

Specific Proton-Donor Properties of Glycine Betaine. Metric Parameters and Enthalpy of Noncovalent Interactions in its Dimer, Water Complexes and Crystalline Hydrate

Abstract
:1. Introduction
2. Results
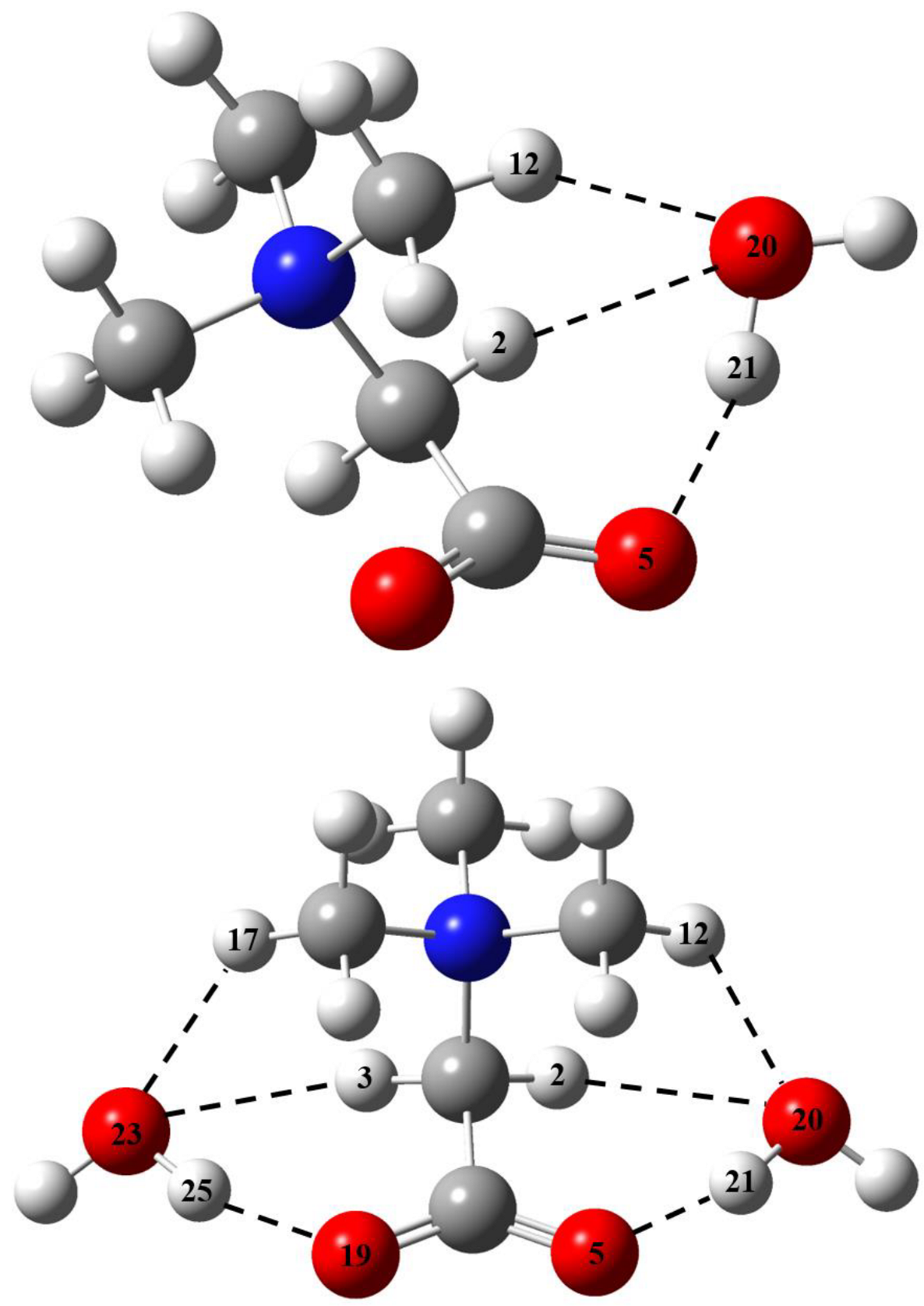
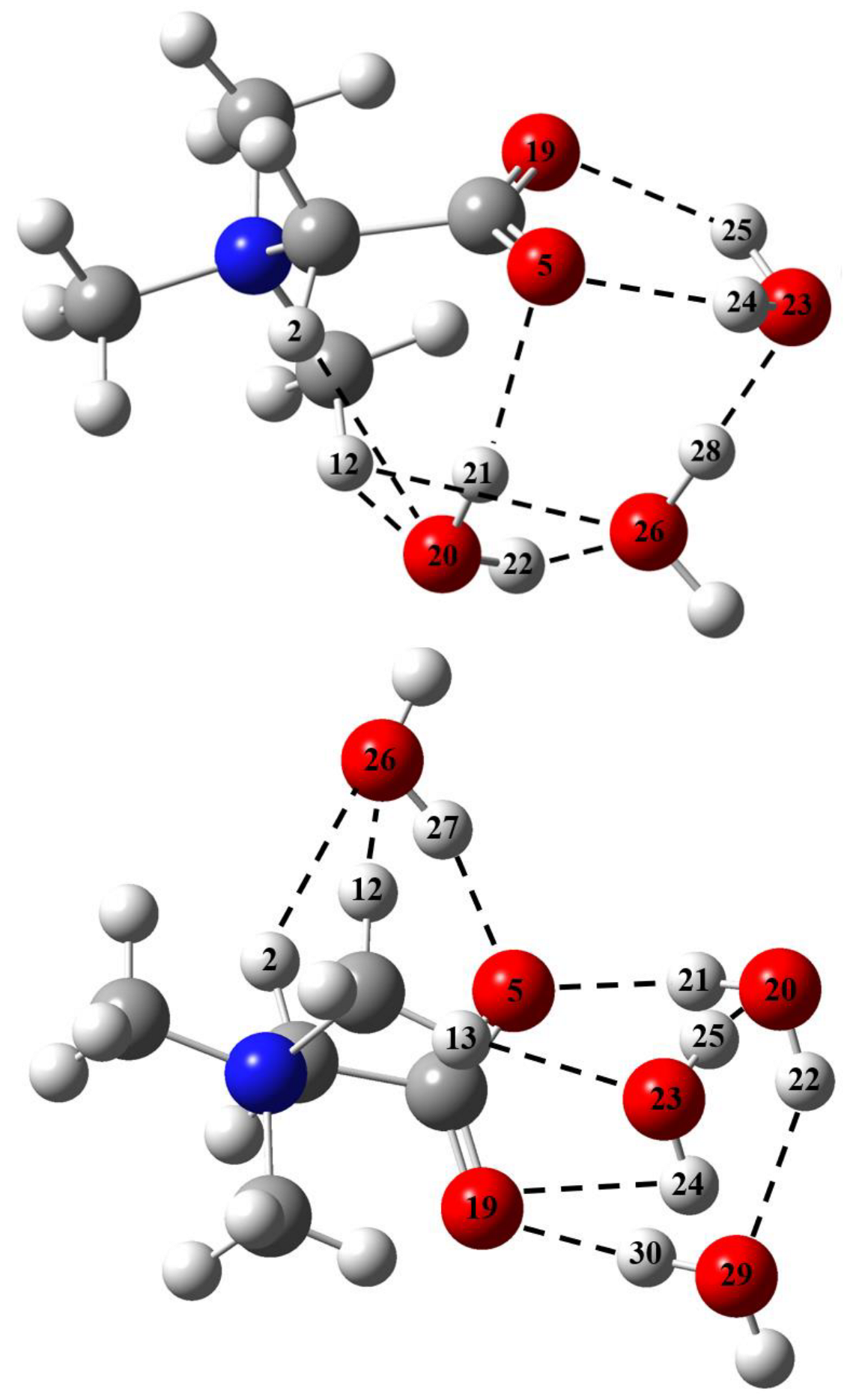
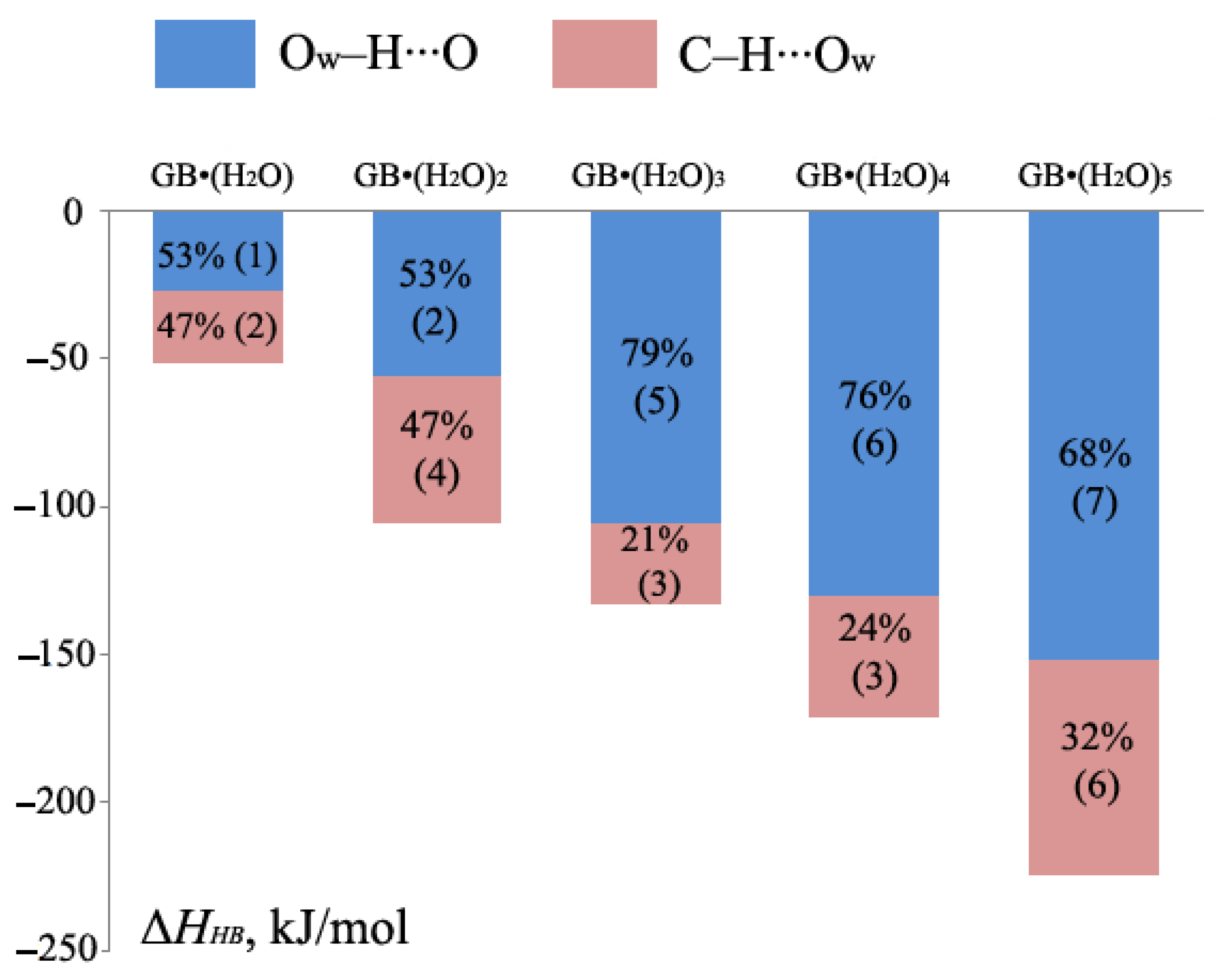
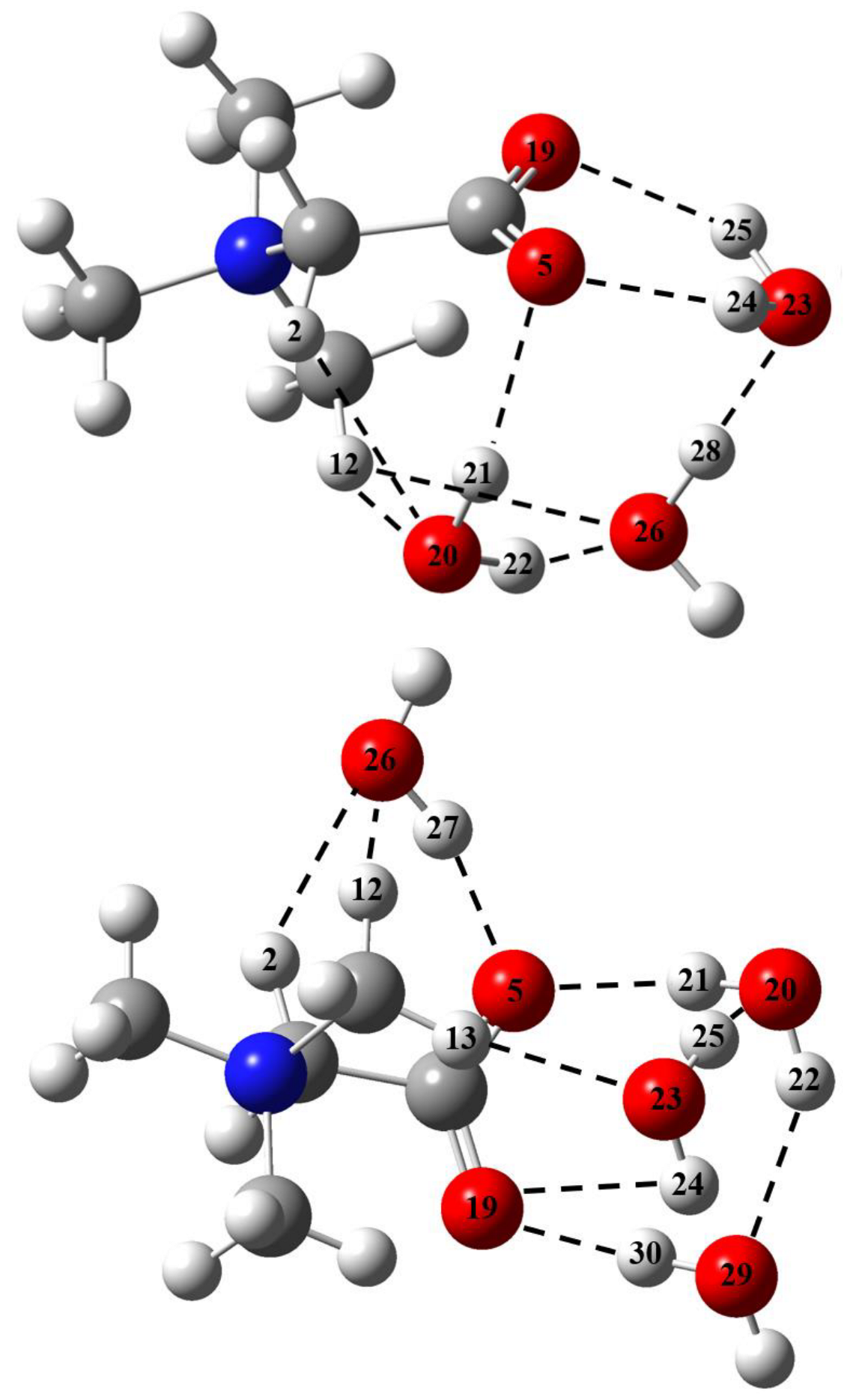
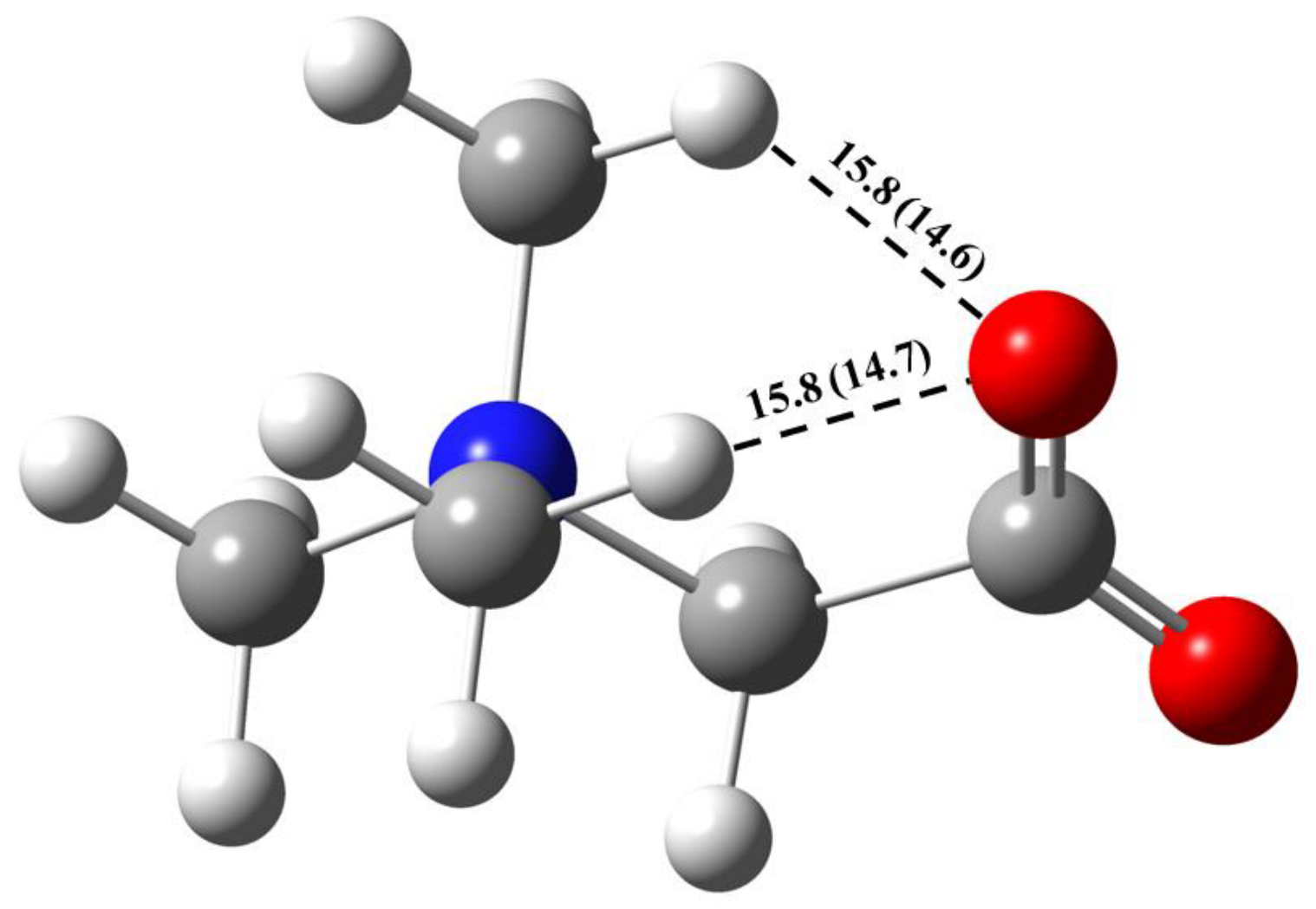
2.1. Metric Parameters and Enthalpy of Intermolecular H-Bonds in Complexes of GB with Water
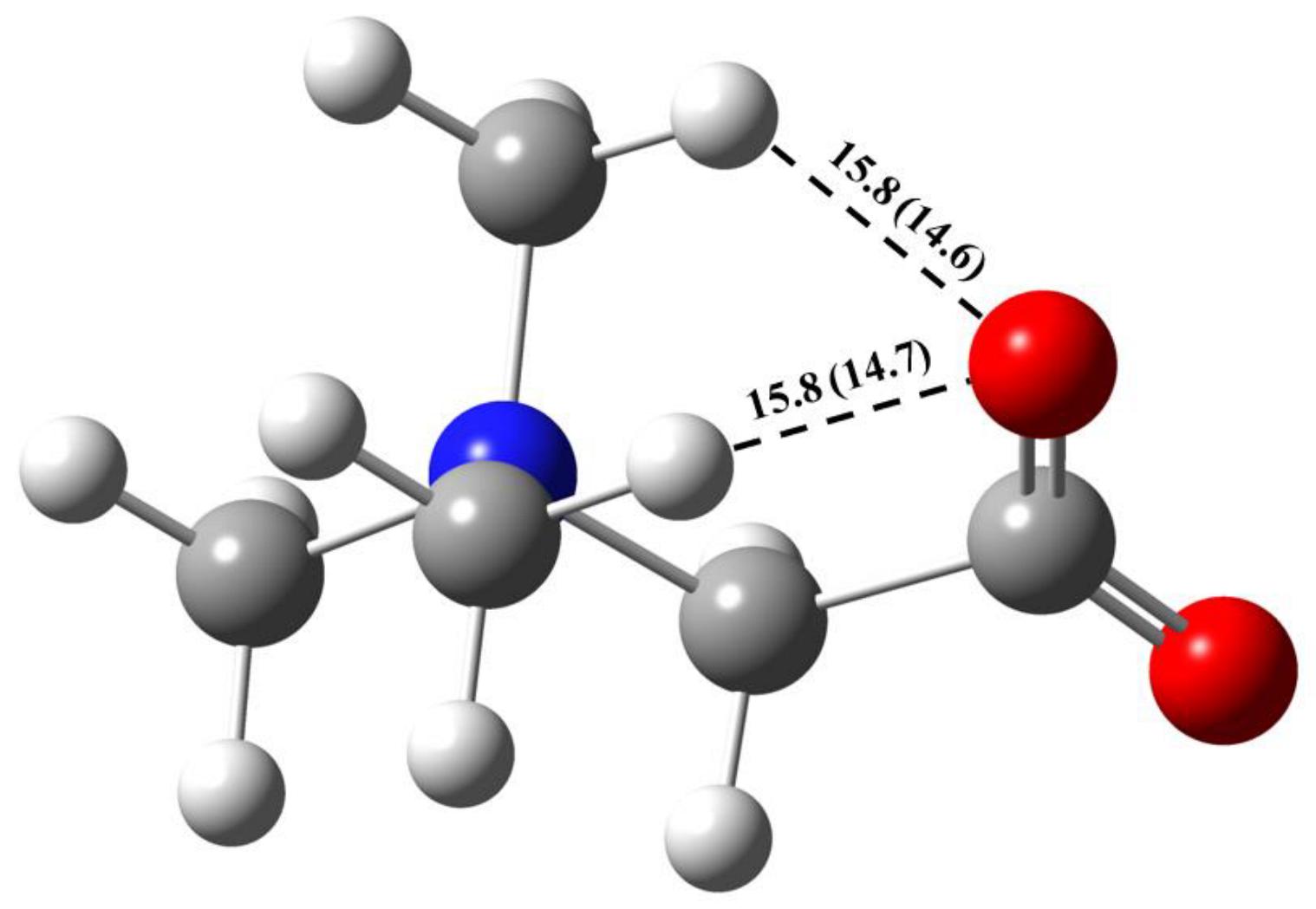
2.2. GB Dimer in Vacuum
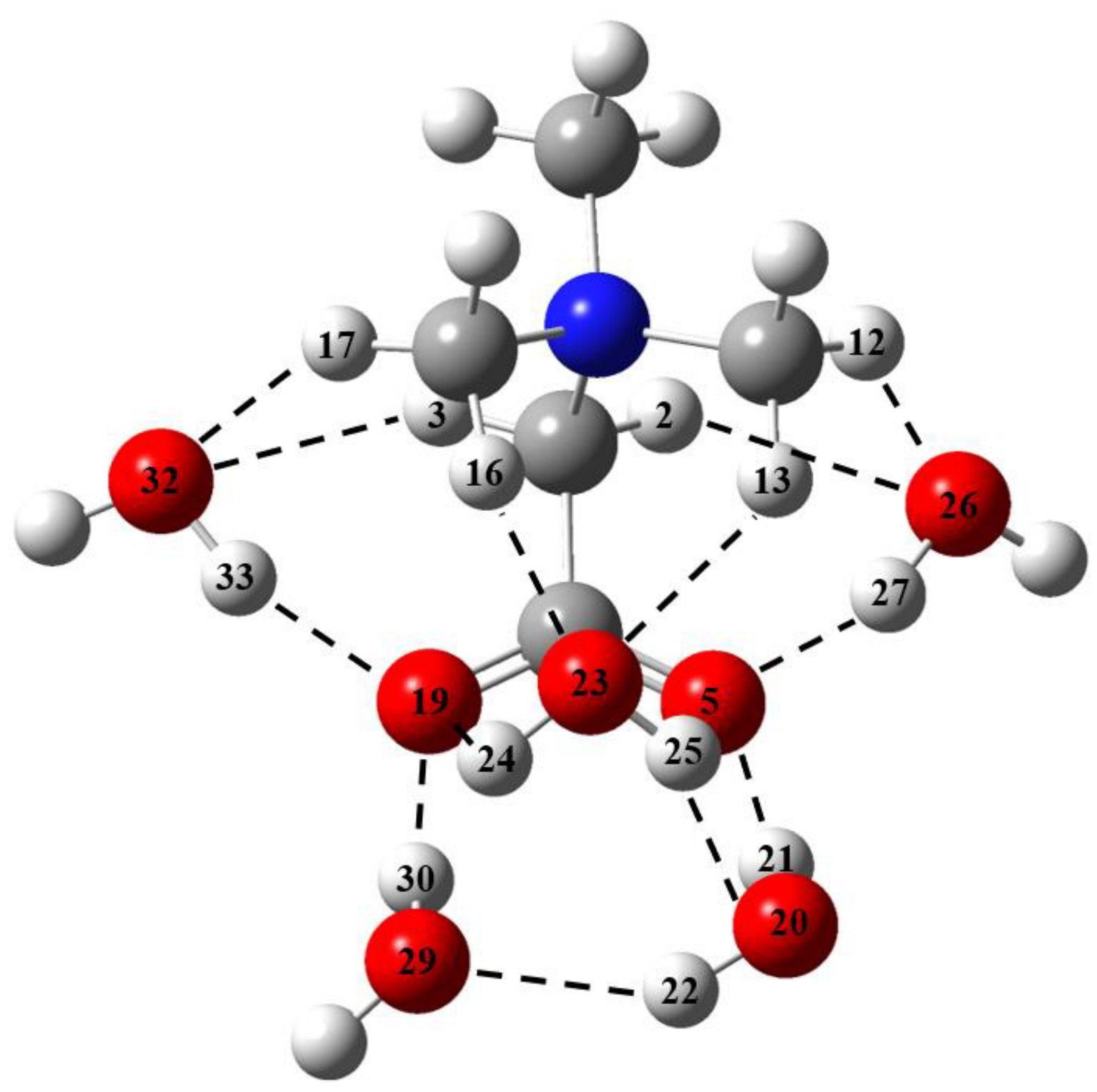
2.3. The GB Crystalline Monohydrate
- (1)
- In the GB crystalline monohydrate, there is a significant strengthening of classical H-bonds in comparison with GB–water complexes in the gas phase;
- (2)
- The number of C–H∙∙∙Ow bonds in the crystalline monohydrate also increases in comparison with the GB–water complexes in the gas phase.
- (1)
- In the GB crystalline monohydrate, the energy of the C–H∙∙∙O bond does not change significantly compared to the corresponding bonds in the GB dimer in the gas phase;
- (2)
- The number of intermolecular bonds in a crystal is much larger than in a dimer. This is due to the fact that in the considered crystal, the GB and water molecules interact with several neighboring GB molecules.
3. Discussion
4. Materials and Methods
4.1. Non-Periodic DFT Computations
4.2. Periodic DFT Computations
4.3. Evaluation of the Enthalpy/energy of Intermolecular H-Bonds and Noncovalent Interactions
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ashraf, M.; Foolad, M.R. Roles of glycine betaine and proline in improving plant abiotic stress resistance. Environ. Exp. Bot. 2007, 59, 206–216. [Google Scholar] [CrossRef]
- El Achkar, T.; Fourmentin, S.; Greige-Gerges, H. Deep eutectic solvents: An overview on their interactions with water and biochemical compounds. J. Mol. Liq. 2019, 288, 111028. [Google Scholar] [CrossRef]
- Figueroa-Soto, C.G.; Valenzuela-Soto, E.M. Glycine betaine rather than acting only as an osmolyte also plays a role as regulator in cellular metabolism. Biochimie 2018, 147, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Di Gioacchino, M.; Bruni, F.; Ricci, M.A. Aqueous solution of betaine: Hydration and aggregation. J. Mol. Liq. 2020, 318, 114253. [Google Scholar] [CrossRef]
- Ohsawa, S.; Tokushima, T.; Okada, K. Hydration of the zwitterionic and protonated forms of glycine betaine probed by soft X-ray emission spectroscopy coupled with chemometrics. J. Phys. Chem. B 2021, 125, 1881–1887. [Google Scholar] [CrossRef] [PubMed]
- Stasiulewicz, M.; Panuszko, A.; Śmiechowski, M.; Bruździak, P.; Maszota, P.; Stangret, J. Effect of urea and glycine betaine on the hydration sphere of model molecules for the surface features of proteins. J. Mol. Liq. 2021, 324, 115090. [Google Scholar] [CrossRef]
- Novak, I. Photoionization of glycine-betaine: A permanent zwitterion. J. Mol. Liq. 2021, 335, 116558. [Google Scholar] [CrossRef]
- Fedotova, M.V.; Kruchinin, S.E. Hydration and ion-binding of glycine betaine: How they may be involved in the protection of proteins under abiotic stresses. J. Mol. Liq. 2017, 244, 489–498. [Google Scholar] [CrossRef]
- Mojtabavi, S.; Jafari, M.; Samadi, N.; Mehrnejad, F.; Faramarzi, M.A. Insights into the Molecular-Level details of betaine interactions with Laccase under various thermal conditions. J. Mol. Liq. 2021, 339, 116832. [Google Scholar] [CrossRef]
- Jiang, L.; Zheng, K. Electronic structures of zwitterionic and protonated forms of glycine betaine in water: Insights into solvent effects from ab initio simulations. J. Mol. Liq. 2022, 369, 120871. [Google Scholar] [CrossRef]
- Derbel, N.; Hernandez, B.; Pfluger, F.; Liquier, J.; Geinguenaud, F.; Jaidane, N.; Lakhdar, Z.B.; Ghomi, M. Vibrational Analysis of Amino Acids and Short Peptides in Hydrated Media. I. L-glycine and L-leucine. J. Phys. Chem. B 2007, 111, 1470–1477. [Google Scholar] [CrossRef]
- Panuszko, A.; Śmiechowski, M.; Stangret, J. Fourier transform infrared spectroscopic and theoretical study of water interactions with glycine and its N-methylated derivatives. J. Chem. Phys. 2011, 134, 115104. [Google Scholar] [CrossRef]
- Kayi, H.; Kaiser, R.I.; Head, J.D. A theoretical investigation of the relative stability of hydrated glycine and methylcarbamic acid—From water clusters to interstellar ices. Phys. Chem. Chem. Phys. 2012, 14, 4942–4958. [Google Scholar] [CrossRef] [PubMed]
- de Tudela, R.P.; Marx, D. Water-induced Zwitterionization of Glycine: Stabilization Mechanism and Spectral Signatures. J. Phys. Chem. Lett. 2016, 7, 5137–5142. [Google Scholar] [CrossRef] [PubMed]
- Fedotova, M.V.; Kruchinin, S.E. 1D-RISM study of glycine zwitterion hydration and ion-molecular complex formation in aqueous NaCl solutions. J. Mol. Liq. 2012, 169, 1–7. [Google Scholar] [CrossRef]
- Rabdano, S.O.; Donets, A.V.; Vovk, M.A.; Michel, D.; Chizhik, V.I. “Hydration Shells” of CH2 Groups of ω-Amino Acids as Studied by Deuteron NMR Relaxation. J. Phys. Chem. B 2015, 119, 13358–13366. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Gao, J. Monte Carlo simulations of the hydration of ammonium and carboxylate ions. J. Phys. Chem. 1986, 90, 2174–2182. [Google Scholar] [CrossRef]
- Gojło, E.; Śmiechowski, M.; Panuszko, A.; Stangret, J. Hydration of Carboxylate Anions: Infrared Spectroscopy of Aqueous Solutions. J. Phys. Chem. B 2009, 113, 8128–8136. [Google Scholar] [CrossRef]
- Del Val, C.; Bondar, A.N. Charged groups at binding interfaces of the PsbO subunit of photosystem II: A combined bioinformatics and simulation study. Biochim. Biophys. Acta Bioenerg. 2017, 1858, 432–441. [Google Scholar] [CrossRef] [PubMed]
- Levina, E.O.; Penkov, N.V.; Rodionova, N.N.; Tarasov, S.A.; Barykina, D.V.; Vener, M.V. Hydration of the Carboxylate Group in Anti-Inflammatory Drugs: ATR-IR and Computational Studies of Aqueous Solution of Sodium Diclofenac. ACS Omega 2018, 3, 302–313. [Google Scholar] [CrossRef]
- Vener, M.V.; Makhrov, D.E.; Voronin, A.P.; Shalafan, D.R. Molecular Dynamics Simulation of Association Processes in Aqueous Solutions of Maleate Salts of Drug-like Compounds: The Role of Counterion. Int. J. Mol. Sci. 2022, 23, 6302. [Google Scholar] [CrossRef]
- Jiang, L.; Lai, L. C–H∙∙∙O Hydrogen Bonds at Protein-Protein Interfaces. J. Biol. Chem. 2002, 277, 37732–37740. [Google Scholar] [CrossRef]
- McConnell, D.B. Biotin’s Lessons in Drug Design. J. Med. Chem. 2021, 64, 16319–16327. [Google Scholar] [CrossRef] [PubMed]
- Surov, A.O.; Voronin, A.P.; Vener, M.V.; Churakov, A.V.; Perlovich, G.L. Specific features of supramolecular organisation and hydrogen bonding in proline cocrystals: A case study of fenamates and diclofenac. CrystEngComm 2018, 20, 6970–6981. [Google Scholar] [CrossRef]
- Voronin, A.P.; Surov, A.O.; Churakov, A.V.; Parashchuk, O.D.; Rykounov, A.A.; Vener, M.V. Combined X-ray Crystallographic, IR/Raman Spectroscopic, and Periodic DFT Investigations of New Multicomponent Crystalline Forms of Anthelmintic Drugs: A Case Study of Carbendazim Maleate. Molecules 2020, 25, 2386. [Google Scholar] [CrossRef] [PubMed]
- Červinka, C.; Fulem, M. Cohesive properties of the crystalline phases of twenty proteinogenic α-aminoacids from first-principles calculations. Phys. Chem. Chem. Phys. 2019, 21, 18501–18515. [Google Scholar] [CrossRef]
- Bader, R.F.W. A Quantum Theory of Molecular Structure and Its Applications. Chem. Rev. 1991, 91, 893–928. [Google Scholar] [CrossRef]
- Rozenberg, M.; Loewenschuss, A.; Marcus, Y. An Empirical Correlation Between Stretching Vibration Redshift and Hydrogen Bond Length. Phys. Chem. Chem. Phys. 2000, 2, 2699–2702. [Google Scholar] [CrossRef]
- Mata, I.; Alkorta, I.; Espinosa, E.; Molins, E. Relationships between Interaction Energy, Intermolecular Distance and Electron Density Properties in Hydrogen Bonded Complexes under External Electric Fields. Chem. Phys. Lett. 2011, 507, 185–189. [Google Scholar] [CrossRef]
- Vener, M.V.; Egorova, A.N.; Churakov, A.V.; Tsirelson, V.G. Intermolecular Hydrogen Bond Energies in Crystals Evaluated Using Electron Density Properties: DFT Computations with Periodic Boundary Conditions. J. Comput. Chem. 2012, 33, 2303–2309. [Google Scholar] [CrossRef]
- Kaczmarek, D.K.; Parus, A.; Łozynski, M.; Pernak, J. Use of ammonium salts or binary mixtures derived from amino acids, glycine betaine, choline and indole-3-butyric acid as plant regulators. RSC Adv. 2020, 10, 43058–43065. [Google Scholar] [CrossRef] [PubMed]
- Aikens, C.M.; Gordon, M.S. Incremental Solvation of Nonionized and Zwitterionic Glycine. J. Am. Chem. Soc. 2006, 128, 12835–12850. [Google Scholar] [CrossRef] [PubMed]
- Markham, G.D.; Bock, C.L.; Book, C.W. Hydration of the Carboxylate Group: An ab Initio Molecular Orbital Study of Acetate-Water Complexes. Struct. Chem. 1997, 8, 293–307. [Google Scholar] [CrossRef]
- Sironi, M.; Fornilia, A.; Fornili, S.L. Water interaction with glycine betaine: A hybrid QM/MM molecular dynamics simulation. Phys. Chem. Chem. Phys. 2001, 3, 1081–1085. [Google Scholar] [CrossRef]
- Lee, H.; Choi, J.H.; Verma, P.K.; Cho, M. Spectral Graph Analyses of Water Hydrogen-Bonding Network and Osmolyte Aggregate Structures in Osmolyte−Water Solutions. J. Phys. Chem. B 2015, 119, 14402–14412. [Google Scholar] [CrossRef] [PubMed]
- Steiner, T. The Hydrogen Bond in the Solid State. Angew. Chem. Int. Ed. 2002, 41, 48–76. [Google Scholar] [CrossRef]
- Gillon, A.L.; Feeder, N.; Davey, R.J.; Storey, R. Hydration in Molecular Crystals-A Cambridge Structural Database Analysis. Cryst. Growth Des. 2003, 3, 663–673. [Google Scholar] [CrossRef]
- Infantes, L.; Fabian, L.; Motherwell, W.D.S. Organic crystal hydrates: What are the important factors for formation. CrystEngComm 2007, 9, 65–71. [Google Scholar] [CrossRef]
- Meot-Ner, M. The Ionic Hydrogen Bond. Chem. Rev. 2005, 105, 213–284. [Google Scholar] [CrossRef] [PubMed]
- Legon, A.C. The Halogen Bond: An Interim Perspective. Phys. Chem. Chem. Phys. 2010, 12, 7736. [Google Scholar] [CrossRef]
- Baiz, C.R.; Błasiak, B.; Bredenbeck, J.; Cho, M.; Choi, J.H.; Corcelli, S.A.; Dijkstra, A.G.; Feng, C.J.; Garrett-Roe, S.; Ge, N.H.; et al. Vibrational Spectroscopic Map, Vibrational Spectroscopy, and Intermolecular Interaction. Chem. Rev. 2020, 120, 7152–7218. [Google Scholar] [CrossRef] [PubMed]
- Sheiner, S. The Pnicogen Bond: Its Relation to Hydrogen, Halogen, and Other Noncovalent Bonds. Acc. Chem. Res. 2012, 46, 280–288. [Google Scholar] [CrossRef] [PubMed]
- Verevkin, S.P.; Kondratev, S.O.; Zaitsau, D.H.; Zherikova, K.V.; Ludwig, R. Quantification and understanding of non-covalent interactions in molecular and ionic systems: Dispersion interactions and hydrogen bonding analysed by thermodynamic methods. J. Mol. Liq. 2021, 343, 117547. [Google Scholar] [CrossRef]
- Vazdar, M.; Heyda, J.; Mason, P.E.; Tesei, G.; Allolio, C.; Lund, M.; Jungwirth, P. Arginine “Magic”: Guanidinium Like-Charge Ion Pairing from Aqueous Salts to Cell Penetrating Peptides. Acc. Chem. Res. 2018, 51, 1455–1464. [Google Scholar] [CrossRef]
- Shenderovich, I. The Partner Does Matter: The Structure of Heteroaggregates of Acridine Orange in Water. Molecules 2019, 24, 2816. [Google Scholar] [CrossRef] [PubMed]
- Inagaki, T.; Aono, S.; Nakano, H.; Yamamoto, T. Like-Charge Attraction of Molecular Cations in Water: Subtle Balance between Interionic Interactions and Ionic Solvation Effect. J. Phys. Chem. B 2014, 118, 5499–5508. [Google Scholar] [CrossRef]
- Kruchinin, S.E.; Fedotova, M.V. Ion Pairing of the Neurotransmitters Acetylcholine and Glutamate in Aqueous Solutions. J. Phys. Chem. B 2021, 125, 11219–11231. [Google Scholar] [CrossRef]
- Shishkina, A.V.; Ksenofontov, A.A.; Penkov, N.V.; Vener, M.V. Diclofenac Ion Hydration: Experimental and Theoretical Search for Anion Pairs. Molecules 2022, 27, 3350. [Google Scholar] [CrossRef]
- Bartashevich, E.V.; Tsirelson, V.G. Interplay between non-covalent interactions in complexes and crystals with halogen bonds. Russ. Chem. Rev. 2014, 83, 1181–1203. [Google Scholar] [CrossRef]
- Medvedev, A.G.; Churakov, A.V.; Navasardyan, M.A.; Prikhodchenko, P.V.; Lev, O.; Vener, M.V. Fast Quantum Approach for Evaluating the Energy of Non-Covalent Interactions in Molecular Crystals: The Case Study of Intermolecular H-Bonds in Crystalline Peroxosolvates. Molecules 2022, 27, 4082. [Google Scholar] [CrossRef]
- Rukenstein, E.; Shulgin, I.L.; Shulgin, L.I. Cooperativity in Ordinary Ice and Breaking of Hydrogen Bonds. J. Phys. Chem. B 2007, 111, 7114–7121. [Google Scholar] [CrossRef]
- Tskhovrebov, A.G.; Novikov, A.S.; Kritchenkov, A.S.; Khrustalev, V.N.; Haukka, M. Attractive halogen···halogen interactions in crystal structure of trans-dibromogold(III) complex. Z. Kristallogr Cryst. Mater. 2020, 235, 477–480. [Google Scholar] [CrossRef]
- Wiscons, R.A.; Bellas, M.K.; Bennion, J.C.; Matzger, A.J. Detonation Performance of Ten Forms of 5,5′-Dinitro-2H,2H′-3,3′-Bi-1,2,4-Triazole (DNBT). Cryst. Growth Des. 2018, 18, 7701–7707. [Google Scholar] [CrossRef]
- Surov, A.O.; Vasilev, N.A.; Churakov, A.V.; Parashchuk, O.D.; Artobolevskii, S.V.; Alatortsev, O.A.; Makhrov, D.E.; Vener, M.V. Two Faces of Water in the Formation and Stabilization of Multicomponent Crystals of Zwitterionic Drug-Like Compounds. Symmetry 2021, 13, 425. [Google Scholar] [CrossRef]
- Rai, S.K.; Gunnam, A.; Beran, G.J.; Kaduk, J.A.; Nangia, A.K. Polymorphs, Solvatomorphs, Hydrate, and Perhydrate of Dabrafenib. Cryst. Growth Des. 2023, 23, 1179–1188. [Google Scholar] [CrossRef]
- Johnstone, R.D.L.; Lennie, A.R.; Parsons, S.; Pidcock, E.; Warren, J.E. Comparison of the effects of pressure on three layered hydrates: A partially successful attempt to predict a high-pressure phase transition. Acta. Crystallogr. B Struct. Sci. Cryst. Eng. Mater. 2009, 65, 731–748. [Google Scholar] [CrossRef]
- Filippov, O.A.; Tsupreva, V.N.; Epstein, L.M.; Lledos, A.; Shubina, E.S. Intermolecular HH Vibrations of Dihydrogen Bonded Complexes H3EH−···HOR in the Low-Frequency Region: Theory and IR Spectra. J. Phys. Chem. A 2008, 112, 8198–8204. [Google Scholar] [CrossRef]
- Verma, P.K.; Lee, H.; Park, J.Y.; Lim, J.H.; Maj, M.; Choi, J.H.; Kwak, K.W.; Cho, M. Modulation of the Hydrogen Bonding Structure of Water by Renal Osmolytes. J. Phys. Chem. Lett. 2015, 6, 2773–2779. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, P.; Dudman, N.P.; Kuchel, P.W.; Wilcken, D.E. 1H NMR determination of urinary betaine in patients with premature vascular disease and mild homocysteinemia. Clin. Chem. 1995, 41, 275–283. [Google Scholar] [CrossRef]
- Guo, J.; Koeppe, B.; Tolstoy, P.M. Trimethylglycine complexes with carboxylic acids and HF: Solvation by a polar aprotic solvent. Phys. Chem. Chem. Phys. 2011, 13, 2335–2341. [Google Scholar] [CrossRef]
- Ilczyszyn, M.M.; Wierzejewska, M.; Ciunik, Z. Crystal structure and vibrational spectra of bis(betaine) sulfamate. Phys. Chem. Chem. Phys. 2000, 2, 3503–3510. [Google Scholar] [CrossRef]
- Shenderovich, I.G. Experimentally Established 15N NMR Absolute Shielding Scale for Theoretical Calculations. J. Phys. Chem. A 2023, 127, 5547–5555. [Google Scholar] [CrossRef] [PubMed]
- Shenderovich, I.G.; Limbach, H.H. Solid State NMR for Non Experts: An Overview of Simple but General Practical Methods. Solids 2021, 2, 139–154. [Google Scholar] [CrossRef]
- Makulski, W.; Jackowski, K. 1H, 13C and 29Si magnetic shielding in gaseous and liquid tetramethylsilane. J. Magn. Reson. 2020, 313, 106716. [Google Scholar] [CrossRef] [PubMed]
- Baldridge, K.K.; Siegel, J.S. Correlation of Empirical δ(TMS) and Absolute NMR Chemical Shifts Predicted by ab Initio Computations. J. Phys. Chem. A 1999, 103, 4038–4042. [Google Scholar] [CrossRef]
- Hu, L.; Staples, R.J.; Shreeve, J.M. Energetic compounds based on a new fused triazolo [4,5-d]pyridazine ring: Nitroimino lights up energetic performance. Chem. Eng. J. 2021, 420, 129839. [Google Scholar] [CrossRef]
- Surov, A.O.; Voronin, A.P.; Drozd, K.V.; Gruzdev, M.S.; Perlovich, G.L.; Prashanth, J.; Balasubramanian, S. Polymorphic forms of antiandrogenic drug nilutamide: Structural and thermodynamic aspects. Phys. Chem. Chem. Phys. 2021, 23, 9695–9708. [Google Scholar] [CrossRef] [PubMed]
- Katsyuba, S.A.; Vener, M.V.; Zvereva, E.E.; Brandenburg, J.G. The role of London dispersion interactions in strong and moderate intermolecular hydrogen bonds in the crystal and in the gas phase. Chem. Phys. Lett. 2017, 672, 124–127. [Google Scholar] [CrossRef]
- Vener, M.V.; Chernyshov, I.Y.; Rykounov, A.A.; Filarowski, A. Structural and spectroscopic features of proton hydrates in the crystalline state. Solid-state DFT study on HCl and triflic acid hydrates. Mol. Phys. 2018, 116, 251–262. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision A.03; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Dennington, R.; Keith, T.A.; Millam, J.M. GaussView, Version 6; Semichem Inc.: Shawnee Mission, KS, USA, 2016. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Vosko, S.H.; Wilk, L.; Nusair, M. Accurate Spin-Dependent Electron Liquid Correlation Energies for Local Spin Density Calculations: A Critical Analysis. Can. J. Phys. 1980, 58, 1200–1211. [Google Scholar] [CrossRef]
- Chaia, J.-D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef]
- Boys, S.F.; Bernardi, F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Shishkina, A.V.; Zhurov, V.V.; Stash, A.I.; Vener, M.V.; Pinkerton, A.A.; Tsirelson, V.G. Noncovalent Interactions in Crystalline Picolinic Acid N-Oxide: Insights from Experimental and Theoretical Charge Density Analysis. Cryst. Growth Des. 2013, 13, 816–828. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comp. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Dovesi, R.; Erba, A.; Orlando, R.; Zicovich-Wilson, C.M.; Civalleri, B.; Maschio, L.; Rérat, M.; Casassa, S.; Baima, J.; Salustro, S.; et al. Quantum-mechanical Condensed Matter Simulations with CRYSTAL. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2018, 8, e1360. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the Damping Function in Dispersion Corrected Density Functional Theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Mohaček-Grošev, V.; Grdadolnik, J.; Stare, J.; Hadži, D. Identification of hydrogen bond modes in polarized Raman spectra of single crystals of α-oxalic acid dihydrate. J. Raman Spectrosc. 2009, 40, 1605–1614. [Google Scholar] [CrossRef]
- Cutini, M.; Civalleri, B.; Corno, M.; Orlando, R.; Brandenburg, J.G.; Maschio, L.; Ugliengo, P. Assessment of Different Quantum Mechanical Methods for the Prediction of Structure and Cohesive Energy of Molecular Crystals. J. Chem. Theory Comput. 2016, 12, 3340–3352. [Google Scholar] [CrossRef] [PubMed]
- Vener, M.V.; Kharlanov, O.G.; Sosorev, A.Y. High-Mobility Naphthalene Diimide Derivatives Revealed by Raman-Based In Silico Screening. Int. J. Mol. Sci. 2022, 23, 13305. [Google Scholar] [CrossRef]
- Rogers, F.J.M.; Radhanpura, K.; Horvat, J.; Farrant, D. On the use of a volume constraint to account for thermal expansion effects on the low-frequency vibrations of molecular crystals. Phys. Chem. Chem. Phys. 2022, 24, 10408–10419. [Google Scholar] [CrossRef]
- Vener, M.V.; Churakov, A.V.; Voronin, A.P.; Parashchuk, O.D.; Artobolevskii, S.V.; Alatortsev, O.A.; Makhrov, D.E.; Medvedev, A.G.; Filarowski, A. Comparison of Proton Acceptor and Proton Donor Properties of H2O and H2O2 in Organic Crystals of Drug-like Compounds: Peroxosolvates vs. Crystallohydrates. Molecules 2022, 27, 717. [Google Scholar] [CrossRef] [PubMed]
- Remoto, P.J.G., III; Berzinš, K.; Fraser-Miller, S.J.; Korter, T.M.; Rades, T.; Rantanen, J.; Gordon, K.C. Exploring the Solid-State Landscape of Carbamazepine during Dehydration: A Low Frequency Raman Spectroscopy Perspective. Pharmaceutics 2023, 15, 1526. [Google Scholar] [CrossRef] [PubMed]
- Voronin, A.P.; Surov, A.O.; Churakov, A.V.; Vener, M.V. Supramolecular Organization in Salts of Riluzole with Dihydroxybenzoic Acids—The Key Role of the Mutual Arrangement of OH Groups. Pharmaceutics 2023, 15, 878. [Google Scholar] [CrossRef] [PubMed]
- Keith, T.A. AIMAll, Version 19.10.12; TK Gristmill Software: Overland Park, KS, USA, 2019. [Google Scholar]
- Gatti, C. TOPOND98 User’s Manual; CNR-CSRSRC: Milano, Italy, 1999. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fragment a | R(H∙∙∙O), Å | –ΔHHB, kJ/mol | ||
|---|---|---|---|---|
| B3LYP/6-31G** | wB97XD/aug-cc-pVDZ | B3LYP/6-31G** | wB97XD/aug-cc-pVDZ | |
| C–H36∙∙∙O2 | 2.435 (139.6) | 2.347 (143.2) | 10.0 | 11.1 |
| C–H32∙∙∙O2 | 2.250 (144.7) | 2.386 (141.2) | 12.7 | 10.6 |
| C–H37∙∙∙O2 | 2.142 (147.5) | 2.175 (148.7) | 14.7 | 14.0 |
| C–H19∙∙∙O21 | 2.142 (147.5) | 2.176 (148.7) | 14.7 | 14.0 |
| C–H11∙∙∙O21 | 2.250 (144.7) | 2.386 (141.2) | 12.7 | 10.6 |
| C–H15∙∙∙O21 | 2.435 (139.6) | 2.347 (143.2) | 10.0 | 11.1 |
| Total enthalpy | 74.8 | 71.5 | ||
| Fragment a | R(H∙∙∙O), Å | –ΔHHB, kJ/mol | ||
|---|---|---|---|---|
| B3LYP | PBE-D3 | B3LYP | PBE-D3 | |
| Water–GB interactions | ||||
| O–H12∙∙∙O1 | 1.676 | 1.656 | 31.1 | 32.3 |
| O–H13∙∙∙O2 | 1.649 | 1.633 | 32.7 | 33.7 |
| O3∙∙∙H11–C | 1.989 | 1.964 | 18.5 | 19.2 |
| O3∙∙∙H2–C | 2.277 | 2.258 | 12.2 | 12.5 |
| O3∙∙∙H4–C | 2.442 | 2.459 | 9.9 | 9.7 |
| O3∙∙∙H8–C | 2.481 | 2.475 | 9.4 | 9.5 |
| O3∙∙∙H3–C | 2.543 | 2.528 | 8.7 | 8.9 |
| O3∙∙∙H7–C | 2.665 | 2.531 | 7.6 | 8.9 |
| O3∙∙∙H9–C | 2.671 | 2.663 | 7.5 | 7.6 |
| Total enthalpy | 137.6 | 142.3 | ||
| GB–GB interactions | ||||
| O1∙∙∙H3–C | 2.188 | 2.175 | 13.8 | 14.1 |
| O1∙∙∙H5–C | 2.522 | 2.486 | 8.9 | 9.4 |
| O1∙∙∙H6–C | 2.647 | 2.641 | 7.7 | 7.8 |
| O1∙∙∙H7–C | 2.527 | 2.518 | 8.9 | 9.0 |
| O1∙∙∙H9–C | 2.275 | 2.263 | 12.3 | 12.5 |
| O1∙∙∙H10–C | 2.205 | 2.201 | 13.5 | 13.6 |
| O2∙∙∙H6–C | 2.168 | 2.158 | 14.2 | 14.4 |
| O2∙∙∙H8–C | 2.630 | 2.628 | 7.9 | 7.9 |
| O2∙∙∙H10–C | 2.253 | 2.228 | 12.6 | 13.1 |
| Total enthalpy | 99.8 | 101.8 | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Frolov, N.E.; Shishkina, A.V.; Vener, M.V. Specific Proton-Donor Properties of Glycine Betaine. Metric Parameters and Enthalpy of Noncovalent Interactions in its Dimer, Water Complexes and Crystalline Hydrate. Int. J. Mol. Sci. 2023, 24, 12971. https://doi.org/10.3390/ijms241612971
Frolov NE, Shishkina AV, Vener MV. Specific Proton-Donor Properties of Glycine Betaine. Metric Parameters and Enthalpy of Noncovalent Interactions in its Dimer, Water Complexes and Crystalline Hydrate. International Journal of Molecular Sciences. 2023; 24(16):12971. https://doi.org/10.3390/ijms241612971
Chicago/Turabian StyleFrolov, Nikita E., Anastasia V. Shishkina, and Mikhail V. Vener. 2023. "Specific Proton-Donor Properties of Glycine Betaine. Metric Parameters and Enthalpy of Noncovalent Interactions in its Dimer, Water Complexes and Crystalline Hydrate" International Journal of Molecular Sciences 24, no. 16: 12971. https://doi.org/10.3390/ijms241612971
APA StyleFrolov, N. E., Shishkina, A. V., & Vener, M. V. (2023). Specific Proton-Donor Properties of Glycine Betaine. Metric Parameters and Enthalpy of Noncovalent Interactions in its Dimer, Water Complexes and Crystalline Hydrate. International Journal of Molecular Sciences, 24(16), 12971. https://doi.org/10.3390/ijms241612971