
Dorsal–Ventral Gradient of Activin Regulates Strength of GABAergic Inhibition along Longitudinal Axis of Mouse Hippocampus in an Activity-Dependent Fashion


Abstract
1. Introduction
2. Results
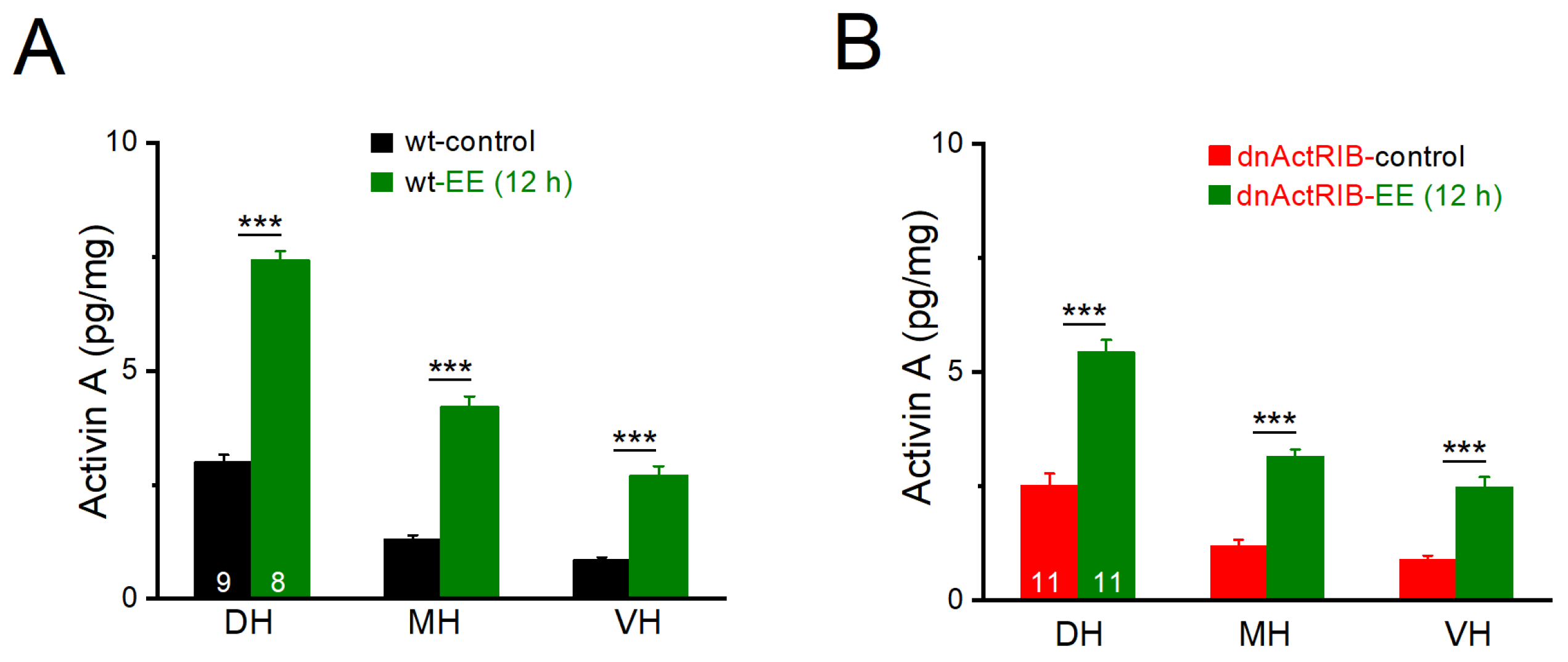
2.1. Activin A Levels Exhibit Dorsal–Ventral Gradient, Which Is Strongly Potentiated by Enriched Environment
2.2. Loss of Activin Signaling Enhances Tonic GABA Inhibition in DH > VH
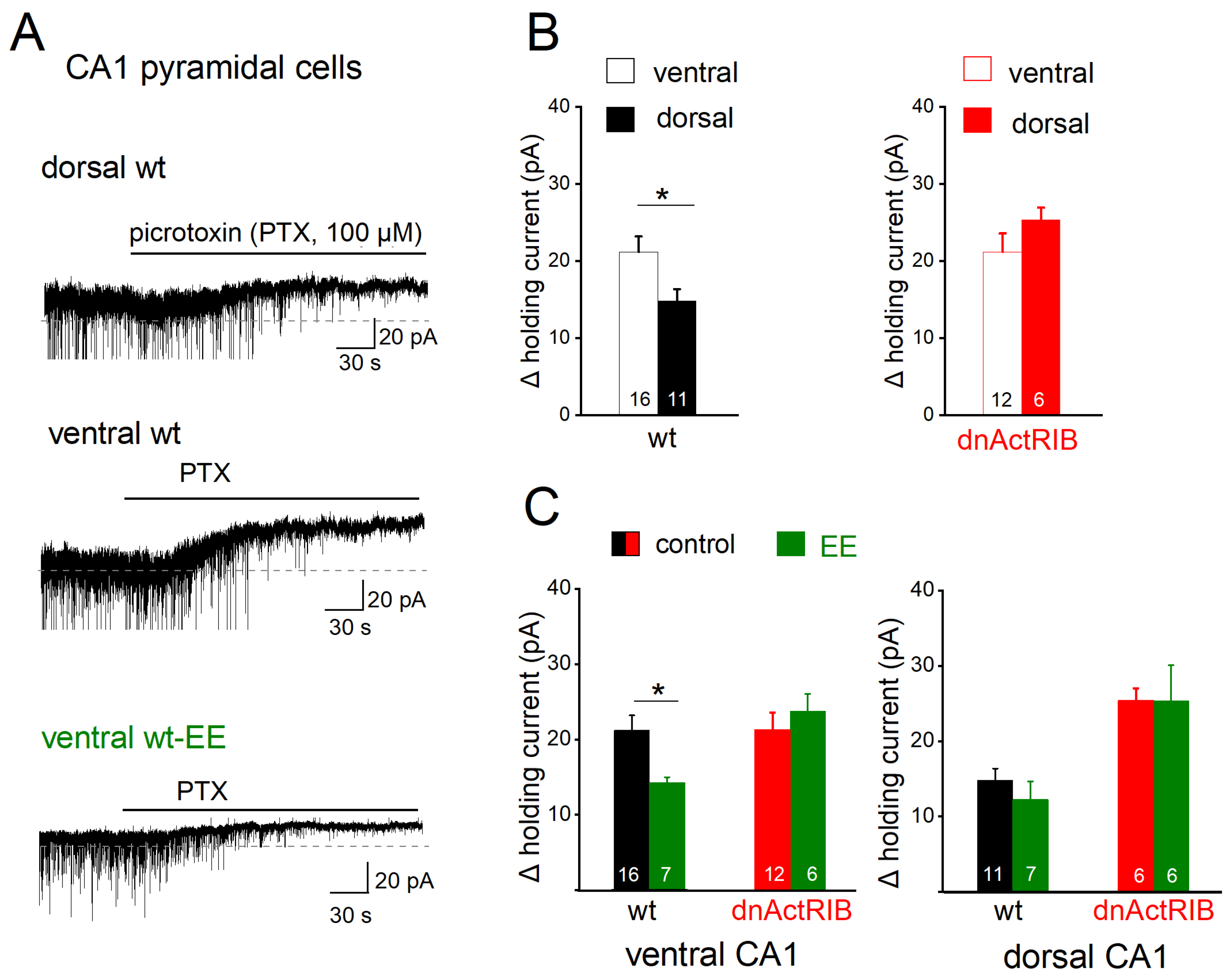
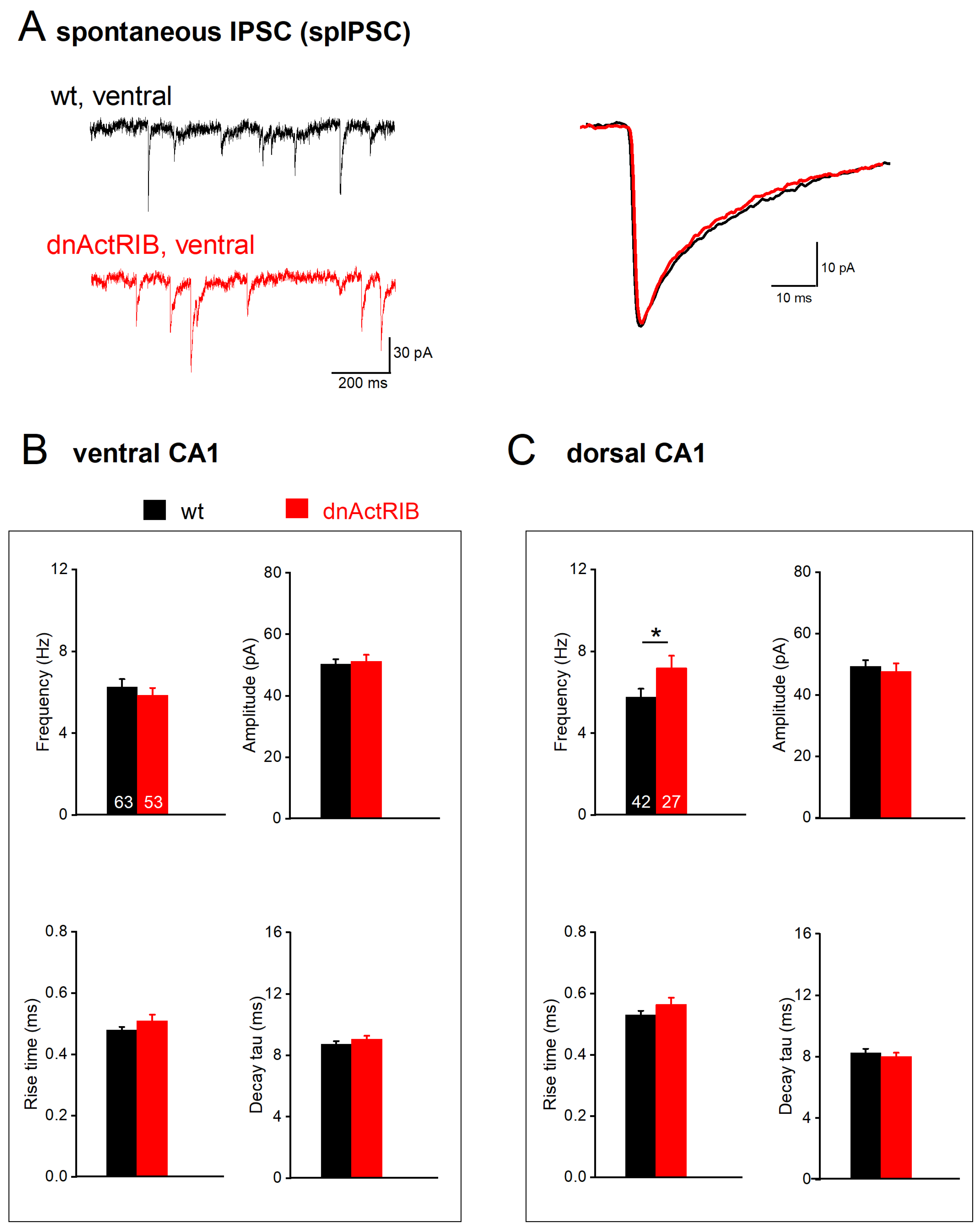
2.3. Disruption of Activin Signaling Facilitates Phasic Inhibition Mainly in Dorsal CA1 Pyramidal Cells
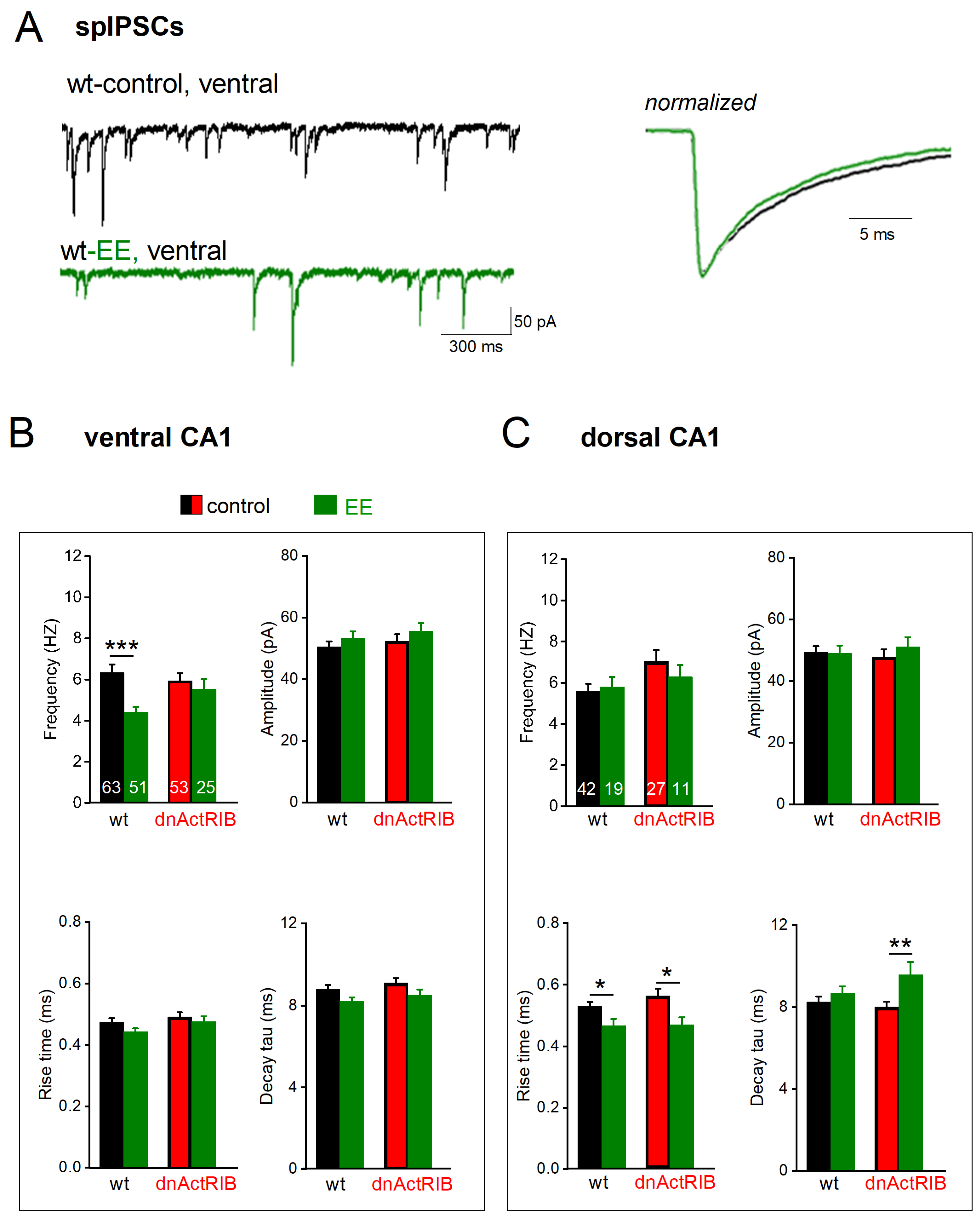
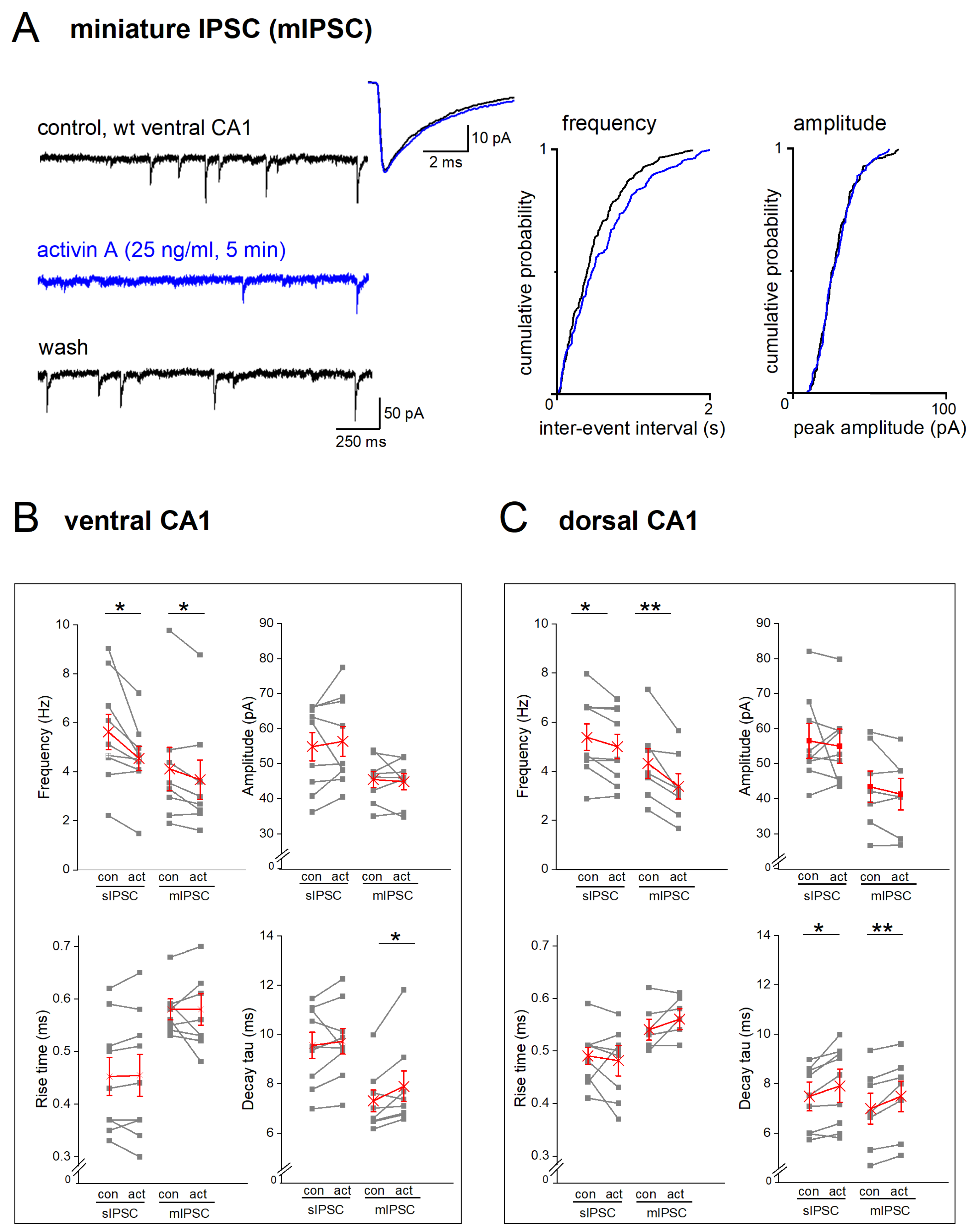
2.4. EE-Induced Activin Signaling Selectively Attenuates Phasic Inhibition in Ventral Hippocampus
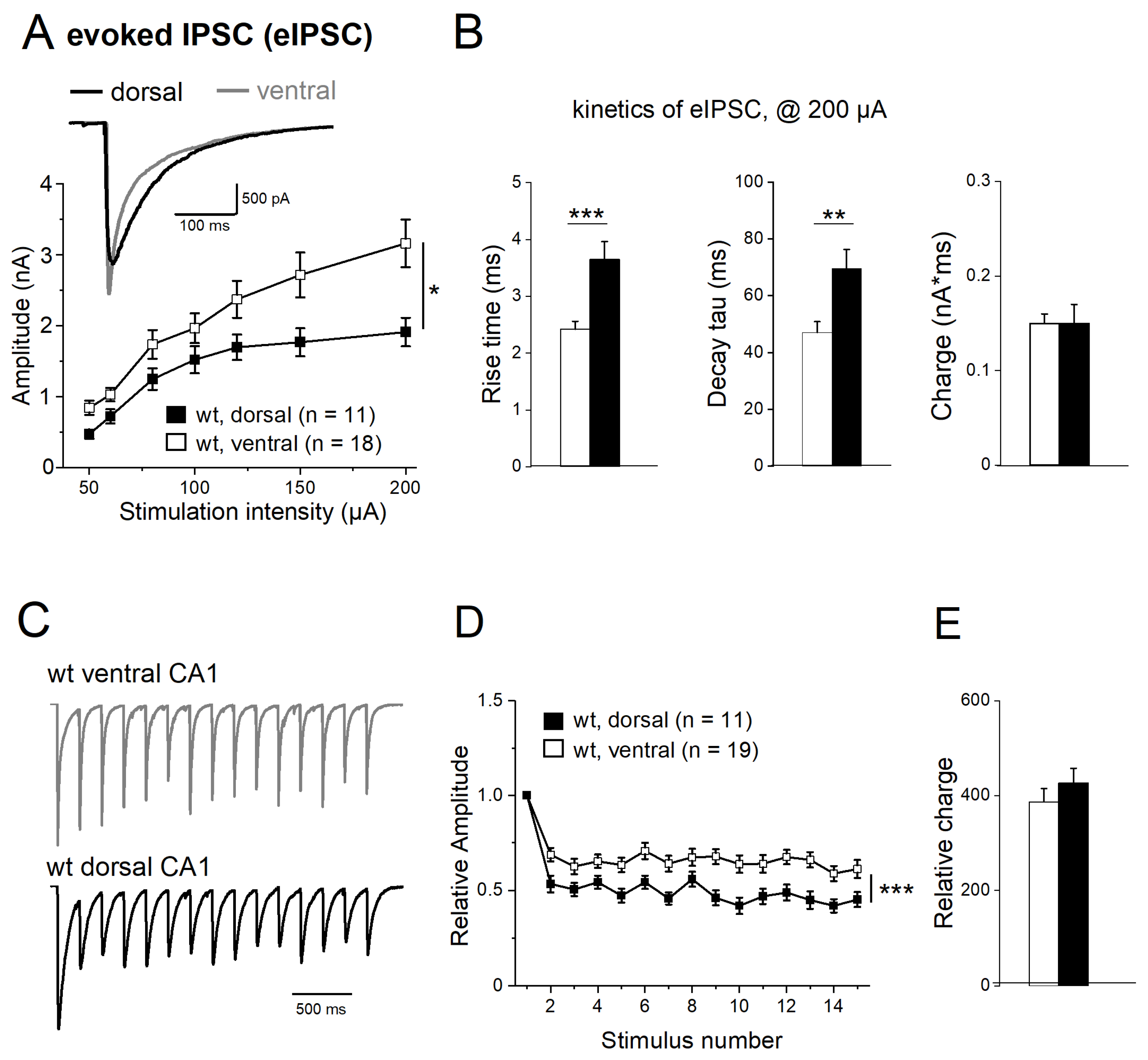
2.5. Properties of Evoked IPSCs along Longitudinal Axis of Hippocampus
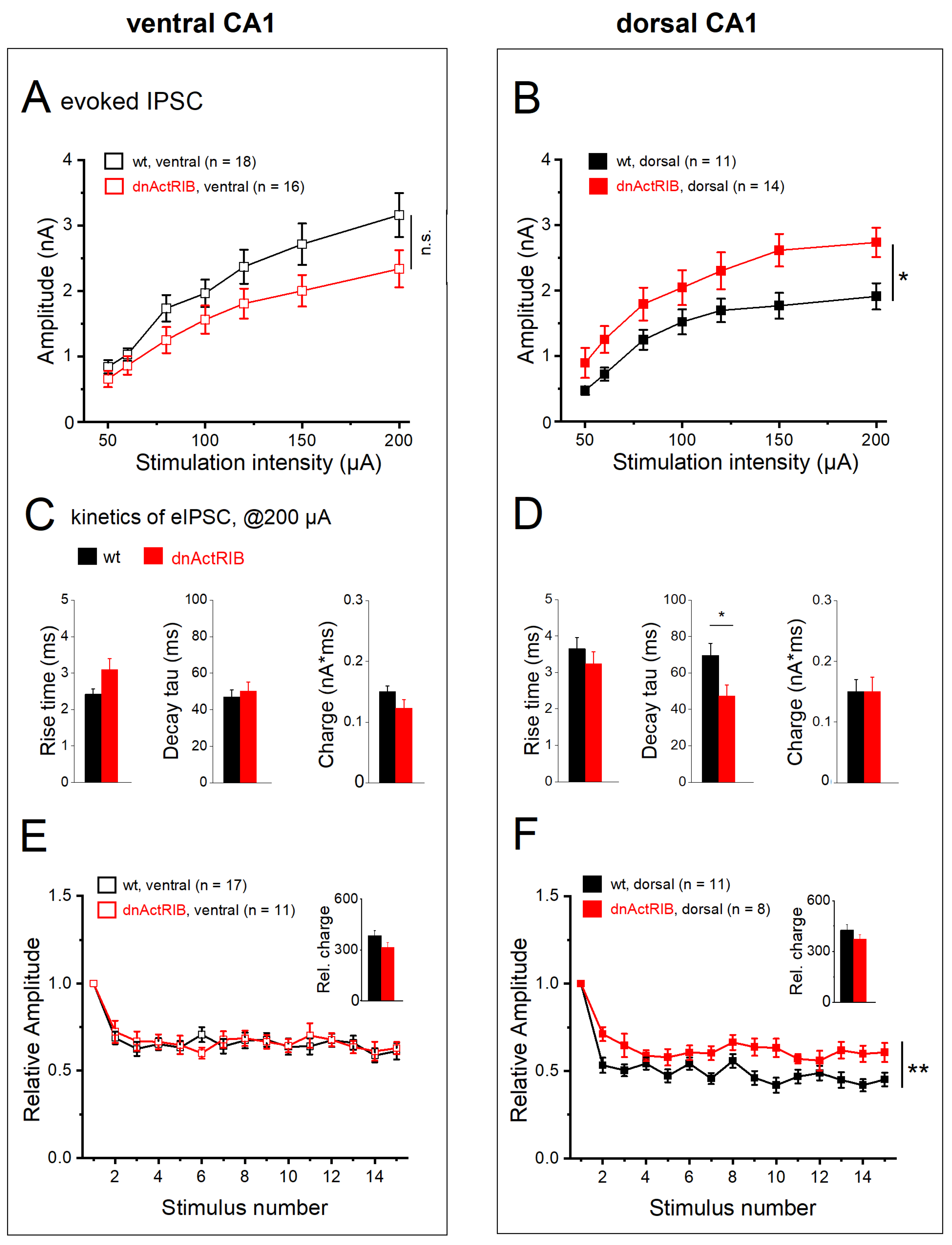
2.6. Activin Signaling Redistributes Weight of eIPSCs between DH and VH
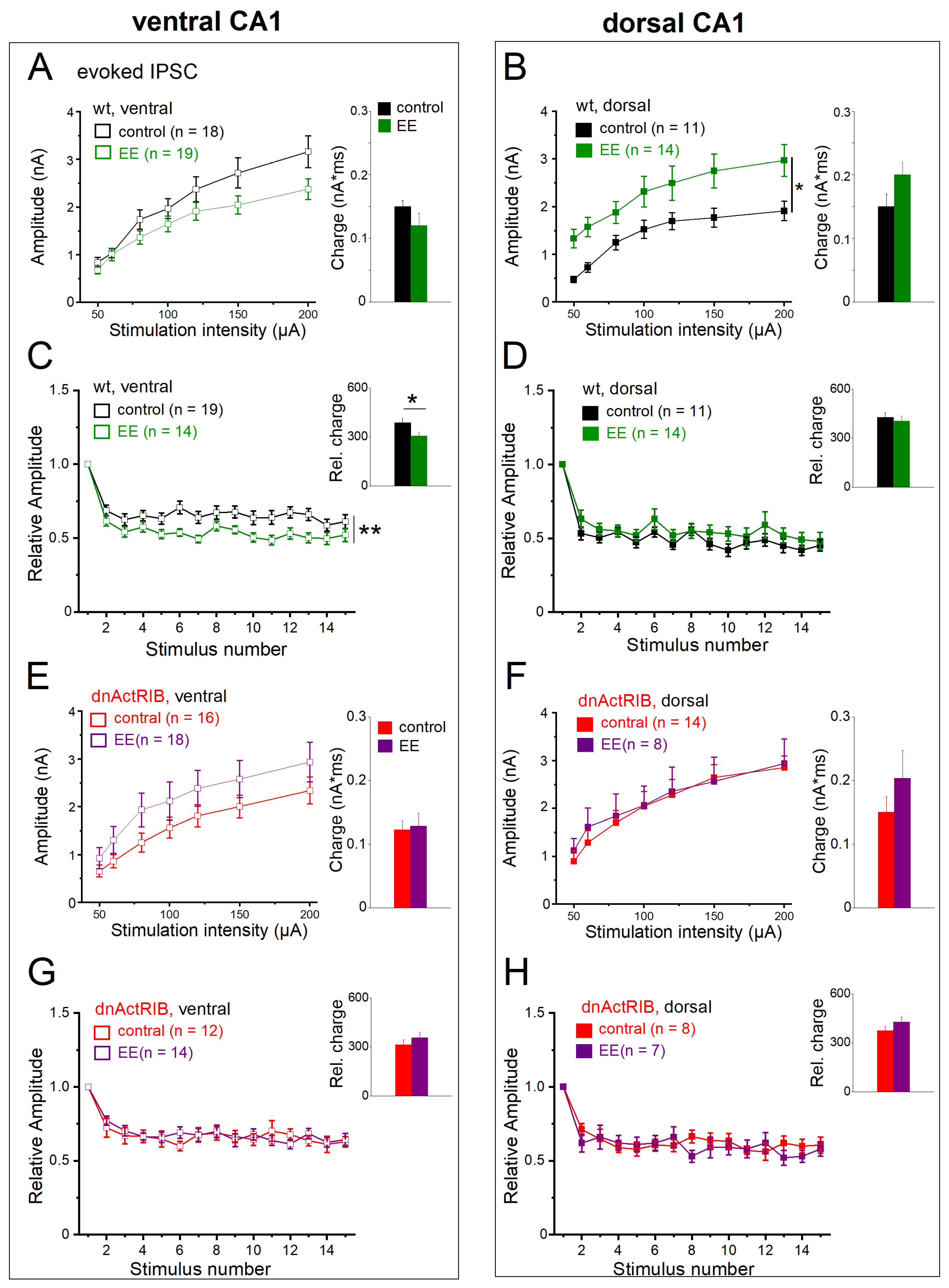
2.7. Activin Signaling Is Critically Involved in the Effects of EE on eIPSCs
3. Discussion
3.1. Hippocampus Exhibits Regional Differences in GABAergic Inhibition
3.2. Effect of Activin in Dorsal and Ventral Hippocampus
4. Materials and Methods
4.1. Animals
4.2. Enriched Environment
4.3. Activin Enzyme-Linked Immunosorbent Assay (ELISA)
4.4. Slice Preparation and Electrophysiological Recordings
4.5. Data Analysis
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Werner, S.; Alzheimer, C. Roles of activin in tissue repair, fibrosis, and inflammatory disease. Cytokine Growth Factor Rev. 2006, 17, 157–171. [Google Scholar] [CrossRef] [PubMed]
- Hedger, M.P.; Winnall, W.R.; Phillips, D.J.; de Kretser, D.M. The regulation and functions of activin and follistatinin inflammation and immunity. Vitam. Horm. 2011, 85, 255–297. [Google Scholar] [CrossRef]
- Andreasson, K.; Worley, P.F. Induction of beta-A activin expression by synaptic activity and during neocortical development. Neuroscience 1995, 69, 781–796. [Google Scholar] [CrossRef] [PubMed]
- Tretter, Y.P.; Hertel, M.; Munz, B.; ten Bruggencate, G.; Werner, S.; Alzheimer, C. Induction of activin A is essential for the neuroprotective action of basic fibroblast growth factor in vivo. Nat. Med. 2000, 6, 812–815. [Google Scholar] [CrossRef]
- Mukerji, S.S.; Rainey, R.N.; Rhodes, J.L.; Hall, A.K. Delayed activin A administration attenuates tissue death after transient focal cerebral ischemia and is associated with decreased stress-responsive kinase activation. J. Neurochem. 2009, 111, 1138–1148. [Google Scholar] [CrossRef] [PubMed]
- Buchthal, B.; Weiss, U.; Bading, H. Post-injury Nose-to-Brain Delivery of Activin A and SerpinB2 Reduces Brain Damage in a Mouse Stroke Model. Mol. Ther. 2018, 26, 2357–2365. [Google Scholar] [CrossRef]
- Müller, M.R.; Zheng, F.; Werner, S.; Alzheimer, C. Transgenic Mice Expressing Dominant-negative Activin Receptor IB in Forebrain Neurons Reveal Novel Functions of Activin at Glutamatergic Synapses. J. Biol. Chem. 2006, 281, 29076–29084. [Google Scholar] [CrossRef]
- Hasegawa, Y.; Mukai, H.; Asashima, M.; Hojo, Y.; Ikeda, M.; Komatsuzaki, Y.; Ooishi, Y.; Kawato, S. Acute modulation of synaptic plasticity of pyramidal neurons by activin in adult hippocampus. Front. Neural Circuits 2014, 8, 56. [Google Scholar] [CrossRef]
- Bloise, E.; Ciarmela, P.; Dela Cruz, C.; Luisi, S.; Petraglia, F.; Reis, F.M. Activin A in Mammalian Physiology. Physiol. Rev. 2019, 99, 739–780. [Google Scholar] [CrossRef]
- Dahlmanns, M.; Dahlmanns, J.K.; Schmidt, C.C.; Valero-Aracama, M.J.; Zheng, F.; Alzheimer, C. Environmental enrichment recruits activin A to recalibrate neural activity in mouse hippocampus. Cereb. Cortex 2023, 33, 663–675. [Google Scholar] [CrossRef]
- Inokuchi, K.; Kato, A.; Hiraia, K.; Hishinuma, F.; Inoue, M.; Ozawa, F. Increase in activin beta A mRNA in rat hippocampus during long-term potentiation. FEBS Lett. 1996, 382, 48–52. [Google Scholar] [CrossRef] [PubMed]
- Link, A.S.; Kurinna, S.; Havlicek, S.; Lehnert, S.; Reichel, M.; Kornhuber, J.; Winner, B.; Huth, T.; Zheng, F.; Werner, S.; et al. Kdm6b and Pmepa1 as Targets of Bioelectrically and Behaviorally Induced Activin A Signaling. Mol. Neurobiol. 2016, 53, 4210–4225. [Google Scholar] [CrossRef] [PubMed]
- Dow, A.L.; Russell, D.S.; Duman, R.S. Regulation of activin mRNA and Smad2 phosphorylation by antidepressant treatment in the rat brain: Effects in behavioral models. J. Neurosci. 2005, 25, 4908–4916. [Google Scholar] [CrossRef][Green Version]
- Ageta, H.; Tsuchida, K. Multifunctional roles of activins in the brain. Vitam. Horm. 2011, 85, 185–206. [Google Scholar] [CrossRef]
- Krieglstein, K.; Zheng, F.; Unsicker, K.; Alzheimer, C. More than being protective: Functional roles for TGF-β/activin signaling pathways at central synapses. Trends Neurosci. 2011, 34, 421–429. [Google Scholar] [CrossRef] [PubMed]
- Ganea, K.; Menke, A.; Schmidt, M.V.; Lucae, S.; Rammes, G.; Liebl, C.; Harbich, D.; Sterlemann, V.; Storch, C.; Uhr, M.; et al. Convergent animal and human evidence suggests the activin/inhibin pathway to be involved in antidepressant response. Transl. Psychiatry 2012, 2, e177. [Google Scholar] [CrossRef]
- Link, A.S.; Zheng, F.; Alzheimer, C. Activin Signaling in the Pathogenesis and Therapy of Neuropsychiatric Diseases. Front. Cell. Neurosci. 2016, 9, 32. [Google Scholar] [CrossRef]
- Gergues, M.M.; Yohn, C.N.; Bharadia, A.; Levinstein, M.R.; Samuels, B.A. Dentate gyrus activin signaling mediates the antidepressant response. Transl. Psychiatry 2021, 11, 7. [Google Scholar] [CrossRef]
- Mitra, S.; Werner, C.; Dietz, D.M. Neuroadaptations and TGF-β signaling: Emerging role in models of neuropsychiatric disorders. Mol. Psychiatry 2022, 27, 296–306. [Google Scholar] [CrossRef]
- Zheng, F.; Adelsberger, H.; Müller, M.; Fritschy, J.M.; Werner, S.; Alzheimer, C. Activin tunes GABAergic neurotransmission and modulates anxiety-like behavior. Mol. Psychiatry 2009, 14, 332–346. [Google Scholar] [CrossRef]
- Zheng, F.; Puppel, A.; Huber, S.E.; Link, A.S.; Eulenburg, V.; van Brederode, J.F.; Müller, C.P.; Alzheimer, C. Activin Controls Ethanol Potentiation of Inhibitory Synaptic Transmission Through GABAA Receptors and Concomitant Behavioral Sedation. Neuropsychopharmacology 2016, 41, 2024–2033. [Google Scholar] [CrossRef] [PubMed]
- Zheng, F.; Valero-Aracama, M.J.; Schaefer, N.; Alzheimer, C. Activin A reduces GIRK current to excite dentate gyrus granule cells. Front. Cell. Neurosci. 2022, 16, 920388. [Google Scholar] [CrossRef] [PubMed]
- Ageta, H.; Murayama, A.; Migishima, R.; Kida, S.; Tsuchida, K.; Yokoyama, M.; Inokuchi, K. Activin in the brain modulates anxiety-related behavior and adult neurogenesis. PLoS ONE 2008, 3, e1869. [Google Scholar] [CrossRef] [PubMed]
- Strange, B.A.; Witter, M.P.; Lein, E.S.; Moser, E.I. Functional organization of the hippocampal longitudinal axis. Nat. Rev. Neurosci. 2014, 15, 655–669. [Google Scholar] [CrossRef] [PubMed]
- Bannerman, D.M.; Rawlins, J.N.; McHugh, S.B.; Deacon, R.M.; Yee, B.K.; Bast, T.; Zhang, W.N.; Pothuizen, H.H.J.; Feldon, J. Regional dissociations within the hippocampus—Memory and anxiety. Neurosci. Biobehav. Rev. 2004, 28, 273–283. [Google Scholar] [CrossRef]
- Fanselow, M.S.; Dong, H.W. Are the dorsal and ventral hippocampus functionally distinct structures? Neuron 2010, 65, 7–19. [Google Scholar] [CrossRef]
- Dougherty, K.A.; Islam, T.; Johnston, D. Intrinsic excitability of CA1 pyramidal neurones from the rat dorsal and ventral hippocampus. J. Physiol. 2012, 590, 5707–5722. [Google Scholar] [CrossRef]
- Malik, R.; Dougherty, K.A.; Parikh, K.; Byrne, C.; Johnston, D. Mapping the electrophysiological and morphological properties of CA1 pyramidal neurons along the longitudinal hippocampal axis. Hippocampus 2016, 26, 341–361. [Google Scholar] [CrossRef]
- Milior, G.; Di Castro, M.A.; Sciarria, L.P.; Garofalo, S.; Branchi, I.; Ragozzino, D.; Limatola, C.; Maggi, L. Electrophysiological Properties of CA1 Pyramidal Neurons along the Longitudinal Axis of the Mouse Hippocampus. Sci. Rep. 2016, 6, 38242. [Google Scholar] [CrossRef]
- Dubovyk, V.; Manahan-Vaughan, D. Less means more: The magnitude of synaptic plasticity along the hippocampal dorso-ventral axis is inversely related to the expression levels of plasticity-related neurotransmitter receptors. Hippocampus 2018, 28, 136–150. [Google Scholar] [CrossRef]
- Ordemann, G.J.; Apgar, C.J.; Brager, D.H. D-type potassium channels normalize action potential firing between dorsal and ventral CA1 neurons of the mouse hippocampus. J. Neurophysiol. 2019, 121, 983–995. [Google Scholar] [CrossRef]
- Toyoda, I.; Bower, M.R.; Leyva, F.; Buckmaster, P.S. Early activation of ventral hippocampus and subiculum during spontaneous seizures in a rat model of temporal lobe epilepsy. J. Neurosci. 2013, 33, 11100–11115. [Google Scholar] [CrossRef] [PubMed]
- Buckmaster, P.S.; Reyes, B.; Kahn, T.; Wyeth, M. Ventral hippocampal formation is the primary epileptogenic zone in a rat model of temporal lobe epilepsy. J. Neurosci. 2022, 42, 7482–7495. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.Y.; Keown, C.L.; Wen, X.; Li, J.; Vousden, D.A.; Anacker, C.; Bhattacharyya, U.; Ryan, R.; Diorio, J.; O’Toole, N.; et al. Environmental enrichment increases transcriptional and epigenetic differentiation between mouse dorsal and ventral dentate gyrus. Nat. Commun. 2018, 9, 298. [Google Scholar] [CrossRef] [PubMed]
- Hörtnagl, H.; Berger, M.L.; Sperk, G.; Pifl, C. Regional heterogeneity in the distribution of neurotransmitter markers in the rat hippocampus. Neuroscience 1991, 45, 261–272. [Google Scholar] [CrossRef]
- Sotiriou, E.; Papatheodoropoulos, C.; Angelatou, F. Differential expression of gamma-aminobutyric acid—A receptor subunits in rat dorsal and ventral hippocampus. J. Neurosci. Res. 2005, 82, 690–700. [Google Scholar] [CrossRef]
- Lothmann, K.; Deitersen, J.; Zilles, K.; Amunts, K.; Herold, C. New boundaries and dissociation of the mouse hippocampus along the dorsal-ventral axis based on glutamatergic, GABAergic and catecholaminergic receptor densities. Hippocampus 2021, 31, 56–78. [Google Scholar] [CrossRef]
- Farrant, M.; Nusser, Z. Variation on an inhibitory theme: Phasic and tonic activation of GABAA receptors. Nat. Rev. Neurosci. 2005, 6, 215–229. [Google Scholar] [CrossRef]
- Mody, I. Aspects of the homeostaic plasticity of GABAA receptor-mediated inhibition. J. Physiol. 2005, 562, 37–46. [Google Scholar] [CrossRef]
- Glykys, J.; Mann, E.O.; Mody, I. Which GABA(A) receptor subunits are necessary for tonic inhibition in the hippocampus? J. Neurosci. 2008, 28, 1421–1426. [Google Scholar] [CrossRef]
- Maggio, N.; Segal, M. Differential corticosteroid modulation of inhibitory synaptic currents in the dorsal and ventral hippocampus. J. Neurosci. 2009, 29, 2857–2866. [Google Scholar] [CrossRef]
- Papatheodoropolus, C.; Asprodini, E.; Nikita, I.; Koutsona, C.; Kostopoulos, G. Weaker synaptic inhibition in CA1 region of ventral compared to dorsal rat hippocampal slices. Brain Res. 2002, 948, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Mody, I.; De Koninck, Y.; Otis, T.S.; Soltesz, I. Bridging the cleft at GABA synapses in the brain. Trends Neurosci. 1994, 17, 517–525. [Google Scholar] [CrossRef] [PubMed]
- Roepstorff, A.; Lambert, J.D. Factors contributing to the decay of the stimulus-evoked IPSC in rat hippocampal CA1 neurons. J. Neurophysiol. 1994, 72, 2911–2926. [Google Scholar] [CrossRef][Green Version]
- Overstreet, L.S.; Jones, M.V.; Westbrook, G.L. Slow desensitization regulates the availability of synaptic GABAA receptors. J. Neurosci. 2000, 20, 7914–7921. [Google Scholar] [CrossRef] [PubMed]
- Sale, A.; Berardi, N.; Maffei, L. Environment and brain plasticity: Towards an endogenous pharmacotherapy. Physiol. Rev. 2014, 94, 189–234. [Google Scholar] [CrossRef]
- Valero-Aracama, M.J.; Sauvage, M.M.; Yoshida, M. Environmental enrichment modulates intrinsic cellular excitability of hippocampal CA1 pyramidal cells in a housing duration and anatomical location-dependent manner. Behav. Brain Res. 2015, 292, 209–218. [Google Scholar] [CrossRef]
- Ohline, S.M.; Abraham, W.C. Environmental enrichment effects on synaptic and cellular physiology of hippocampal neurons. Neuropharmacology 2019, 145, 3–12. [Google Scholar] [CrossRef]
- Kuzumaki, N.; Ikegami, D.; Tamura, R.; Hareyama, N.; Imai, S.; Narita, M.; Torigoe, K.; Niikura, K.; Takeshima, H.; Ando, T.; et al. Hippocampal epigenetic modification at the brain-derived neurotrophic factor gene induced by an enriched environment. Hippocampus 2011, 21, 127–132. [Google Scholar] [CrossRef]
- Jaeger, B.N.; Linker, S.B.; Parylak, S.L.; Barron, J.J.; Gallina, I.S.; Saavedra, C.D.; Fitzpatrick, C.; Kim, C.K.; Schafer, S.T.; Lacar, B.; et al. A novel environment-evoked transcriptional signature predicts reactivity in single dentate granule neurons. Nat. Commun. 2018, 9, 3084. [Google Scholar] [CrossRef]
- Colino-Oliveira, M.; Rombo, D.M.; Dias, R.B.; Ribeiro, J.A.; Sebastiao, A.M. BDNF-induced presynaptic facilitation of GABAergic transmission in the hippocampus of young adults is dependent of TrkB and adenosine A2A receptors. Purinergic Signal. 2016, 12, 283–294. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| spIPSCs | Frequency (Hz) | Amplitude (pA) | Rise Time (ms) | Decay Tau (ms) | Half-Width (ms) | Charge (pA*ms) | |
|---|---|---|---|---|---|---|---|
| wt-con | ventral (63) | 6.26 ± 0.38 | 50.27 ± 1.63 | 0.48 ± 0.01 ** | 8.72 ± 0.21 | 6.03 ± 0.16 | 466.09 ± 17.37 |
| dorsal (42) | 5.77 ± 0.40 | 49.35 ± 2.00 | 0.53 ± 0.01 | 8.26 ± 0.26 | 5.73 ± 0.18 | 457.19 ± 16.79 | |
| dnActRIB-con | ventral (53) | 5.86 ± 0.35 * | 51.19 ± 2.14 | 0.51 ± 0.02 | 9.05 ± 0.23 ** | 6.32 ± 0.22 | 515.19 ± 16.79 |
| dorsal (27) | 7.19 ± 0.60 # | 47.74 ± 2.58 | 0.56 ± 0.02 | 8.02 ± 0.24 | 5.68 ± 0.21 | 447.53 ± 24.78 | |
| wt-EE | ventral (51) | 4.41 ± 0.27 *,§§§ | 53.06 ± 2.37 | 0.45 ± 0.01 | 8.17 ± 0.19 | 5.47 ± 0.14 § | 489.68 ± 238.51 |
| dorsal (19) | 5.81 ± 0.47 | 49.02 ± 2.52 | 0.47 ± 0.02 § | 8.58 ± 0.34 | 5.82 ± 0.25 | 454.92 ± 23.13 | |
| dnActRIB-EE | ventral (25) | 5.53 ± 0.48 | 55.67 ± 2.62 | 0.48 ± 0.02 | 8.56 ± 0.28 | 6.07 ± 0.25 | 506.33 ± 27.64 |
| dorsal (11) | 6.28 ± 0.57 | 51.15 ± 3.07 | 0.47 ± 0.02 § | 9.59 ± 0.61 §§ | 6.30 ± 0.50 | 502.98 ± 13.15 |
| eIPSCs | Amplitude (pA) | Rise Time (10–90%; ms) | Decay Tau (ms) | Half-Width (ms) | Charge (nA*ms) | |
|---|---|---|---|---|---|---|
| wt-con | ventral (18) | 3.16 ± 0.33 * | 2.42 ± 0.14 *** | 47.12 ± 0.37 ** | 28.67 ± 2.71 ** | 0.15 ± 0.01 |
| dorsal (11) | 1.91 ± 0.20 | 3.65 ± 0.31 | 69.61 ± 6.83 | 48.95 ± 5.75 | 0.15 ± 0.02 | |
| dnActRIB-con | ventral (16) | 2.34 ± 0.28 | 3.09 ± 0.31 | 50.33 ± 4.85 | 32.44 ± 3.93 | 0.12 ± 0.01 |
| dorsal (14) | 2.73 ± 0.22 # | 3.24 ± 0.33 | 47.29 ± 6.11 # | 29.36 ± 4.48 # | 0.15 ± 0.02 | |
| wt-EE | ventral (19) | 2.38 ± 0.22 | 2.93 ± 0.28 | 43.22 ± 3.35 ** | 29.06 ± 2.64 ** | 0.12 ± 0.01 ** |
| dorsal (14) | 2.97 ± 0.34 § | 3.75 ± 0.40 | 58.56 ± 4.73 | 44.03 ± 4.34 | 0.20 ± 0.02 | |
| dnActRIB-EE | ventral (18) | 2.94 ± 0.42 | 2.83 ± 0.26 | 42.44 ± 4.23 * | 28.10 ± 3.59 | 0.13 ± 0.02 |
| dorsal (8) | 2.94 ± 0.51 | 3.28 ± 0.34 | 56.68 ± 4.67 | 38.67 ± 2.69 | 0.20 ± 0.04 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Valero-Aracama, M.J.; Zheng, F.; Alzheimer, C. Dorsal–Ventral Gradient of Activin Regulates Strength of GABAergic Inhibition along Longitudinal Axis of Mouse Hippocampus in an Activity-Dependent Fashion. Int. J. Mol. Sci. 2023, 24, 13145. https://doi.org/10.3390/ijms241713145
Valero-Aracama MJ, Zheng F, Alzheimer C. Dorsal–Ventral Gradient of Activin Regulates Strength of GABAergic Inhibition along Longitudinal Axis of Mouse Hippocampus in an Activity-Dependent Fashion. International Journal of Molecular Sciences. 2023; 24(17):13145. https://doi.org/10.3390/ijms241713145
Chicago/Turabian StyleValero-Aracama, Maria Jesus, Fang Zheng, and Christian Alzheimer. 2023. "Dorsal–Ventral Gradient of Activin Regulates Strength of GABAergic Inhibition along Longitudinal Axis of Mouse Hippocampus in an Activity-Dependent Fashion" International Journal of Molecular Sciences 24, no. 17: 13145. https://doi.org/10.3390/ijms241713145
APA StyleValero-Aracama, M. J., Zheng, F., & Alzheimer, C. (2023). Dorsal–Ventral Gradient of Activin Regulates Strength of GABAergic Inhibition along Longitudinal Axis of Mouse Hippocampus in an Activity-Dependent Fashion. International Journal of Molecular Sciences, 24(17), 13145. https://doi.org/10.3390/ijms241713145