T.; Obara, Y.; Yamauchi, A.; Couvillon, A.D.; Mason, J.J.; Ishii, K.; Nakahata, N. Phosphorylation of ERK5 on Thr732 Is Associated with ERK5 Nuclear Localization and ERK5-Dependent Transcription. PLoS ONE 2015, 10, e0117914. [Google Scholar] [CrossRef]
- Rao-Ruiz, P.; Couey, J.J.; Marcelo, I.M.; Bouwkamp, C.G.; Slump, D.E.; Matos, M.R.; van der Loo, R.J.; Martins, G.J.; Hout, M.v.D.; van Ijcken, W.F.; et al. Engram-specific transcriptome profiling of contextual memory consolidation. Nat. Commun. 2019, 10, 2232. [Google Scholar] [CrossRef]
- Ribeiro, M.J.; Schofield, M.G.; Kemenes, I.; O’Shea, M.; Kemenes, G.; Benjamin, P.R. Activation of MAPK is necessary for long-term memory consolidation following food-reward conditioning. Learn. Mem. 2005, 12, 538–545. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ventre, E.; Herbach, U.; Espinasse, T.; Benoit, G.; Gandrillon, O. One model fits all: Combining inference and simulation of gene regulatory networks. PLoS Comput. Biol. 2023, 19, e1010962. [Google Scholar] [CrossRef] [PubMed]
- Wolf, F.A.; Angerer, P.; Theis, F.J. SCANPY: Large-scale single-cell gene expression data analysis. Genome Biol. 2018, 19, 15. [Google Scholar] [CrossRef] [PubMed]
- Ianevski, A.; Giri, A.K.; Aittokallio, T. Fully-automated and ultra-fast cell-type identification using specific marker combinations from single-cell transcriptomic data. Nat. Commun. 2022, 13, 1246. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.B.; Jiang, X.; Quake, S.R.; Südhof, T.C. Persistent transcriptional programmes are associated with remote memory. Nature 2020, 587, 437–442. [Google Scholar] [CrossRef]
- Gonit, M. The Androgen Receptor as a Transcriptional Co-Activator: Implications in the Growth and Progression of Prostate Cancer; University of Toledo: Toledo, OH, USA, 2011; p. 233. [Google Scholar]
- Chikarmane, R. Coordinated Activation of the Androgen Receptor Transcriptional Program in Prostate Cancer; Johns Hopkins University: Baltimore, MD, USA, 2022; p. 96. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | Category | Directed | Signed | Additional Required Input |
|---|---|---|---|---|
| scTIGER | Corr | ✓ | ✓ | - |
| PIDC | MI | ✘ | ✘ | - |
| SINCERITIES | Reg | ✓ | ✓ | - |
| GRNBoost2 | RF | ✓ | ✘ | - |
| PPCOR | Corr | ✘ | ✓ | - |
| SCODE | ODE + Reg | ✓ | ✓ | ODE Parameters |
| Method | Criteria | LI | CY | HSC | mCAD |
|---|---|---|---|---|---|
| scTIGER | Recall | 1 | 1 | 0.667 | 0.667 |
| Precision | 1 | 0.75 | 0.56 | 0.85 | |
| Specificity | 1 | 0.778 | 0.676 | 0 | |
| F1 Score | 1 | 0.857 | 0.609 | 0.75 | |
| PIDC | Recall | 1 | 1 | 0.619 | 0.667 |
| Precision | 0.583 | 0.75 | 0.52 | 0.85 | |
| Specificity | 0.643 | 0.778 | 0.647 | 0 | |
| F1 Score | 0.636 | 0.857 | 0.565 | 0.75 | |
| SINCERITIES | Recall | 0.286 | 0.667 | 0.667 | 1 |
| Precision | 0.25 | 0.4 | 0.452 | 0.9 | |
| Specificity | 0.571 | 0.333 | 0.5 | 0 | |
| F1 Score | 0.267 | 0.5 | 0.538 | 0.947 | |
| GRNBoost2 | Recall | 0.857 | 1 | 0.667 | 1 |
| Precision | 0.667 | 0.75 | 0.56 | 0.9 | |
| Specificity | 0.786 | 0.778 | 0.676 | 0 | |
| F1 Score | 0.75 | 0.857 | 0.609 | 0.947 | |
| PPCOR | Recall | 0.857 | 0.667 | 0.190 | 0.444 |
| Precision | 1 | 0.667 | 0.571 | 0.8 | |
| Specificity | 1 | 0.778 | 0.912 | 0 | |
| F1 Score | 0.923 | 0.667 | 0.286 | 0.571 | |
| SCODE | Recall | 0.571 | 0.333 | 0.667 | 1 |
| Precision | 0.5 | 0.25 | 0.452 | 0.9 | |
| Specificity | 0.714 | 0.333 | 0.5 | 0 | |
| F1 Score | 0.533 | 0.286 | 0.538 | 0.947 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dautle, M.; Zhang, S.; Chen, Y. scTIGER: A Deep-Learning Method for Inferring Gene Regulatory Networks from Case versus Control scRNA-seq Datasets. Int. J. Mol. Sci. 2023, 24, 13339. https://doi.org/10.3390/ijms241713339
Dautle M, Zhang S, Chen Y. scTIGER: A Deep-Learning Method for Inferring Gene Regulatory Networks from Case versus Control scRNA-seq Datasets. International Journal of Molecular Sciences. 2023; 24(17):13339. https://doi.org/10.3390/ijms241713339
Chicago/Turabian StyleDautle, Madison, Shaoqiang Zhang, and Yong Chen. 2023. "scTIGER: A Deep-Learning Method for Inferring Gene Regulatory Networks from Case versus Control scRNA-seq Datasets" International Journal of Molecular Sciences 24, no. 17: 13339. https://doi.org/10.3390/ijms241713339
APA StyleDautle, M., Zhang, S., & Chen, Y. (2023). scTIGER: A Deep-Learning Method for Inferring Gene Regulatory Networks from Case versus Control scRNA-seq Datasets. International Journal of Molecular Sciences, 24(17), 13339. https://doi.org/10.3390/ijms241713339