

Lipid Mediators and Cytokines/Chemokines Display Differential Profiles in Severe versus Mild/Moderate COVID-19 Patients
 , ,
, ,  and
and 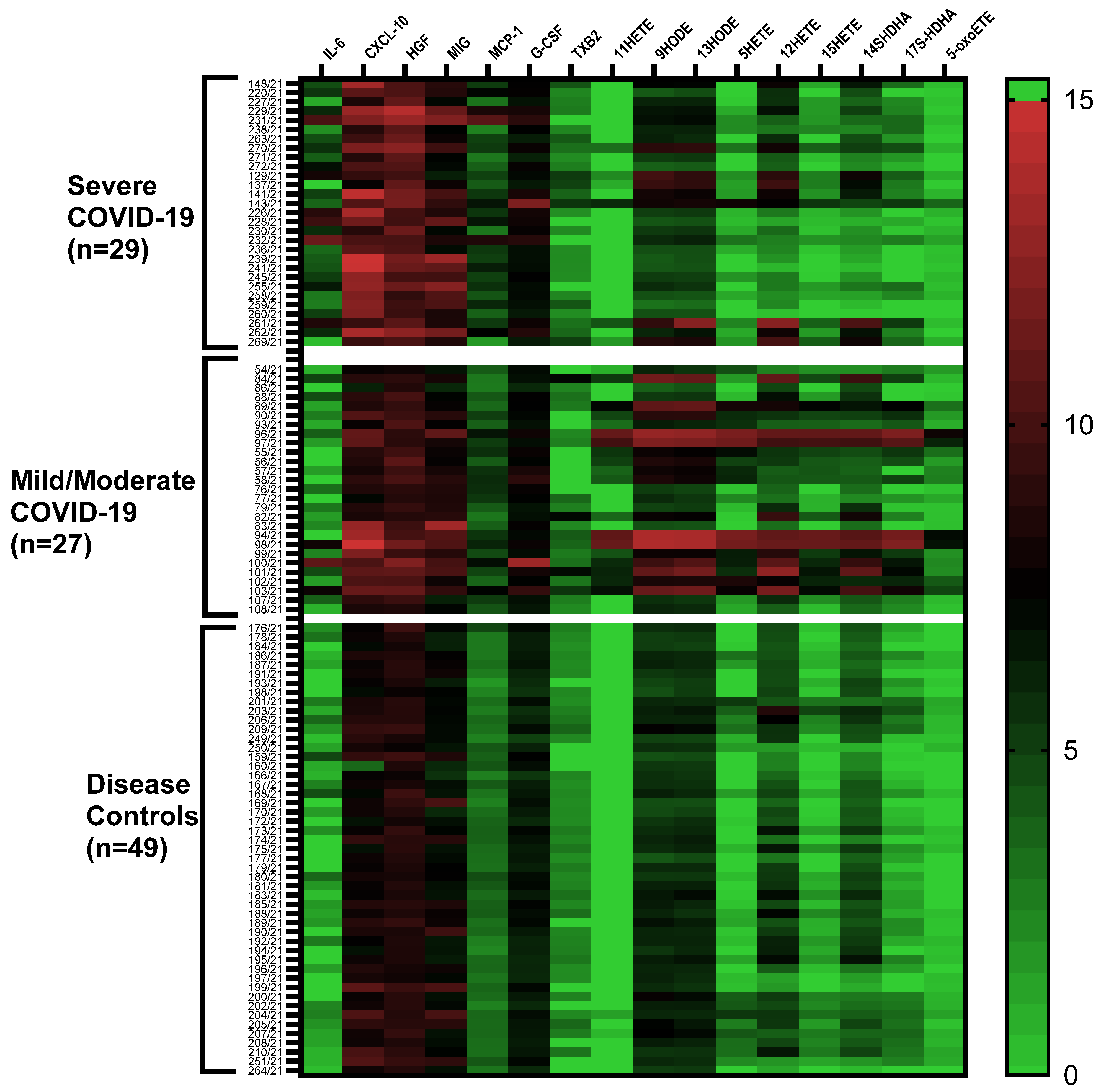{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Cytokine/Chemokine and Lipid Mediator Profiles
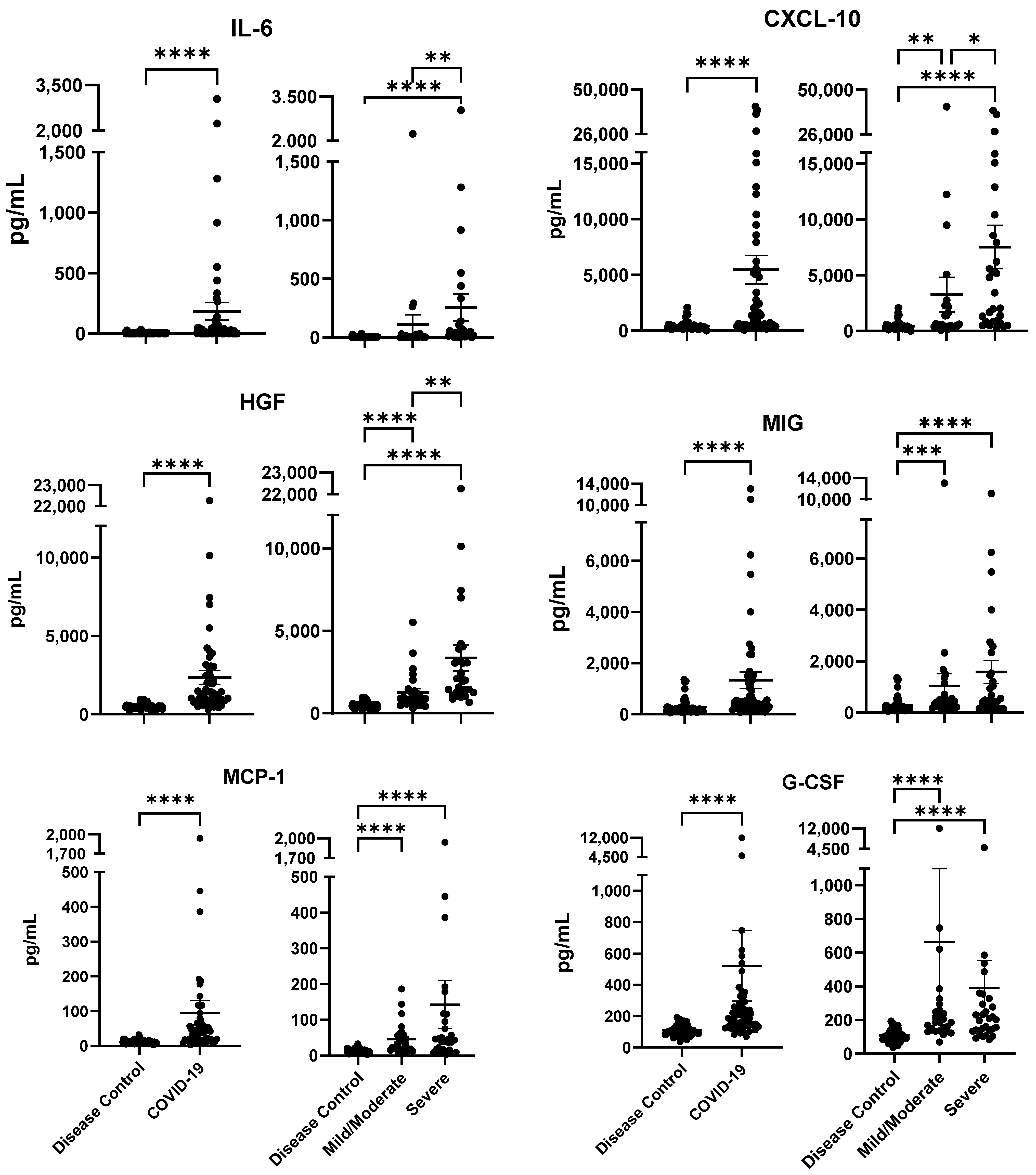
2.2. Cytokine/Chemokine Profiles Associated with COVID-19 Severity
2.3. Lipid Mediator Profiles Related to COVID-19 Severity
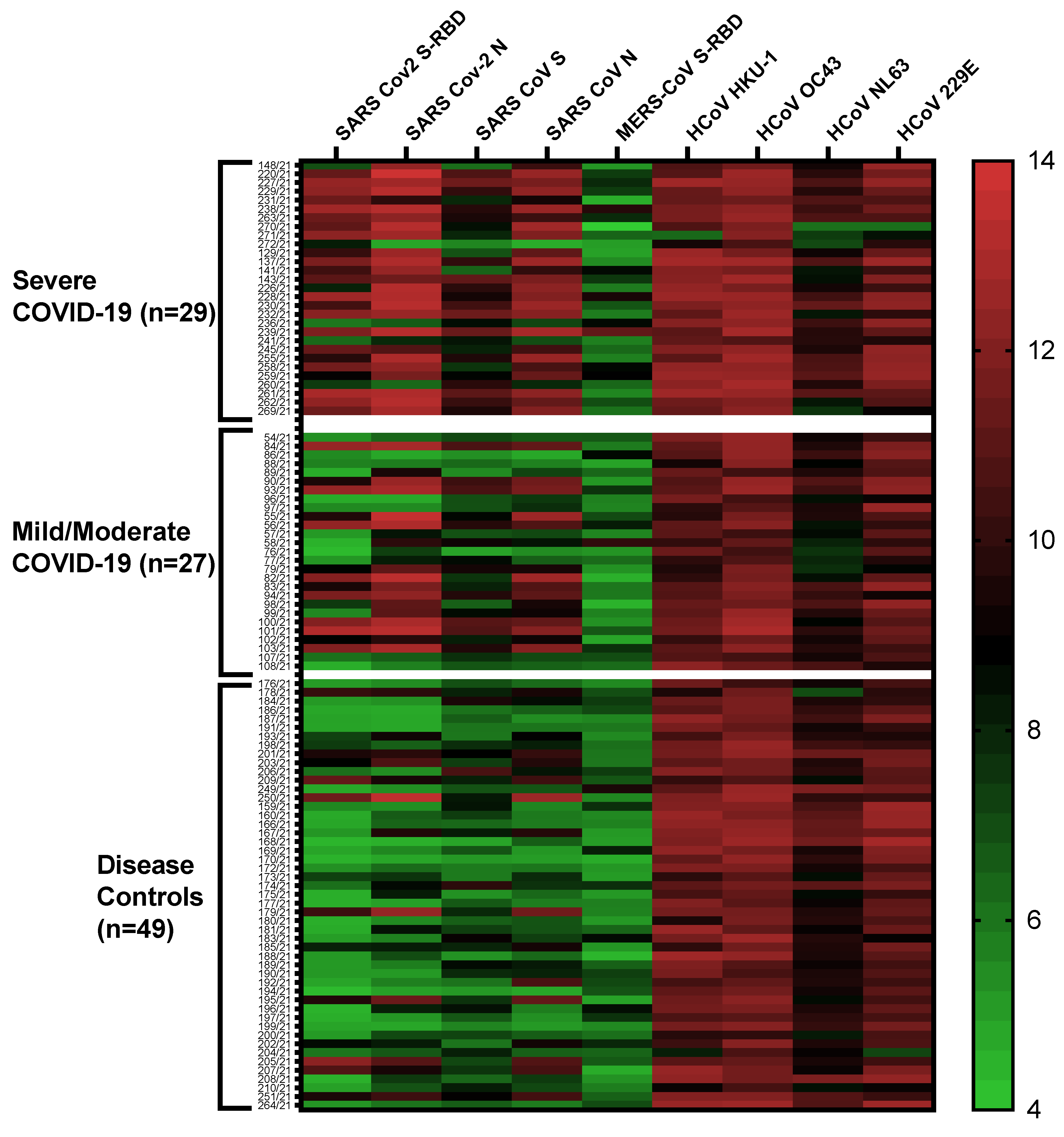
2.4. Multiplex Antibody Profiles in COVID-19 Patients and Disease Controls
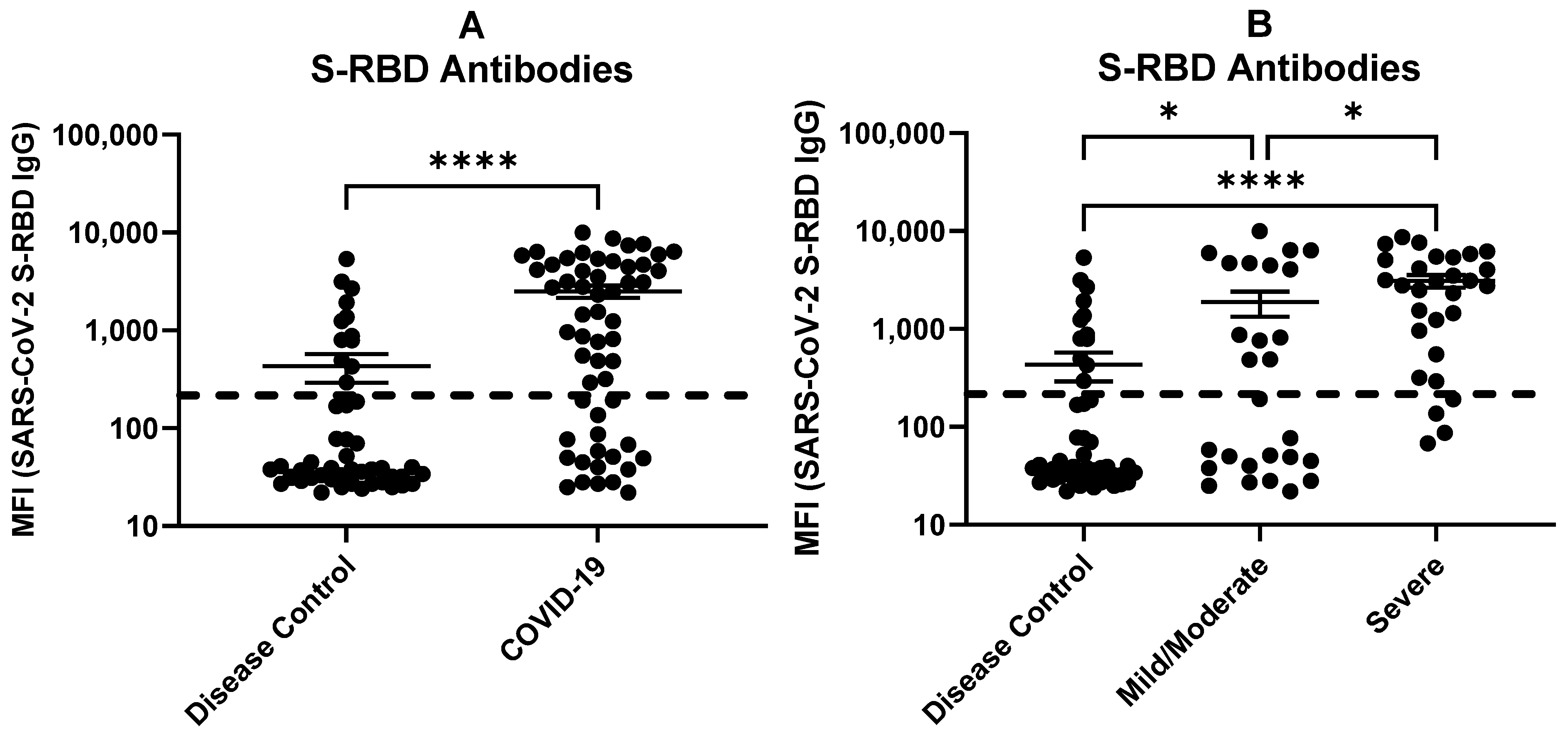
2.5. Antibodies against SARS-CoV-2 S-RBD Related to COVID-19 Severity
3. Discussion
4. Materials and Methods
4.1. Ethics Statement
4.2. COVID-19 Patient Plasma Samples
4.3. Respiratory Disease Control Patient Samples (Other Than COVID-19)
4.4. Healthy Control Group
4.5. Multiplex Antibody Assay
4.6. Multiplex Cytokine/Chemokine Assay
4.7. Lipidomics
4.8. Data Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Organization WHO. WHO Coronavirus Disease (COVID-19) Dashboard with Vaccination Data|WHO Coronavirus (COVID-19) Dashboard With Vaccination Data. 2023. Available online: https://covid19.who.int/ (accessed on 6 July 2023).
- Estenssoro, E.; Loudet, C.I.; Dubin, A.; Edul, V.S.K.; Plotnikow, G.; Andrian, M.; Sagardía, J.; Bezzi, M.; Mandich, V.; Groer, C.; et al. Clinical characteristics, respiratory management, and determinants of oxygenation in COVID-19 ARDS: A prospective cohort study. J. Crit. Care 2022, 71, 154021. [Google Scholar] [CrossRef]
- Alsafi, R.T. Lessons from SARS-CoV, MERS-CoV, and SARS-CoV-2 Infections: What We Know So Far. Can. J. Infect. Dis. Med. Microbiol. 2022, 2022, 1156273. [Google Scholar] [CrossRef]
- Dinarello, C.A. Proinflammatory cytokines. Chest 2000, 118, 503–508. [Google Scholar] [CrossRef]
- Wright, T.M.; Feghali, C.A. Cytokines in acute and chronic inflammation. Front. Biosci. 1997, 2, d12–d26. [Google Scholar] [CrossRef]
- Kimura, H.; Yoshizumi, M.; Ishii, H.; Oishi, K.; Ryo, A. Cytokine production and signaling pathways in respiratory virus infection. Front. Microbiol. 2013, 4, 276. [Google Scholar] [CrossRef]
- Menachery, V.D.; Eisfeld, A.J.; Schäfer, A.; Josset, L.; Sims, A.C.; Proll, S.; Fan, S.; Li, C.; Neumann, G.; Tilton, S.C.; et al. Pathogenic Influenza Viruses and Coronaviruses Utilize Similar and Contrasting Approaches to Control Interferon-Stimulated Gene Responses. mBio 2014, 5, e01174-14. [Google Scholar] [CrossRef] [PubMed]
- Soy, M.; Keser, G.; Atagündüz, P.; Tabak, F.; Atagündüz, I.; Kayhan, S. Cytokine storm in COVID-19: Pathogenesis and overview of anti-inflammatory agents used in treatment. Clin. Rheumatol. 2020, 39, 2085–2094. [Google Scholar] [CrossRef] [PubMed]
- Min, C.-K.; Cheon, S.; Ha, N.-Y.; Sohn, K.M.; Kim, Y.; Aigerim, A.; Shin, H.M.; Choi, J.Y.; Inn, K.S.; Kim, J.H.; et al. Comparative and kinetic analysis of viral shedding and immunological responses in MERS patients representing a broad spectrum of disease severity. Sci. Rep. 2016, 6, 25359. [Google Scholar] [CrossRef] [PubMed]
- Giamarellos-Bourboulis, E.J.; Netea, M.G.; Rovina, N.; Akinosoglou, K.; Antoniadou, A.; Antonakos, N.; Damoraki, G.; Gkavogianni, T.; Adami, M.E.; Katsaounou, P.; et al. Complex Immune Dysregulation in COVID-19 Patients with Severe Respiratory Failure. Cell Host Microbe 2020, 27, 992–1000.e3. [Google Scholar] [CrossRef] [PubMed]
- Lucas, C.; Wong, P.; Klein, J.; Castro, T.B.R.; Silva, J.; Sundaram, M.; Ellingson, M.K.; Mao, T.; Oh, J.E.; Israelow, B.; et al. Longitudinal analyses reveal immunological misfiring in severe COVID-19. Nature 2020, 584, 463–469. [Google Scholar] [CrossRef]
- Mehta, P.; McAuley, D.F.; Brown, M.; Sanchez, E.; Tattersall, R.S.; Manson, J.J. COVID-19: Consider cytokine storm syndromes and immunosuppression. Lancet 2020, 395, 1033–1034. [Google Scholar] [CrossRef] [PubMed]
- Ravindran, R.; McReynolds, C.; Yang, J.; Hammock, B.D.; Ikram, A.; Ali, A.; Bashir, A.; Zohra, T.; Chang, W.L.W.; Hartigan-O’Connor, D.J.; et al. Immune response dynamics in COVID-19 patients to SARS-CoV-2 and other human coronaviruses. PLoS ONE 2021, 16, e0254367. [Google Scholar] [CrossRef] [PubMed]
- Dennis, E.A.; Norris, P.C. Eicosanoid storm in infection and inflammation. Nat. Rev. Immunol. 2015, 15, 511–523. [Google Scholar] [CrossRef] [PubMed]
- Litwack, G. Eicosanoids. In Hormones; Academic Press: Cambridge, MA, USA, 2022; pp. 195–212. [Google Scholar]
- Meriwether, D.; Sulaiman, D.; Volpe, C.; Dorfman, A.; Grijalva, V.; Dorreh, N.; Solorzano-Vargas, R.S.; Wang, J.; O’Connor, E.; Papesh, J.; et al. Apolipoprotein A-I mimetics mitigate intestinal inflammation in COX2-dependent inflammatory bowel disease model. J. Clin. Investig. 2019, 129, 3670–3685. [Google Scholar] [CrossRef]
- Sharma, S.; Ruffenach, G.; Umar, S.; Motayagheni, N.; Reddy, S.T.; Eghbali, M. Role of Oxidized Lipids in Pulmonary Arterial Hypertension. Pulm. Circ. 2016, 6, 261–273. [Google Scholar] [CrossRef] [PubMed]
- Ruffenach, G.; O’connor, E.; Vaillancourt, M.; Hong, J.; Cao, N.; Sarji, S.; Moazeni, S.; Papesh, J.; Grijalva, V.; Cunningham, C.M.; et al. Oral 15-Hydroxyeicosatetraenoic Acid Induces Pulmonary Hypertension in Mice by Triggering T Cell–Dependent Endothelial Cell Apoptosis. Hypertension 2020, 76, 985–996. [Google Scholar] [CrossRef]
- Serhan, C.N. Pro-resolving lipid mediators are leads for resolution physiology. Nature 2014, 510, 92–101. [Google Scholar] [CrossRef]
- Archambault, A.S.; Zaid, Y.; Rakotoarivelo, V.; Turcotte, C.; Doré, É.; Dubuc, I.; Martin, C.; Flamand, O.; Amar, Y.; Cheikh, A.; et al. High levels of eicosanoids and docosanoids in the lungs of intubated COVID-19 patients. FASEB J. 2021, 35, e21666. [Google Scholar] [CrossRef]
- Palmas, F.; Clarke, J.; Colas, R.A.; Gomez, E.A.; Keogh, A.; Boylan, M.; McEvoy, N.; McElvaney, O.J.; McElvaney, O.; Alalqam, R.; et al. Dysregulated plasma lipid mediator profiles in critically ill COVID-19 patients. PLoS ONE 2021, 16, e0256226. [Google Scholar] [CrossRef]
- Turnbull, J.; Jha, R.R.; Ortori, C.A.; Lunt, E.; Tighe, P.J.; Irving, W.L.; Gohir, S.A.; Kim, D.H.; Valdes, A.M.; Tarr, A.W.; et al. Serum Levels of Proinflammatory Lipid Mediators and Specialized Proresolving Molecules Are Increased in Patients With Severe Acute Respiratory Syndrome Coronavirus 2 and Correlate with Markers of the Adaptive Immune Response. J. Infect. Dis. 2022, 225, 2142–2154. [Google Scholar] [CrossRef]
- Hammock, B.D.; Wang, W.; Gilligan, M.M.; Panigrahy, D. Eicosanoids: The Overlooked Storm in Coronavirus Disease 2019 (COVID-19)? Am. J. Pathol. 2020, 190, 1782–1788. [Google Scholar] [CrossRef] [PubMed]
- Jaimes, J.A.; André, N.M.; Chappie, J.S.; Millet, J.K.; Whittaker, G.R. Phylogenetic Analysis and Structural Modeling of SARS-CoV-2 Spike Protein Reveals an Evolutionary Distinct and Proteolytically Sensitive Activation Loop. J. Mol. Biol. 2020, 432, 3309–3325. [Google Scholar] [CrossRef] [PubMed]
- Dutta, N.K.; Mazumdar, K.; Gordy, J.T. The Nucleocapsid Protein of SARS-CoV-2: A Target for Vaccine Development. J. Virol. 2020, 94, e00647-20. [Google Scholar] [CrossRef] [PubMed]
- Montazersaheb, S.; Khatibi, S.M.H.; Hejazi, M.S.; Tarhriz, V.; Farjami, A.; Sorbeni, F.G.; Farahzadi, R.; Ghasemnejad, T. COVID-19 infection: An overview on cytokine storm and related interventions. Virol. J. 2022, 19, 92. [Google Scholar] [CrossRef] [PubMed]
- Shivshankar, P.; Karmouty-Quintana, H.; Mills, T.; Doursout, M.F.; Wang, Y.; Czopik, A.K.; Evans, S.E.; Eltzschig, H.K.; Yuan, X. SARS-CoV-2 Infection: Host Response, Immunity, and Therapeutic Targets. Inflammation 2022, 45, 1430–1449. [Google Scholar] [CrossRef]
- Park, A.; Iwasaki, A. Type I and Type III Interferons—Induction, Signaling, Evasion, and Application to Combat COVID-19. Cell Host Microbe 2020, 27, 870–878. [Google Scholar] [CrossRef]
- Ravindran, R.; Krishnan, V.V.; Khanum, A.; Luciw, P.A.; Khan, I.H. Exploratory study on plasma immunomodulator and antibody profiles in tuberculosis patients. Clin. Vaccine Immunol. 2013, 20, 1283–1290. [Google Scholar] [CrossRef][Green Version]
- Chavez, K.; Ravindran, R.; Dehnad, A.; Khan, I.H. Gender biased immune-biomarkers in active tuberculosis and correlation of their profiles to efficacy of therapy. Tuberculosis 2016, 99, 17–24. [Google Scholar] [CrossRef]
- Sharma, S.; Umar, S.; Potus, F.; Iorga, A.; Wong, G.; Meriwether, D.; Breuils-Bonnet, S.; Mai, D.; Navab, K.; Ross, D.; et al. Apolipoprotein A-I Mimetic Peptide 4F Rescues Pulmonary Hypertension by Inducing MicroRNA-193-3p. Circulation 2014, 130, 776–785. [Google Scholar] [CrossRef]
- Chattopadhyay, A.; Yang, X.; Mukherjee, P.; Sulaiman, D.; Fogelman, H.R.; Grijalva, V.; Dubinett, S.; Wasler, T.C.; Manash, K.; Salehi-Rad, P.R.; et al. Treating the Intestine with Oral ApoA-I Mimetic Tg6F Reduces Tumor Burden in Mouse Models of Metastatic Lung Cancer. Sci. Rep. 2018, 8, 9032. [Google Scholar] [CrossRef]
- Wong, L.-Y.R.; Zheng, J.; Wilhelmsen, K.; Li, K.; Ortiz, M.E.; Schnicker, N.J.; Thurman, A.; Pezzulo, A.A.; Szachowicz, P.J.; Li, P.; et al. Eicosanoid signalling blockade protects middle-aged mice from severe COVID-19. Nature 2022, 605, 146–151. [Google Scholar] [CrossRef] [PubMed]
- Kelesidis, T.; Madhav, S.; Petcherski, A.; Cristelle, H.; O’Connor, E.; Hultgren, N.W.; Ritou, E.; Williams, D.S.; Shirihai, O.S.; Reddy, S.T. The ApoA-I mimetic peptide 4F attenuates in vitro replication of SARS-CoV-2, associated apoptosis, oxidative stress and inflammation in epithelial cells. Virulence 2021, 12, 2214–2227. [Google Scholar] [CrossRef]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef]
- Chang, S.; Minn, D.; Kim, S.-W.; Kim, Y. Inflammatory Markers and Cytokines in Moderate and Critical Cases of COVID-19. Clin. Lab. 2021, 67. [Google Scholar] [CrossRef]
- Kunnumakkara, A.B.; Rana, V.; Parama, D.; Banik, K.; Girisa, S.; Henamayee, S.; Thakur, K.K.; Dutta, U.; Garodia, P.; Gupta, S.C.; et al. COVID-19, cytokines, inflammation, and spices: How are they related? Life Sci. 2021, 284, 119201. [Google Scholar] [CrossRef] [PubMed]
- Perreau, M.; Suffiotti, M.; Marques-Vidal, P.; Wiedemann, A.; Levy, Y.; Laouénan, C.; Ghosn, J.; Fenwick, C.; Comte, D.; Roger, T.; et al. The cytokines HGF and CXCL13 predict the severity and the mortality in COVID-19 patients. Nat. Commun. 2021, 12, 4888. [Google Scholar] [CrossRef] [PubMed]
- Shrotri, M.; van Schalkwyk, M.C.I.; Post, N.; Eddy, D.; Huntley, C.; Leeman, D.; Rigby, S.; Williams, S.V.; Bermingham, W.H.; Kellam, P.; et al. T cell response to SARS-CoV-2 infection in humans: A systematic review. PLoS ONE 2021, 16, e0245532. [Google Scholar] [CrossRef]
- McGregor, R.; Chauss, D.; Freiwald, T.; Yan, B.; Wang, L.; Nova-Lamperti, E.; Zhang, Z.; Teague, H.; West, E.E.; Bibby, J.; et al. An autocrine Vitamin D-driven Th1 shutdown program can be exploited for COVID-19. bioRxiv 2020. [Google Scholar] [CrossRef]
- Aleebrahim-Dehkordi, E.; Molavi, B.; Mokhtari, M.; Deravi, N.; Fathi, M.; Fazel, T.; Mohebalizadeh, M.; Koochaki, P.; Shobeiri, P. Hasanpour-Dehkordi AT helper type (Th1/Th2) responses to SARS-CoV-2 and influenza A (H1N1) virus: From cytokines produced to immune responses. Transpl. Immunol. 2022, 70, 101495. [Google Scholar] [CrossRef]
- Park, M.D. Macrophages: A Trojan horse in COVID-19? Nat. Rev. Immunol. 2020, 20, 351. [Google Scholar] [CrossRef]
- Roncati, L.; Ligabue, G.; Fabbiani, L.; Malagoli, C.; Gallo, G.; Lusenti, B.; Nasillo, V.; Manenti, A.; Maiorana, A. Type 3 hypersensitivity in COVID-19 vasculitis. Clin. Immunol. 2020, 217, 108487. [Google Scholar] [CrossRef]
- Rocha, V.Z.; Folco, E.J. Inflammatory Concepts of Obesity. Int. J. Inflamm. 2011, 2011, 529061. [Google Scholar] [CrossRef]
- El-Kadre, L.J.; Tinoco, A.C. Interleukin-6 and obesity: The crosstalk between intestine, pancreas and liver. Curr. Opin. Clin. Nutr. Metab. Care. 2013, 16, 564–568. [Google Scholar] [CrossRef] [PubMed]
- Marin, V.; Montero-Julian, F.A.; Grès, S.; Boulay, V.; Bongrand, P.; Farnarier, C.; Kaplanski, G. The IL-6-soluble IL-6Ralpha autocrine loop of endothelial activation as an intermediate between acute and chronic inflammation: An experimental model involving thrombin. J. Immunol. 2001, 167, 3435–3442. [Google Scholar] [CrossRef] [PubMed]
- Galimi, F.; Cottone, E.; Vigna, E.; Arena, N.; Boccaccio, C.; Giordano, S.; Naldini, L.; Comoglio, P.M. Hepatocyte Growth Factor Is a Regulator of Monocyte-Macrophage Function. J. Immunol. 2001, 166, 1241–1247. [Google Scholar] [CrossRef] [PubMed]
- van der Voort, R.; Taher, T.E.; Keehnen, R.M.; Smit, L.; Groenink, M.; Pals, S.T. Paracrine Regulation of Germinal Center B Cell Adhesion through the c-Met–Hepatocyte Growth Factor/Scatter Factor Pathway. J. Exp. Med. 1997, 185, 2121–2131. [Google Scholar] [CrossRef]
- Singhal, E.; Kumar, P.; Sen, P. A novel role for Bruton’s tyrosine kinase in hepatocyte growth factor-mediated immunoregulation of dendritic cells. J. Biol. Chem. 2011, 286, 32054–32063. [Google Scholar] [CrossRef]
- Zhang, N.; Zhao, Y.-D.; Wang, X.-M. CXCL10 an important chemokine associated with cytokine storm in COVID-19 infected patients. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 7497–7505. [Google Scholar] [CrossRef]
- Blot, M.; Jacquier, M.; Glele, L.-S.A.; Beltramo, G.; Nguyen, M.; Bonniaud, P.; Prin, S.; Andreu, P.; Bouhemad, B.; Bour, J.-B.; et al. CXCL10 could drive longer duration of mechanical ventilation during COVID-19 ARDS. Crit. Care 2020, 24, 632. [Google Scholar] [CrossRef]
- Ochoa-Ramirez, L.A.; Ramos-Payan, R.; Jimenez-Gastelum, G.R.; Rodriguez-Millan, J.; Aguilar-Medina, M.; Rios-Tostado, J.J.; Ayala-Ham, A.; Bermudez, M.; Osuna-Ramos, J.F.; Olimon-Andalon, V.; et al. The Chemokine MIG is Associated with an Increased Risk of COVID-19 Mortality in Mexican Patients. Iran. J. Immunol. 2022, 19, 311–320. [Google Scholar] [CrossRef]
- Tapela, K.; Ochieng’ Olwal, C.; Quaye, O. Parallels in the pathogenesis of SARS-CoV-2 and M. tuberculosis: A synergistic or antagonistic alliance? Future Microbiol. 2020, 15, 1691–1695. [Google Scholar] [CrossRef]
- Biringer, R.G. The enzymology of human eicosanoid pathways: The lipoxygenase branches. Mol. Biol. Rep. 2020, 47, 7189–7207. [Google Scholar] [CrossRef]
- Yan, X.; Chen, G.; Jin, Z.; Zhang, Z.; Zhang, B.; He, J.; Yin, S.; Huang, J.; Fan, M.; Li, Z.; et al. Anti-SARS-CoV-2 IgG levels in relation to disease severity of COVID-19. J. Med. Virol. 2022, 94, 380–383. [Google Scholar] [CrossRef]
- Garcia-Beltran, W.F.; Lam, E.C.; Astudillo, M.G.; Yang, D.; Miller, T.E.; Feldman, J.; Hauser, B.M.; Caradonna, T.M.; Clayton, K.L.; Nitido, A.D.; et al. COVID-19-neutralizing antibodies predict disease severity and survival. Cell 2021, 184, 476–488.e11. [Google Scholar] [CrossRef] [PubMed]
- Long, Q.-X.; Liu, B.-Z.; Deng, H.-J.; Wu, G.-C.; Deng, K.; Chen, Y.-K.; Liao, P.; Qiu, J.F.; Lin, Y.; Cai, X.F.; et al. Antibody responses to SARS-CoV-2 in patients with COVID-19. Nat. Med. 2020, 26, 845–848. [Google Scholar] [CrossRef] [PubMed]
- Petersen, L.R.; Sami, S.; Vuong, N.; Pathela, P.; Weiss, D.; Morgenthau, B.M.; Henseler, R.A.; Daskalakis, D.C.; Atas, J.; Patel, A.; et al. Lack of Antibodies to Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) in a Large Cohort of Previously Infected Persons. Clin. Infect. Dis. 2020, 73, e3066–e3073. [Google Scholar] [CrossRef]
- Wellinghausen, N.; Voss, M.; Ivanova, R.; Deininger, S. Evaluation of the SARS-CoV-2-IgG response in outpatients by five commercial immunoassays. GMS Infect. Dis. 2020, 8, Doc22. [Google Scholar] [PubMed]
- Robbiani, D.F.; Gaebler, C.; Muecksch, F.; Lorenzi, J.C.C.; Wang, Z.; Cho, A.; Agudelo, M.; Barnes, C.O.; Gazumyan, A.; Finkin, S.; et al. Convergent antibody responses to SARS-CoV-2 in convalescent individuals. Nature 2020, 584, 437–442. [Google Scholar] [CrossRef] [PubMed]
- Vetter, P.; Eckerle, I.; Kaiser, L. COVID-19: A puzzle with many missing pieces. BMJ 2020, 368, m627. [Google Scholar] [CrossRef]
- Arevalo-Rodriguez, I.; Buitrago-Garcia, D.; Simancas-Racines, D.; Zambrano-Achig, P.; Del Campo, R.; Ciapponi, A.; Sued, O.; Martinez-García, L.; Rutjes, A.W.; Low, N.; et al. False-negative results of initial RT-PCR assays for COVID-19: A systematic review. PLoS ONE 2020, 15, e0242958. [Google Scholar] [CrossRef]
- Khan, I.H.; Ravindran, R.; Krishnan, V.V.; Awan, I.N.; Rizvi, S.K.; Saqib, M.A.; Shahzad, M.I.; Tahseen, S.; Ireton, G.; Goulding, C.W.; et al. Plasma antibody profiles as diagnostic biomarkers for tuberculosis. Clin. Vaccine Immunol. 2011, 18, 2148–2153. [Google Scholar] [CrossRef] [PubMed]
- Khaliq, A.; Ravindran, R.; Hussainy, S.F.; Krishnan, V.V.; Ambreen, A.; Yusuf, N.W.; Irum, S.; Rashid, A.; Jamil, M.; Zaffar, F.; et al. Field evaluation of a blood based test for active tuberculosis in endemic settings. PLoS ONE 2017, 12, e0173359. [Google Scholar] [CrossRef] [PubMed]
- Benson, D.A.; Cavanaugh, M.; Clark, K.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Sayers, E.W. GenBank. Nucleic Acids Res. 2013, 41, D36–D42. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ravindran, R.; O’Connor, E.; Gupta, A.; Luciw, P.A.; Khan, A.I.; Dorreh, N.; Chiang, K.; Ikram, A.; Reddy, S. Lipid Mediators and Cytokines/Chemokines Display Differential Profiles in Severe versus Mild/Moderate COVID-19 Patients. Int. J. Mol. Sci. 2023, 24, 13054. https://doi.org/10.3390/ijms241713054
Ravindran R, O’Connor E, Gupta A, Luciw PA, Khan AI, Dorreh N, Chiang K, Ikram A, Reddy S. Lipid Mediators and Cytokines/Chemokines Display Differential Profiles in Severe versus Mild/Moderate COVID-19 Patients. International Journal of Molecular Sciences. 2023; 24(17):13054. https://doi.org/10.3390/ijms241713054
Chicago/Turabian StyleRavindran, Resmi, Ellen O’Connor, Ajay Gupta, Paul A. Luciw, Aleena I. Khan, Nasrin Dorreh, Kate Chiang, Aamer Ikram, and Srinivasa Reddy. 2023. "Lipid Mediators and Cytokines/Chemokines Display Differential Profiles in Severe versus Mild/Moderate COVID-19 Patients" International Journal of Molecular Sciences 24, no. 17: 13054. https://doi.org/10.3390/ijms241713054
APA StyleRavindran, R., O’Connor, E., Gupta, A., Luciw, P. A., Khan, A. I., Dorreh, N., Chiang, K., Ikram, A., & Reddy, S. (2023). Lipid Mediators and Cytokines/Chemokines Display Differential Profiles in Severe versus Mild/Moderate COVID-19 Patients. International Journal of Molecular Sciences, 24(17), 13054. https://doi.org/10.3390/ijms241713054