
Tryptophan Modulation in Cancer-Associated Cachexia Mouse Models
 , ,
, ,
Abstract
1. Introduction
2. Results
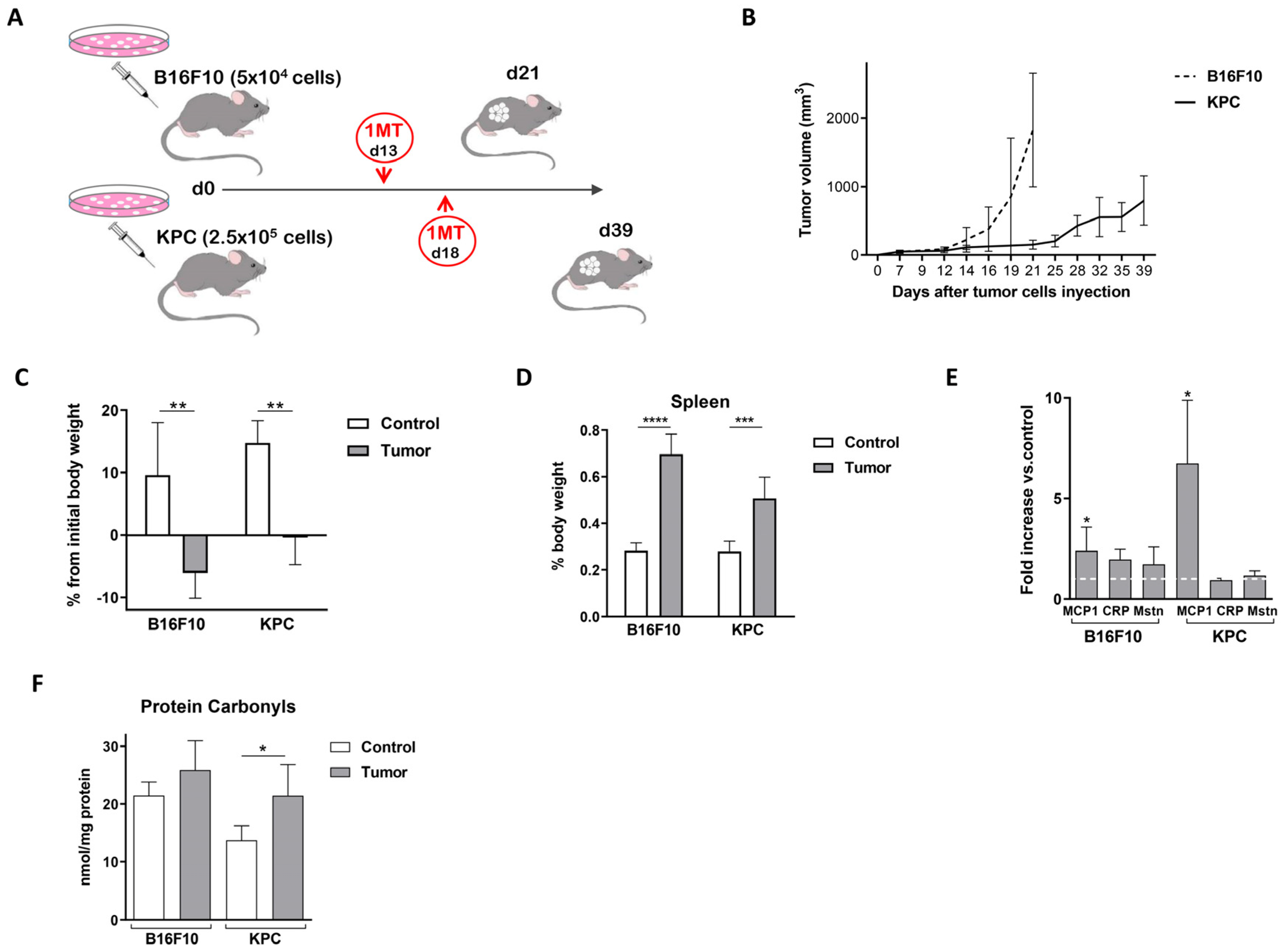
2.1. Murine Models for the Study of Cancer-Associated Cachexia Recapitulate Human Disease
2.2. Plasma of Cachectic Cancer Patients and Tumor-Bearing Mice Showed Reduced Tryptophan Levels
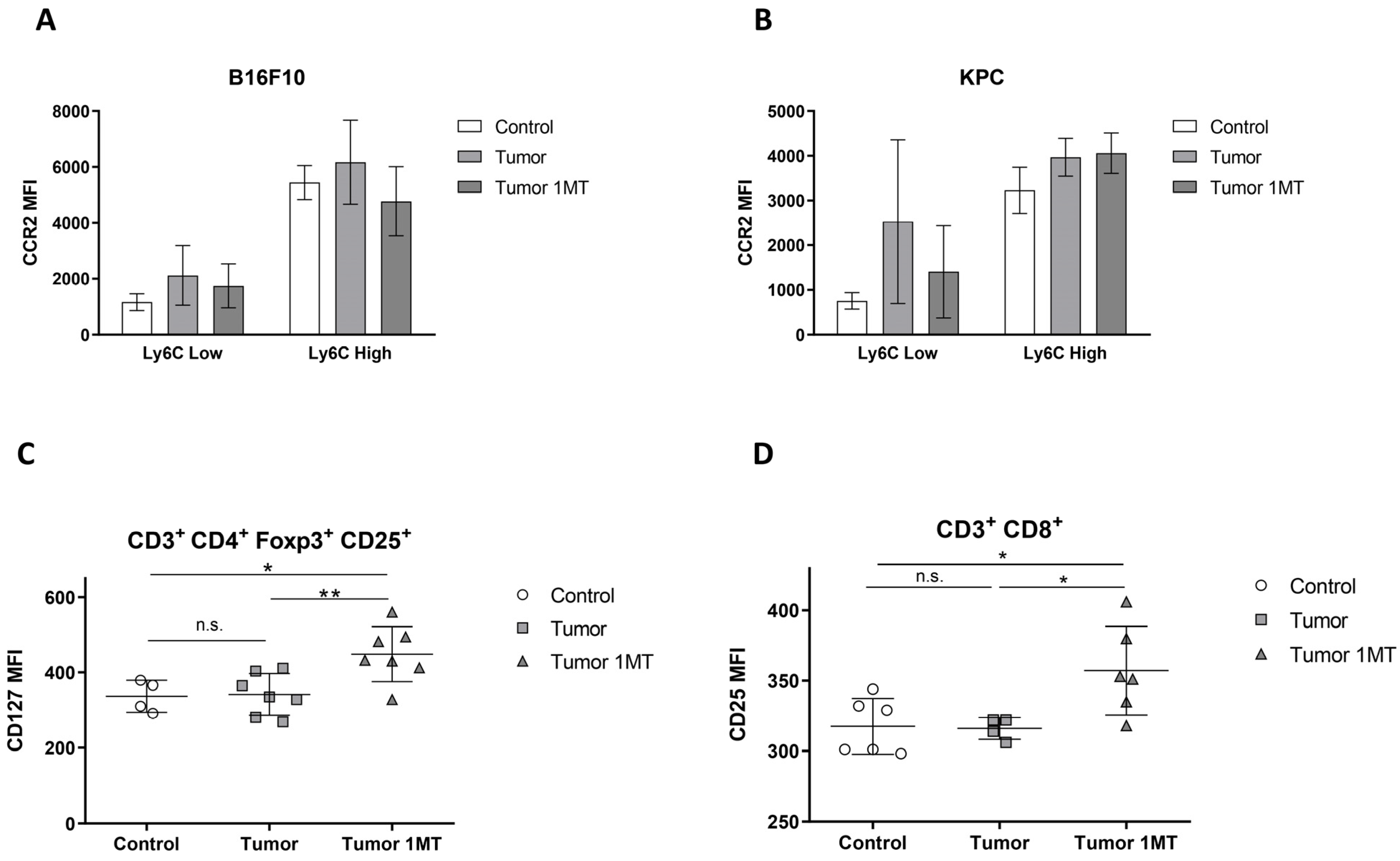
2.3. 1-Methyl-Tryptophan Treatment Modifies Tryptophan Metabolism and Ameliorates Inflammation Signs in Tumor-Bearing Mice
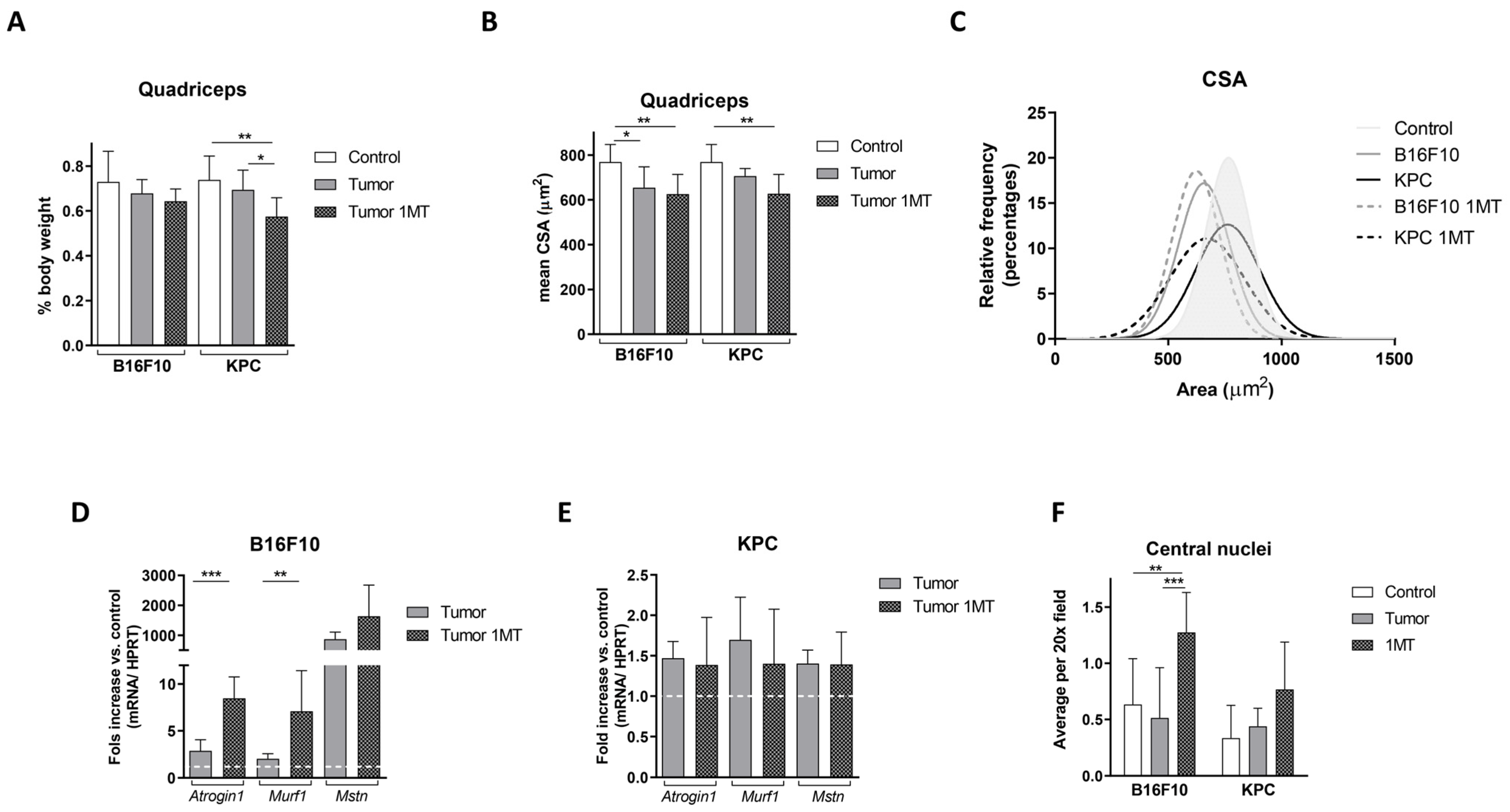
2.4. Muscle Deteriorates with 1-MT Treatment
3. Discussion
4. Materials and Methods
4.1. Patient Cohorts and Samples
4.2. Cell Culture
4.3. Animal Experiments
4.4. 1-Methyl-Tryptophan Administration
4.5. Enzyme-Linked Immunosorbent Assay (ELISA)
4.6. Oxidative Stress Assays
4.7. Reverse Transcriptase Quantitative PCR
4.8. Histology
4.9. Flow Cytometry
4.10. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Baracos, V.E.; Martin, L.; Korc, M.; Guttridge, D.C.; Fearon, K.C.H. Cancer-associated cachexia. Nat. Rev. Dis. Primers 2018, 4, 17105. [Google Scholar] [CrossRef] [PubMed]
- Dolly, A.; Dumas, J.F.; Servais, S. Cancer cachexia and skeletal muscle atrophy in clinical studies: What do we really know? J. Cachexia Sarcopenia Muscle 2020, 11, 1413–1428. [Google Scholar] [CrossRef]
- Marceca, G.P.; Londhe, P.; Calore, F. Management of Cancer Cachexia: Attempting to Develop New Pharmacological Agents for New Effective Therapeutic Options. Front. Oncol. 2020, 10, 298. [Google Scholar] [CrossRef] [PubMed]
- Siff, T.; Parajuli, P.; Razzaque, M.S.; Atfi, A. Cancer-Mediated Muscle Cachexia: Etiology and Clinical Management. Trends Endocrinol. Metab. 2021, 32, 382–402. [Google Scholar] [CrossRef]
- Fearon, K.C. Cancer cachexia and fat-muscle physiology. N. Engl. J. Med. 2011, 365, 565–567. [Google Scholar] [CrossRef]
- Argiles, J.M.; Lopez-Soriano, F.J.; Stemmler, B.; Busquets, S. Therapeutic strategies against cancer cachexia. Eur. J. Transl. Myol. 2019, 29, 7960. [Google Scholar] [CrossRef] [PubMed]
- McKeaveney, C.; Maxwell, P.; Noble, H.; Reid, J. A Critical Review of Multimodal Interventions for Cachexia. Adv. Nutr. 2021, 12, 523–532. [Google Scholar] [CrossRef]
- Argiles, J.M.; Lopez-Soriano, F.J.; Stemmler, B.; Busquets, S. Cancer-associated cachexia—Understanding the tumour macroenvironment and microenvironment to improve management. Nat. Rev. Clin. Oncol. 2023, 20, 250–264. [Google Scholar] [CrossRef]
- Cala, M.P.; Agullo-Ortuno, M.T.; Prieto-Garcia, E.; Gonzalez-Riano, C.; Parrilla-Rubio, L.; Barbas, C.; Díaz-García, C.V.; García, A.; Pernaut, C.; Adeva, J.; et al. Multiplatform plasma fingerprinting in cancer cachexia: A pilot observational and translational study. J. Cachexia Sarcopenia Muscle 2018, 9, 348–357. [Google Scholar] [CrossRef]
- Platten, M.; Nollen, E.A.A.; Rohrig, U.F.; Fallarino, F.; Opitz, C.A. Tryptophan metabolism as a common therapeutic target in cancer, neurodegeneration and beyond. Nat. Rev. Drug Discov. 2019, 18, 379–401. [Google Scholar] [CrossRef]
- Lanser, L.; Kink, P.; Egger, E.M.; Willenbacher, W.; Fuchs, D.; Weiss, G.; Kurz, K. Inflammation-Induced Tryptophan Breakdown is Related With Anemia, Fatigue, and Depression in Cancer. Front. Immunol. 2020, 11, 249. [Google Scholar] [CrossRef] [PubMed]
- Laviano, A.; Meguid, M.M.; Cascino, A.; Molfino, A.; Rossi Fanelli, F. Tryptophan in wasting diseases: At the crossing between immune function and behaviour. Curr. Opin. Clin. Nutr. Metab. Care 2009, 12, 392–397. [Google Scholar] [CrossRef] [PubMed]
- Ninomiya, S.; Nakamura, N.; Nakamura, H.; Mizutani, T.; Kaneda, Y.; Yamaguchi, K.; Matsumoto, T.; Kitagawa, J.; Kanemura, N.; Shiraki, M.; et al. Low Levels of Serum Tryptophan Underlie Skeletal Muscle Atrophy. Nutrients 2020, 12, 978. [Google Scholar] [CrossRef] [PubMed]
- Le Naour, J.; Galluzzi, L.; Zitvogel, L.; Kroemer, G.; Vacchelli, E. Trial watch: IDO inhibitors in cancer therapy. Oncoimmunology 2020, 9, 1777625. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Wang, J.; Yu, S. IDO in Colorectal Tumorigenesis: Involvement of Immune Tolerance and Significance in Prevention and Therapy. Cell. Mol. Gastroenterol. Hepatol. 2021, 12, 1503–1504. [Google Scholar] [CrossRef]
- Long, G.V.; Dummer, R.; Hamid, O.; Gajewski, T.F.; Caglevic, C.; Dalle, S.; Arance, A.; Carlino, M.S.; Grob, J.-J.; Kim, T.M.; et al. Epacadostat plus pembrolizumab versus placebo plus pembrolizumab in patients with unresectable or metastatic melanoma (ECHO-301/KEYNOTE-252): A phase 3, randomised, double-blind study. Lancet Oncol. 2019, 20, 1083–1097. [Google Scholar] [CrossRef]
- Talbert, E.; Lewis, H.L.; Farren, M.R.; Ramsey, M.L.; Chakedis, J.M.; Rajasekera, P.; Haverick, E.; Sarna, A.; Bloomston, M.; Pawlik, T.M.; et al. Circulating monocyte chemoattractant protein-1 (MCP-1) is associated with cachexia in treatment-naive pancreatic cancer patients. J. Cachexia Sarcopenia Muscle 2018, 9, 358–368. [Google Scholar] [CrossRef]
- Ju, J.E.; Kim, M.-S.; Kang, J.H.; Lee, J.Y.; Lee, M.S.; Kim, E.H.; Chung, N.; Jeong, Y.K. Potential role of immunological factors in early diagnosis of cancer cachexia in C26 tumor-bearing mice. Appl. Biol. Chem. 2019, 62, 1–9. [Google Scholar] [CrossRef]
- Baczek, J.; Silkiewicz, M.; Wojszel, Z.B. Myostatin as a Biomarker of Muscle Wasting and other Pathologies-State of the Art and Knowledge Gaps. Nutrients 2020, 12, 2401. [Google Scholar] [CrossRef]
- Martin, A.; Gallot, Y.S.; Freyssenet, D. Molecular mechanisms of cancer cachexia-related loss of skeletal muscle mass: Data analysis from preclinical and clinical studies. J. Cachexia Sarcopenia Muscle 2023, 14, 1150–1167. [Google Scholar] [CrossRef]
- Akagawa, M. Protein carbonylation: Molecular mechanisms, biological implications, and analytical approaches. Free. Radic. Res. 2021, 55, 307–320. [Google Scholar] [CrossRef] [PubMed]
- Ábrigo, J.; Elorza, A.A.; Riedel, C.A.; Vilos, C.; Simon, F.; Cabrera, D.; Estrada, L.; Cabello-Verrugio, C. Role of Oxidative Stress as Key Regulator of Muscle Wasting during Cachexia. Oxidative Med. Cell. Longev. 2018, 2018, 2063179. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.; Schweikart, K.; Tomaszewski, J.; Page, J.G.; Noker, P.E.; Buhrow, S.A.; Reid, J.M.; Ames, M.M.; Munn, D.H. Toxicology and pharmacokinetics of 1-methyl-d-tryptophan: Absence of toxicity due to saturating absorption. Food Chem. Toxicol. 2008, 46, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Divanovic, S.; Sawtell, N.M.; Trompette, A.; Warning, J.I.; Dias, A.; Cooper, A.M.; Yap, G.S.; Arditi, M.; Shimada, K.; DuHadaway, J.B.; et al. Opposing Biological Functions of Tryptophan Catabolizing Enzymes During Intracellular Infection. J. Infect. Dis. 2011, 205, 152–161. [Google Scholar] [CrossRef]
- Chiarella, P.; Vulcano, M.; Bruzzo, J.; Vermeulen, M.; Vanzulli, S.; Maglioco, A.; Camerano, G.; Palacios, V.; Fernández, G.; Brando, R.F.; et al. Anti-inflammatory pretreatment enables an efficient dendritic cell-based immunotherapy against established tumors. Cancer Immunol. Immunother. 2007, 57, 701–718. [Google Scholar] [CrossRef]
- Taillandier, D.; Polge, C. Skeletal muscle atrogenes: From rodent models to human pathologies. Biochimie 2019, 166, 251–269. [Google Scholar] [CrossRef]
- Cervenka, I.; Agudelo, L.Z.; Ruas, J.L. Kynurenines: Tryptophan’s metabolites in exercise, inflammation, and mental health. Science 2017, 357, eaaf9794. [Google Scholar] [CrossRef]
- Holmgaard, R.B.; Zamarin, D.; Munn, D.H.; Wolchok, J.D.; Allison, J.P. Indoleamine 2,3-dioxygenase is a critical resistance mechanism in antitumor T cell immunotherapy targeting CTLA-4. J. Exp. Med. 2013, 210, 1389–1402. [Google Scholar] [CrossRef]
- Takamatsu, M.; Hirata, A.; Ohtaki, H.; Hoshi, M.; Ando, T.; Ito, H.; Hatano, Y.; Tomita, H.; Kuno, T.; Saito, K.; et al. Inhibition of indoleamine 2,3-dioxygenase 1 expression alters immune response in colon tumor microenvironment in mice. Cancer Sci. 2015, 106, 1008–1015. [Google Scholar] [CrossRef]
- Kiank, C.; Zeden, J.-P.; Drude, S.; Domanska, G.; Fusch, G.; Otten, W.; Schuett, C. Psychological Stress-Induced, IDO1-Dependent Tryptophan Catabolism: Implications on Immunosuppression in Mice and Humans. PLoS ONE 2010, 5, e11825. [Google Scholar] [CrossRef]
- Gomez-Cabrera, M.-C.; Domenech, E.; Viña, J. Moderate exercise is an antioxidant: Upregulation of antioxidant genes by training. Free. Radic. Biol. Med. 2008, 44, 126–131. [Google Scholar] [CrossRef] [PubMed]
- Ristow, M.; Zarse, K.; Oberbach, A.; Klöting, N.; Birringer, M.; Kiehntopf, M.; Stumvoll, M.; Kahn, C.R.; Blüher, M. Antioxidants prevent health-promoting effects of physical exercise in humans. Proc. Natl. Acad. Sci. USA 2009, 106, 8665–8670. [Google Scholar] [CrossRef] [PubMed]
- Laviano, A.; Meguid, M.M.; Preziosa, I.; Fanelli, F.R. Oxidative stress and wasting in cancer. Curr. Opin. Clin. Nutr. Metab. Care 2007, 10, 449–456. [Google Scholar] [CrossRef] [PubMed]
- Petermann, M.; Orfanos, Z.; Sellau, J.; Gharaibeh, M.; Lotter, H.; Fleischer, B.; Keller, C. CCR2 Deficiency Impairs Ly6Clo and Ly6Chi Monocyte Responses in Orientia tsutsugamushi Infection. Front. Immunol. 2021, 12, 670219. [Google Scholar] [CrossRef] [PubMed]
- Hao, Q.; Vadgama, J.V.; Wang, P. CCL2/CCR2 signaling in cancer pathogenesis. Cell Commun. Signal. 2020, 18, 82. [Google Scholar] [CrossRef] [PubMed]
- Kadomoto, S.; Izumi, K.; Mizokami, A. Roles of CCL2-CCR2 Axis in the Tumor Microenvironment. Int. J. Mol. Sci. 2021, 22, 8530. [Google Scholar] [CrossRef]
- Burfeind, K.G.; Zhu, X.; Norgard, M.A.; Levasseur, P.R.; Huisman, C.; Buenafe, A.C.; Olson, B.; Michaelis, K.A.; Torres, E.R.S.; Jeng, S.; et al. Circulating myeloid cells invade the central nervous system to mediate cachexia during pancreatic cancer. eLife 2020, 9, e54095. [Google Scholar] [CrossRef]
- Ohue, Y.; Nishikawa, H. Regulatory T (Treg) cells in cancer: Can Treg cells be a new therapeutic target? Cancer Sci. 2019, 110, 2080–2089. [Google Scholar] [CrossRef]
- Liu, W.; Putnam, A.L.; Xu-Yu, Z.; Szot, G.L.; Lee, M.R.; Zhu, S.; Gottlieb, P.A.; Kapranov, P.; Gingeras, T.R.; de St Groth, B.F.; et al. CD127 expression inversely correlates with FoxP3 and suppressive function of human CD4+ T reg cells. J. Exp. Med. 2006, 203, 1701–1711. [Google Scholar] [CrossRef]
- Létourneau, S.; Krieg, C.; Pantaleo, G.; Boyman, O. IL-2– and CD25-dependent immunoregulatory mechanisms in the homeostasis of T-cell subsets. J. Allergy Clin. Immunol. 2009, 123, 758–762. [Google Scholar] [CrossRef]
- Hawkins, C.L.; Morgan, P.E.; Davies, M.J. Quantification of protein modification by oxidants. Free. Radic. Biol. Med. 2009, 46, 965–988. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| qPCR Primers | Forward | Reverse |
|---|---|---|
| HPRT | GGCCAGACTTTGTTGGATTTG | TGCGCTATCTTAGGCTTTGT |
| Atrogin1 | AGATTCGCAAGCGTTTGATC | GGGAAAGTGAGACGGAGCAG |
| Murf1 | ATTGTAGAAGCCTCCAAGGG | GGTGTTCTTCTTTACCCTCTGTG |
| Myostatin | AGTGGATCTAAATGAGGGCAGT | GTTTCCAGGCGCAGCTTAC |
| Monocytes Panel | Manufacturer | Lymphocytes Panel | Manufacturer |
|---|---|---|---|
| anti-CD11b-APC Fire | BioLegend, San Diego, CA, USA | anti-CD4-APC Fire | BioLegend |
| anti-Ly6C-PerCP | BioLegend | anti-CD3-PE | BioLegend |
| anti-Ly6G-FITC | BioLegend | anti-CD25-APC | BioLegend |
| anti-CD43-APC | BioLegend | anti-CD127/IL7Rα BV421 | BioLegend |
| anti-CCR2-PE | BioLegend | anti-CD8-PerCP | BioLegend |
| anti-CX3CR1-BV421 | Miltenyi Biotech, San Diego, CA, USA | anti-Foxp3-FITC | eBioscience, Waltham, MA, USA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Agulló-Ortuño, M.T.; Mancebo, E.; Grau, M.; Núñez Sobrino, J.A.; Paz-Ares, L.; López-Martín, J.A.; Flández, M. Tryptophan Modulation in Cancer-Associated Cachexia Mouse Models. Int. J. Mol. Sci. 2023, 24, 13005. https://doi.org/10.3390/ijms241613005
Agulló-Ortuño MT, Mancebo E, Grau M, Núñez Sobrino JA, Paz-Ares L, López-Martín JA, Flández M. Tryptophan Modulation in Cancer-Associated Cachexia Mouse Models. International Journal of Molecular Sciences. 2023; 24(16):13005. https://doi.org/10.3390/ijms241613005
Chicago/Turabian StyleAgulló-Ortuño, M. Teresa, Esther Mancebo, Montserrat Grau, Juan Antonio Núñez Sobrino, Luis Paz-Ares, José A. López-Martín, and Marta Flández. 2023. "Tryptophan Modulation in Cancer-Associated Cachexia Mouse Models" International Journal of Molecular Sciences 24, no. 16: 13005. https://doi.org/10.3390/ijms241613005
APA StyleAgulló-Ortuño, M. T., Mancebo, E., Grau, M., Núñez Sobrino, J. A., Paz-Ares, L., López-Martín, J. A., & Flández, M. (2023). Tryptophan Modulation in Cancer-Associated Cachexia Mouse Models. International Journal of Molecular Sciences, 24(16), 13005. https://doi.org/10.3390/ijms241613005