
Anti-Glycolipid Antibody Examination in Five EAE Models and Theiler’s Virus Model of Multiple Sclerosis: Detection of Anti-GM1, GM3, GM4, and Sulfatide Antibodies in Relapsing-Remitting EAE

 ,
,  ,
,  ,
, 

Abstract
1. Introduction
2. Results
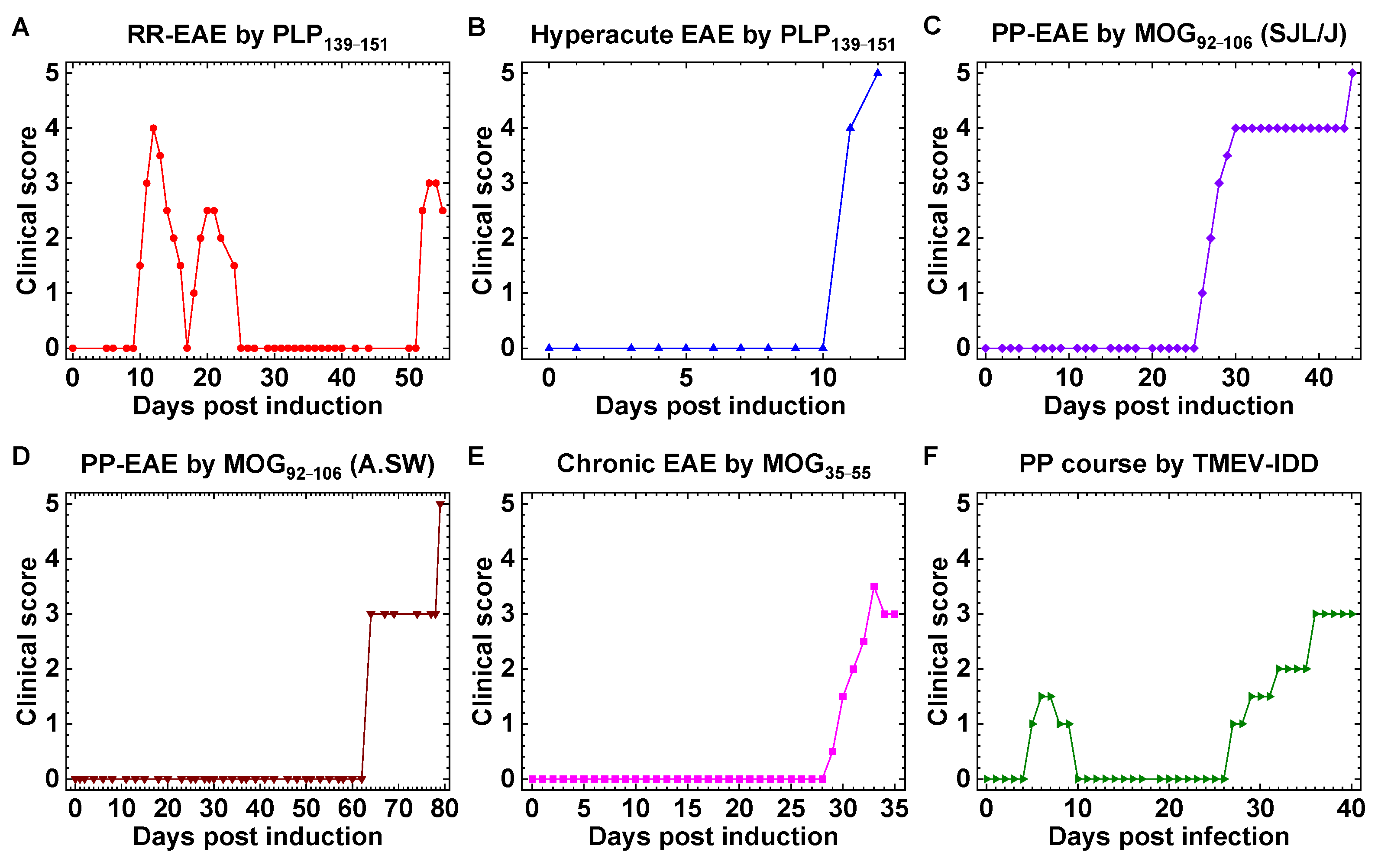
2.1. Mouse Models of MS with Distinct Clinical Courses
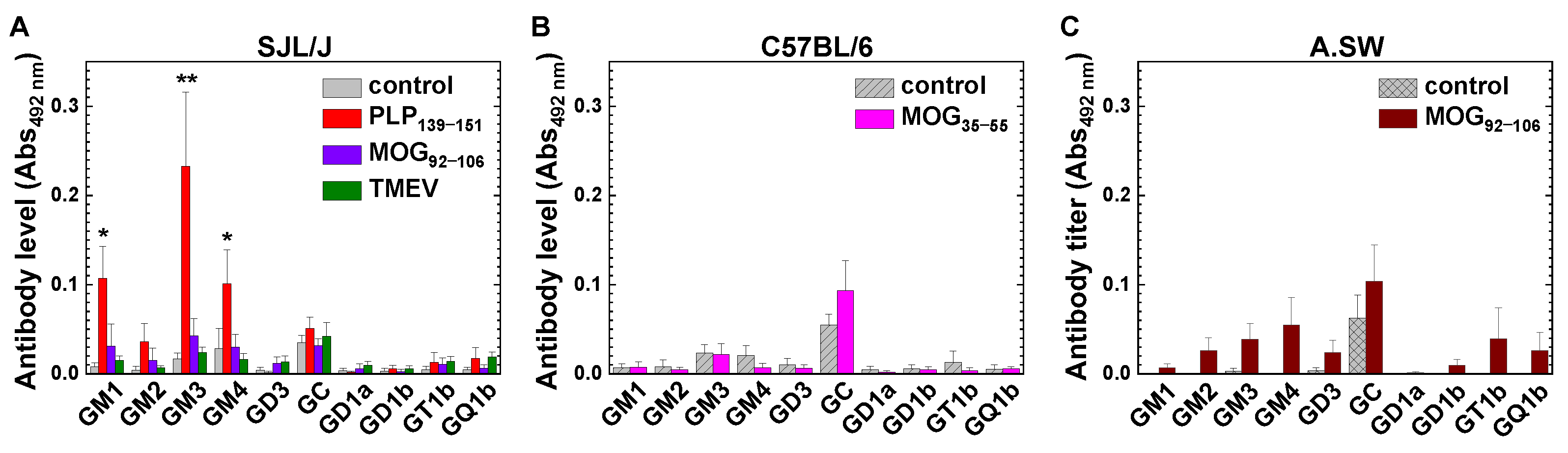
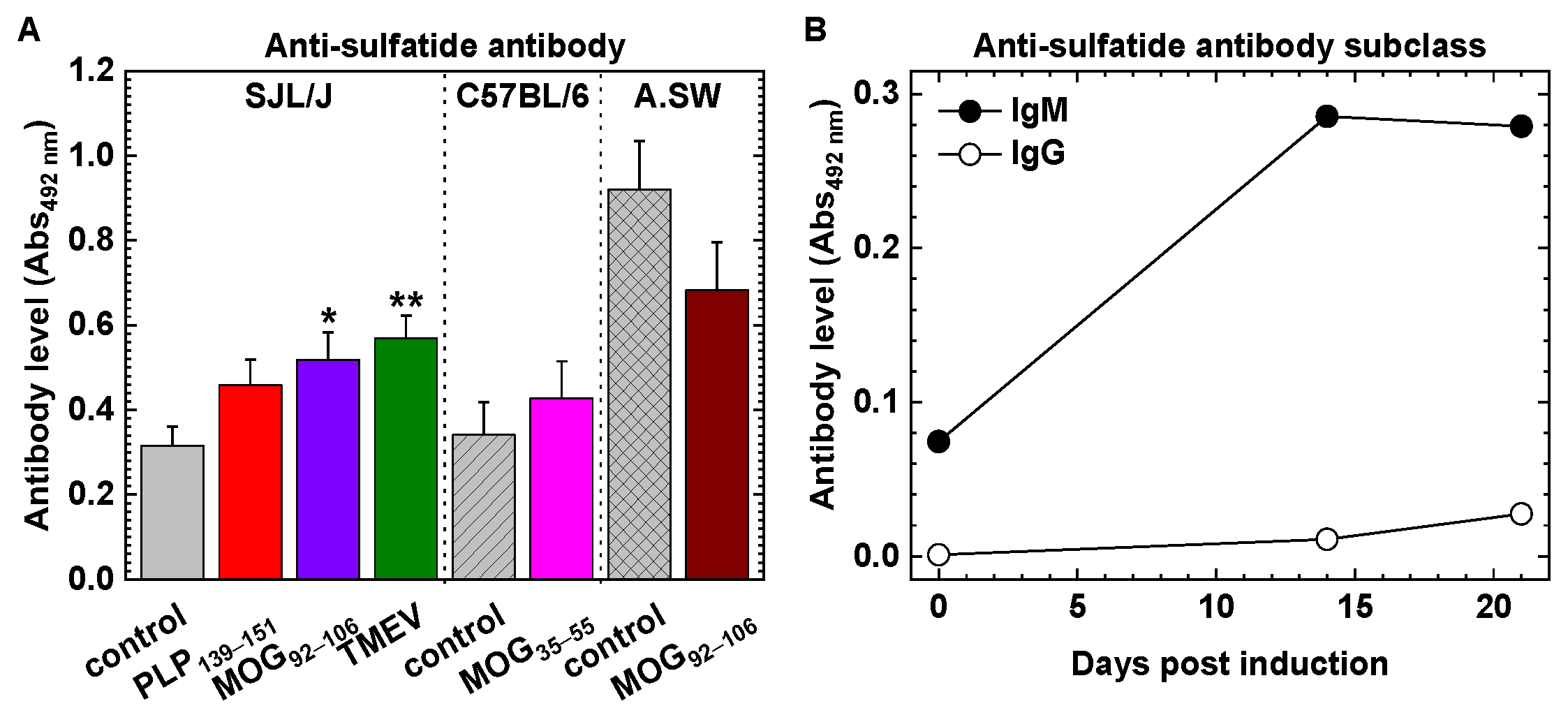
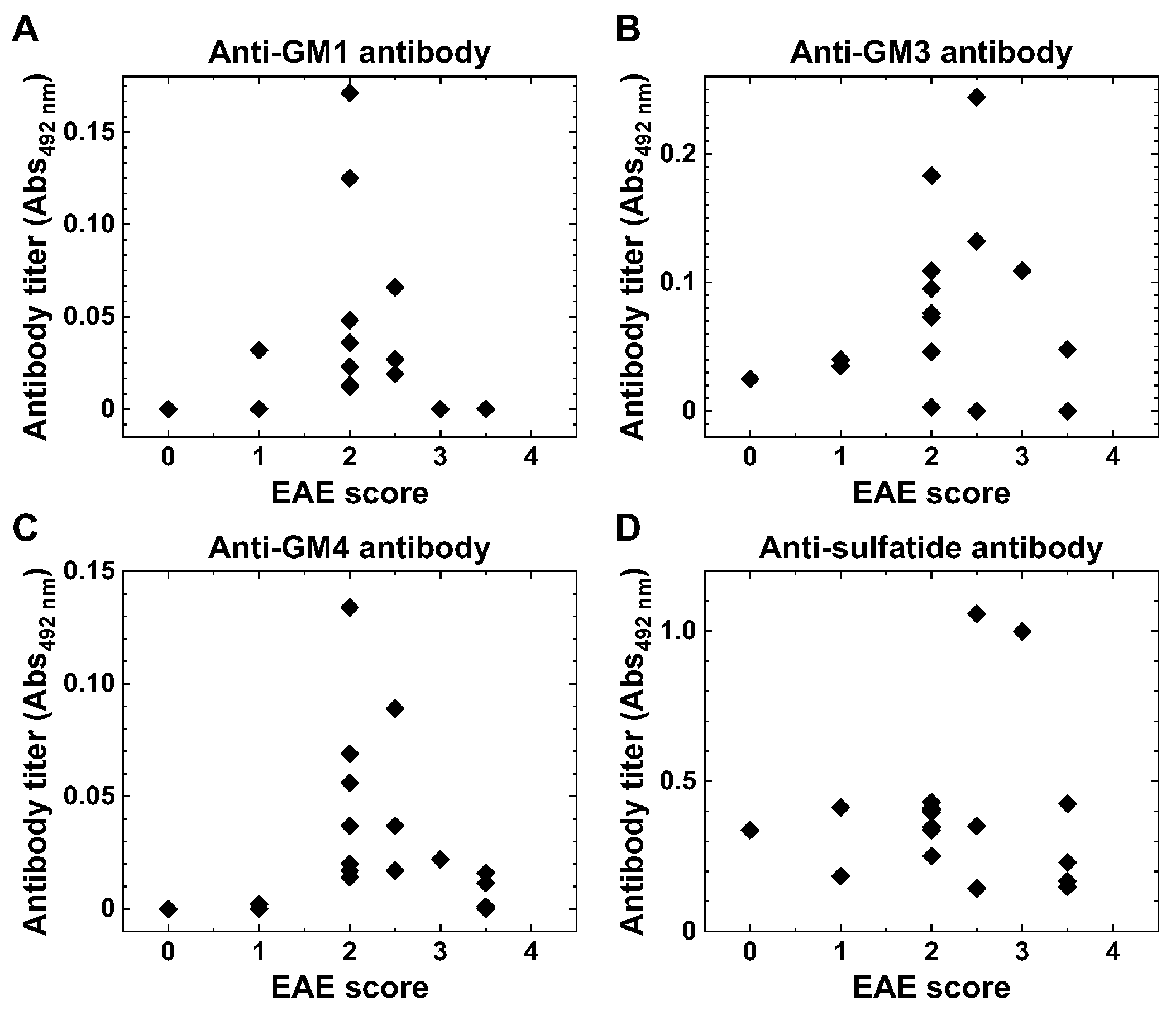
2.2. Anti-Glycolipid Antibodies in PLP139–151-Sensitized SJL/J Mice with RR-EAE
2.3. Anti-Glycolipid Antibodies in the Initial Stage of PLP139–151-Induced EAE
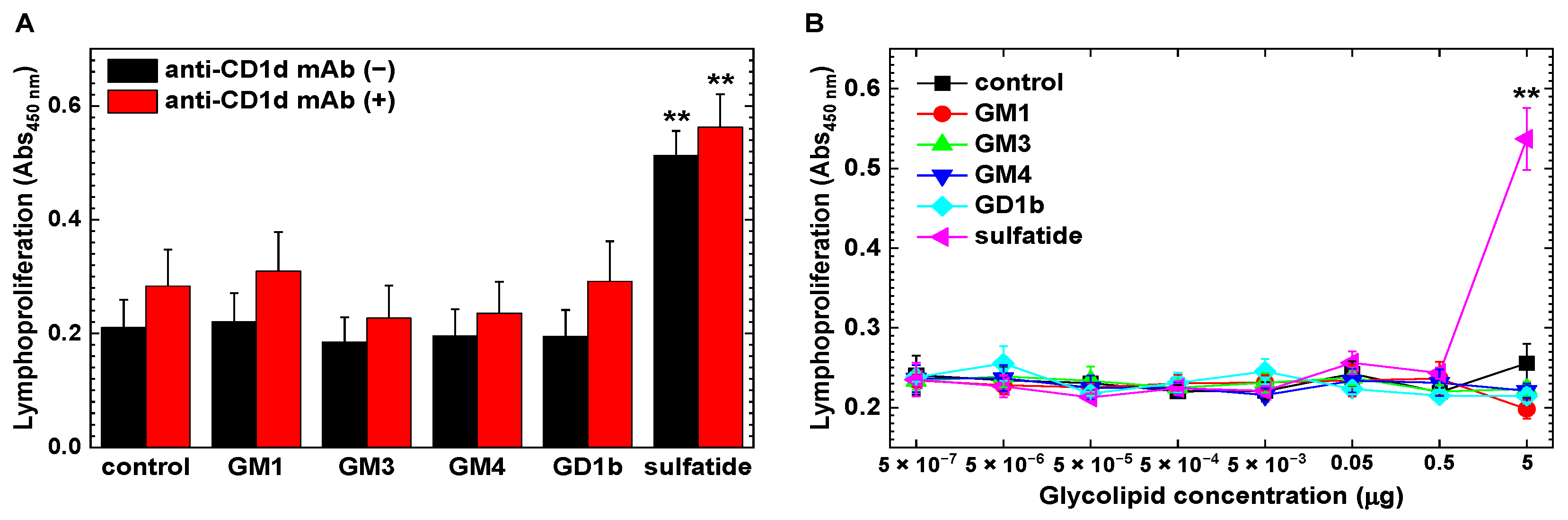
2.4. Increased CD1d-Independent Anti-Sulfatide Lymphoproliferative Responses
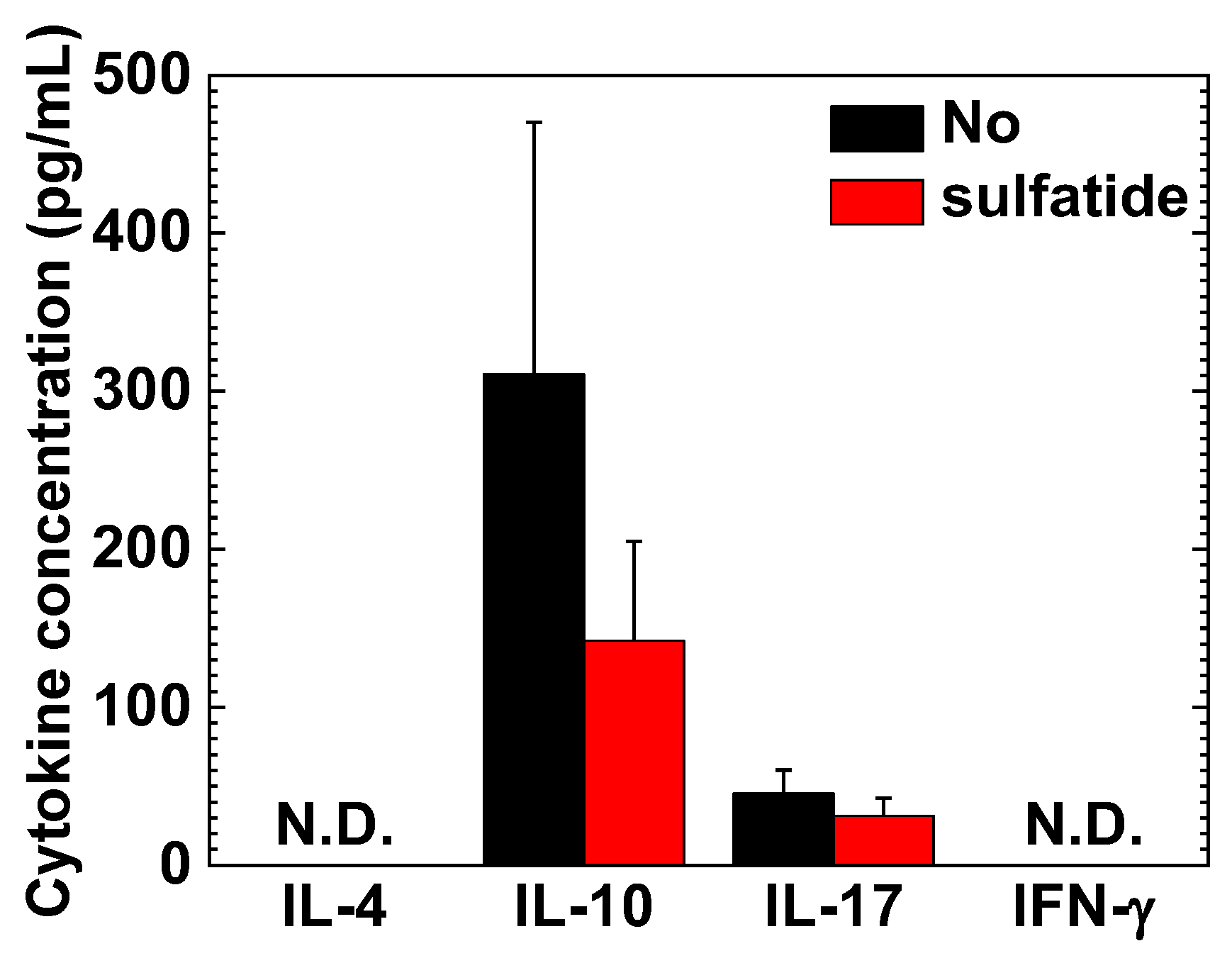
2.5. Interleukin (IL)-10 and IL-17A in MNC Cultures
3. Discussion
3.1. Antibodies against GM1, GM3, GM4 and Sulfatide in RR-EAE and Human Diseases
3.2. Implications of Anti-Glycolipid Antibodies for Remissions/Relapses in RR-EAE
3.3. Detrimental Roles of Anti-Glycolipid mAbs in TMEV-IDD and MOG92–106-Induced EAE
3.4. Potential Beneficial Roles of Natural mAbs in MS and MS Models
3.5. Roles of Epitope Spreading for Induction of Anti-Glycolipids Antibodies
4. Materials and Methods
4.1. Mice
4.2. Induction and Evaluation of EAE
4.3. Induction and Evaluation of TMEV-IDD
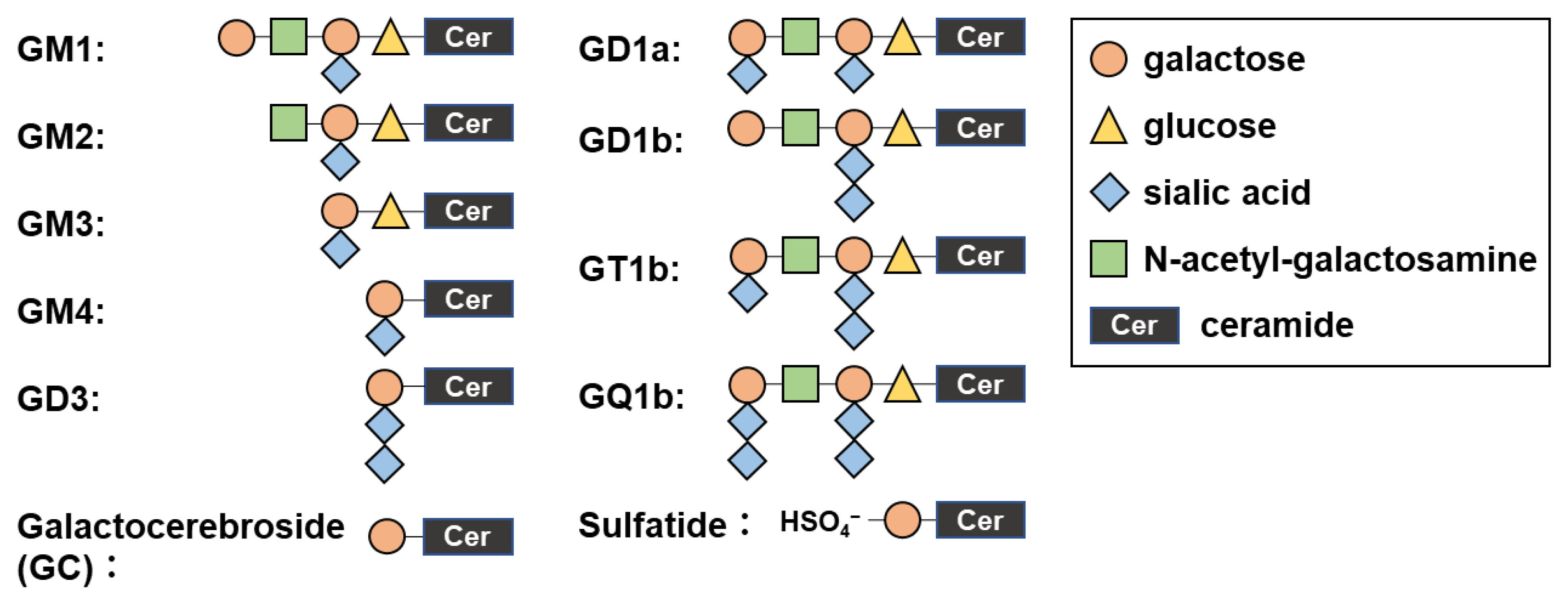
4.4. ELISAs for Antibodies against Glycolipids
4.5. Lymphoproliferative Responses and Cytokine ELISAs
4.6. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Merrill, A.H.; Vu, M.N. Glycolipids. In Encyclopedia of Cell Biology; Bradshaw, R.A., Stahl, P.D., Eds.; Academic Press: Waltham, MA, USA, 2016; pp. 180–193. [Google Scholar]
- Yu, R.K.; Tsai, Y.-T.; Ariga, T.; Yanagisawa, M. Structures, biosynthesis, and functions of gangliosides—An overview. J. Oleo Sci. 2011, 60, 537–544. [Google Scholar] [CrossRef]
- Schnaar, R.L. Glycolipid-mediated cell-cell recognition in inflammation and nerve regeneration. Arch. Biochem. Biophys. 2004, 426, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Omura, S.; Shimizu, K.; Kuwahara, M.; Morikawa-Urase, M.; Kusunoki, S.; Tsunoda, I. Exploratory factor analysis determines latent factors in Guillain-Barré syndrome. Sci. Rep. 2022, 12, 21837. [Google Scholar] [CrossRef] [PubMed]
- Prokazova, N.V.; Samovilova, N.N.; Gracheva, E.V.; Golovanova, N.K. Ganglioside GM3 and its biological functions. Biochemistry 2009, 74, 235–249. [Google Scholar] [CrossRef] [PubMed]
- Yu, R.K.; Iqbal, K. Sialosylgalactosyl ceramide as a specific marker for human myelin and oligodendroglial perikarya: Gangliosides of human myelin, oligodendroglia and neurons. J. Neurochem. 1979, 32, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Kawamura, N.; Taketomi, T. A new procedure for the isolation of brain gangliosides, and determination of their long chain base compositions. J. Biochem. 1977, 81, 1217–1225. [Google Scholar] [PubMed]
- Wanleenuwat, P.; Iwanowski, P.; Kozubski, W. Antiganglioside antibodies in neurological diseases. J. Neurol. Sci. 2020, 408, 116576. [Google Scholar] [CrossRef] [PubMed]
- Compston, A.; Coles, A. Multiple sclerosis. Lancet 2008, 372, 1502–1517. [Google Scholar] [CrossRef]
- Klineova, S.; Lublin, F.D. Clinical course of multiple sclerosis. Cold Spring Harb. Perspect. Med. 2018, 8, a028928. [Google Scholar] [CrossRef]
- Lublin, F.D.; Reingold, S.C. Defining the clinical course of multiple sclerosis: Results of an international survey. National Multiple Sclerosis Society (USA) Advisory Committee on Clinical Trials of New Agents in Multiple Sclerosis. Neurology 1996, 46, 907–911. [Google Scholar] [CrossRef]
- Lublin, F.D.; Reingold, S.C.; Cohen, J.A.; Cutter, G.R.; Sørensen, P.S.; Thompson, A.J.; Wolinsky, J.S.; Balcer, L.J.; Banwell, B.; Barkhof, F.; et al. Defining the clinical course of multiple sclerosis: The 2013 revisions. Neurology 2014, 83, 278–286. [Google Scholar] [CrossRef] [PubMed]
- Arnon, R.; Crisp, E.; Kelley, R.; Ellison, G.W.; Myers, L.W.; Tourtellotte, W.W. Anti-ganglioside antibodies in multiple sclerosis. J. Neurol. Sci. 1980, 46, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Fraussen, J.; Claes, N.; de Bock, L.; Somers, V. Targets of the humoral autoimmune response in multiple sclerosis. Autoimmun. Rev. 2014, 13, 1126–1137. [Google Scholar] [CrossRef] [PubMed]
- Sadatipour, B.T.; Greer, J.M.; Pender, M.P. Increased circulating antiganglioside antibodies in primary and secondary progressive multiple sclerosis. Ann. Neurol. 1998, 44, 980–983. [Google Scholar] [CrossRef] [PubMed]
- Owens, T. Animal models for multiple sclerosis. Adv. Neurol. 2006, 98, 77–89. [Google Scholar] [PubMed]
- Terry, R.L.; Ifergan, I.; Miller, S.D. Experimental autoimmune encephalomyelitis in mice. Methods Mol. Biol. 2016, 1304, 145–160. [Google Scholar] [CrossRef] [PubMed]
- Sato, F.; Nakamura, Y.; Katsuki, A.; Khadka, S.; Ahmad, I.; Omura, S.; Martinez, N.E.; Tsunoda, I. Curdlan, a microbial β-glucan, has contrasting effects on autoimmune and viral models of multiple sclerosis. Front. Cell. Infect. Microbiol. 2022, 12, 805302. [Google Scholar] [CrossRef] [PubMed]
- Omura, S.; Sato, F.; Martinez, N.E.; Park, A.-M.; Fujita, M.; Kennett, N.J.; Cvek, U.; Minagar, A.; Alexander, J.S.; Tsunoda, I. Bioinformatics analyses determined the distinct CNS and peripheral surrogate biomarker candidates between two mouse models for progressive multiple sclerosis. Front. Immunol. 2019, 10, 516. [Google Scholar] [CrossRef]
- Tsunoda, I.; Kuang, L.-Q.; Theil, D.J.; Fujinami, R.S. Antibody association with a novel model for primary progressive multiple sclerosis: Induction of relapsing-remitting and progressive forms of EAE in H2s mouse strains. Brain Pathol. 2000, 10, 402–418. [Google Scholar] [CrossRef]
- Daniels, J.B.; Pappenheimer, A.M.; Richardson, S. Observations on encephalomyelitis of mice (DA strain). J. Exp. Med. 1952, 96, 517–530. [Google Scholar] [CrossRef]
- Hofstetter, H.H.; Ibrahim, S.M.; Koczan, D.; Kruse, N.; Weishaupt, A.; Toyka, K.V.; Gold, R. Therapeutic efficacy of IL-17 neutralization in murine experimental autoimmune encephalomyelitis. Cell Immunol. 2005, 237, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Inoue, M.; Chen, P.-H.; Siecinski, S.; Li, Q.-J.; Liu, C.; Steinman, L.; Gregory, S.G.; Benner, E.; Shinohara, M.L. An interferon-β-resistant and NLRP3 inflammasome-independent subtype of EAE with neuronal damage. Nat. Neurosci. 2016, 19, 1599–1609. [Google Scholar] [CrossRef] [PubMed]
- Lipton, H.L.; Dal Canto, M.C. Theiler’s virus-induced demyelination: Prevention by immunosuppression. Science 1976, 192, 62–64. [Google Scholar] [CrossRef] [PubMed]
- Press, R.; Matá, S.; Lolli, F.; Zhu, J.; Andersson, T.; Link, H. Temporal profile of anti-ganglioside antibodies and their relation to clinical parameters and treatment in Guillain-Barré syndrome. J. Neurol. Sci. 2001, 190, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Tsunoda, I.; Tanaka, T.; Fujinami, R.S. Regulatory role of CD1d in neurotropic virus infection. J. Virol. 2008, 82, 10279–10289. [Google Scholar] [CrossRef] [PubMed]
- Peterson, L.K.; Tsunoda, I.; Masaki, T.; Fujinami, R.S. Polyreactive myelin oligodendrocyte glycoprotein antibodies: Implications for systemic autoimmunity in progressive experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2007, 183, 69–80. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kaida, K.; Ariga, T.; Yu, R.K. Antiganglioside antibodies and their pathophysiological effects on Guillain-Barré syndrome and related disorders--a review. Glycobiology 2009, 19, 676–692. [Google Scholar] [CrossRef]
- Aoyama, K.; Ishikura, H.; Mishima, S.; Murai, M.; Tsumura, H.; Kumakura, S.; Kobayashi, S. Guillain-Barré syndrome complicated with hemolytic anemia in association with antiganglioside GM3 antibody. Am. J. Med. 2001, 110, 399–400. [Google Scholar] [CrossRef]
- Cutillo, G.; Saariaho, A.-H.; Meri, S. Physiology of gangliosides and the role of antiganglioside antibodies in human diseases. Cell Mol. Immunol. 2020, 17, 313–322. [Google Scholar] [CrossRef]
- Ilyas, A.A.; Mithen, F.A.; Dalakas, M.C.; Wargo, M.; Chen, Z.-W.; Bielory, L.; Cook, S.D. Antibodies to sulfated glycolipids in Guillain-Barré syndrome. J. Neurol. Sci. 1991, 105, 108–117. [Google Scholar] [CrossRef]
- Fredman, P.; Vedeler, C.A.; Nyland, H.; Aarli, J.A.; Svennerholm, L. Antibodies in sera from patients with inflammatory demyelinating polyradiculoneuropathy react with ganglioside LM1 and sulphatide of peripheral nerve myelin. J. Neurol. 1991, 238, 75–79. [Google Scholar] [CrossRef] [PubMed]
- Giannotta, C.; Di Pietro, D.; Gallia, F.; Nobile-Orazio, E. Anti-sulfatide IgM antibodies in peripheral neuropathy: To test or not to test? Eur. J. Neurol. 2015, 22, 879–882. [Google Scholar] [CrossRef] [PubMed]
- Rinaldi, S.; Brennan, K.M.; Kalna, G.; Walgaard, C.; Van Doorn, P.A.; Jacobs, B.C.; Yu, R.K.; Mansson, J.-E.; Goodyear, C.S.; Willison, H.J. Antibodies to heteromeric glycolipid complexes in Guillain-Barré syndrome. PLoS ONE 2013, 8, e82337. [Google Scholar] [CrossRef]
- Kanter, J.L.; Narayana, S.; Ho, P.P.; Catz, I.; Warren, K.G.; Sobel, R.A.; Steinman, L.; Robinson, W.H. Lipid microarrays identify key mediators of autoimmune brain inflammation. Nat. Med. 2006, 12, 138–143. [Google Scholar] [CrossRef] [PubMed]
- Halder, R.C.; Jahng, A.; Maricic, I.; Kumar, V. Mini review: Immune response to myelin-derived sulfatide and CNS-demyelination. Neurochem. Res. 2007, 32, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Tsunoda, I.; Fujinami, R.S. TMEV and neuroantigens: Myelin genes and proteins, molecular mimicry, epitope spreading and autoantibody-mediated remyelination. In Experimental Models of Multiple Sclerosis; Lavi, E., Constantinescu, C.S., Eds.; Springer: New York, NY, USA, 2005; pp. 593–616. [Google Scholar]
- Jahng, A.; Maricic, I.; Aguilera, C.; Cardell, S.; Halder, R.C.; Kumar, V. Prevention of autoimmunity by targeting a distinct, noninvariant CD1d-reactive T cell population reactive to sulfatide. J. Exp. Med. 2004, 199, 947–957. [Google Scholar] [CrossRef] [PubMed]
- Rosner, B. Regression and correlateion methods. In Fundamentals of Biostatistics, 7th ed.; Brooks/Cole Pub Co: Boston, MA, USA, 2011; pp. 427–515. [Google Scholar]
- Dawson-Saunders, B.; Trapp, R.G. Summarizing data. In Basic and Clinical Biostatistics, 1st ed.; Appleton & Lange: Norwalk, CT, USA, 1990; pp. 43–63. [Google Scholar]
- Vetter, T.R. Magic mirror, on the wall—Which is the right study design of them all?—Part II. Anesth. Analg. 2017, 125, 328–332. [Google Scholar] [CrossRef] [PubMed]
- Kaida, K.; Kusunoki, S. Antibodies to gangliosides and ganglioside complexes in Guillain-Barré syndrome and Fisher syndrome: Mini-review. J. Neuroimmunol. 2010, 223, 5–12. [Google Scholar] [CrossRef]
- Inoue, N.; Kunishige, M.; Yoshida, S.; Oshima, Y.; Ohnishi, Y.; Kuroda, Y.; Asano, A.; Yoshino, H.; Matsumoto, T.; Mitsui, T. Dissociation between titer of anti-ganglioside antibody and severity of symptoms in a case of Guillain-Barré syndrome with treatment-related fluctuation. J. Neurol. Sci. 2003, 210, 105–108. [Google Scholar] [CrossRef]
- Thomma, R.C.M.; Fokke, C.; Walgaard, C.; Vermeulen-de Jongh, D.M.C.; Tio-Gillen, A.; van Rijs, W.; van Doorn, P.A.; Huizinga, R.; Jacobs, B.C. High and persistent anti-GM1 antibody titers are associated with poor clinical recovery in Guillain-Barré syndrome. Neurol. Neuroimmunol. Neuroinflamm. 2023, 10, e200107. [Google Scholar] [CrossRef]
- van Schaik, I.N.; Vermeulen, M.; van Doorn, P.A.; Brand, A. Anti-GM1 antibodies in patients with chronic inflammatory demyelinating polyneuropathy (CIDP) treated with intravenous immunoglobulin (IVIg). J. Neuroimmunol. 1994, 54, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Lardone, R.D.; Yuki, N.; Odaka, M.; Daniotti, J.L.; Irazoqui, F.J.; Nores, G.A. Anti-GM1 IgG antibodies in Guillain-Barré syndrome: Fine specificity is associated with disease severity. J. Neurol. Neurosurg. Psychiatry 2010, 81, 629–633. [Google Scholar] [CrossRef] [PubMed]
- Fujinami, R.S.; Zurbriggen, A.; Powell, H.C. Monoclonal antibody defines determinant between Theiler’s virus and lipid-like structures. J. Neuroimmunol. 1988, 20, 25–32. [Google Scholar] [CrossRef]
- Yamada, M.; Zurbriggen, A.; Fujinami, R.S. Monoclonal antibody to Theiler’s murine encephalomyelitis virus defines a determinant on myelin and oligodendrocytes, and augments demyelination in experimental allergic encephalomyelitis. J. Exp. Med. 1990, 171, 1893–1907. [Google Scholar] [CrossRef]
- Coutinho, A.; Kazatchkine, M.D.; Avrameas, S. Natural autoantibodies. Curr. Opin. Immunol. 1995, 7, 812–818. [Google Scholar] [CrossRef] [PubMed]
- Peterson, L.K.; Masaki, T.; Wheelwright, S.R.; Tsunoda, I.; Fujinami, R.S. Cross-reactive myelin antibody induces renal pathology. Autoimmunity 2008, 41, 526–536. [Google Scholar] [CrossRef]
- Matsumoto, Y.; Yuki, N.; Van Kaer, L.; Furukawa, K.; Hirata, K.; Sugita, M. Cutting edge: Guillain-Barré syndrome-associated IgG responses to gangliosides are generated independently of CD1 function in mice. J. Immunol. 2008, 180, 39–43. [Google Scholar] [CrossRef]
- Pellicci, D.G.; Uldrich, A.P. Unappreciated diversity within the pool of CD1d-restricted T cells. Semin. Cell Dev. Biol. 2018, 84, 42–47. [Google Scholar] [CrossRef]
- Mycko, M.P.; Sliwinska, B.; Cichalewska, M.; Cwiklinska, H.; Raine, C.S.; Selmaj, K.W. Brain glycolipids suppress T helper cells and inhibit autoimmune demyelination. J. Neurosci. 2014, 34, 8646–8658. [Google Scholar] [CrossRef]
- Shamshiev, A.; Donda, A.; Carena, I.; Mori, L.; Kappos, L.; De Libero, G. Self glycolipids as T-cell autoantigens. Eur. J. Immunol. 1999, 29, 1667–1675. [Google Scholar] [CrossRef]
- Asakura, K.; Miller, D.J.; Pogulis, R.J.; Pease, L.R.; Rodriguez, M. Oligodendrocyte-reactive O1, O4, and HNK-1 monoclonal antibodies are encoded by germline immunoglobulin genes. Brain Res. Mol. Brain Res. 1995, 34, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Asakura, K.; Miller, D.J.; Pease, L.R.; Rodriguez, M. Targeting of IgMκ antibodies to oligodendrocytes promotes CNS remyelination. J. Neurosci. 1998, 18, 7700–7708. [Google Scholar] [CrossRef] [PubMed]
- Warrington, A.E.; Asakura, K.; Bieber, A.J.; Ciric, B.; Van Keulen, V.; Kaveri, S.V.; Kyle, R.A.; Pease, L.R.; Rodriguez, M. Human monoclonal antibodies reactive to oligodendrocytes promote remyelination in a model of multiple sclerosis. Proc. Natl. Acad. Sci. USA 2000, 97, 6820–6825. [Google Scholar] [CrossRef]
- Miller, D.J.; Sanborn, K.S.; Katzmann, J.A.; Rodriguez, M. Monoclonal autoantibodies promote central nervous system repair in an animal model of multiple sclerosis. J. Neurosci. 1994, 14, 6230–6238. [Google Scholar] [CrossRef]
- Kirschning, E.; Jensen, K.; Dübel, S.; Rutter, G.; Hohenberg, H.; Will, H. Primary structure of the antigen-binding domains of a human oligodendrocyte-reactive IgM monoclonal antibody derived from a patient with multiple sclerosis. J. Neuroimmunol. 1999, 99, 122–130. [Google Scholar] [CrossRef] [PubMed]
- Matsiota, P.; Blancher, A.; Doyon, B.; Guilbert, B.; Clanet, M.; Kouvelas, E.D.; Avrameas, S. Comparative study of natural autoantibodies in the serum and cerebrospinal fluid of normal individuals and patients with multiple sclerosis and other neurological diseases. Ann. Inst. Pasteur Immunol. 1988, 139, 99–108. [Google Scholar] [CrossRef]
- Vanderlugt, C.J.; Miller, S.D. Epitope spreading. Curr. Opin. Immunol. 1996, 8, 831–836. [Google Scholar] [CrossRef]
- National Research Council (US). Committee for the Update of the Guide for the Care and Use of Laboratory Animals. In Guide for the Care and Use of Laboratory Animals, 8th ed.; National Academies Press: Washington, DC, USA, 2011. [Google Scholar]
- Khadka, S.; Omura, S.; Sato, F.; Nishio, K.; Kakeya, H.; Tsunoda, I. Curcumin β-D-glucuronide modulates an autoimmune model of multiple sclerosis with altered gut microbiota in the ileum and feces. Front. Cell. Infect. Microbiol. 2021, 11, 772962. [Google Scholar] [CrossRef]
- Bajaj, A.; Rosner, B.; Lockley, S.W.; Schernhammer, E.S. Validation of a light questionnaire with real-life photopic illuminance measurements: The Harvard Light Exposure Assessment questionnaire. Cancer Epidemiol. Biomark. Prev. 2011, 20, 1341–1349. [Google Scholar] [CrossRef]
- Mukaka, M.M. Statistics corner: A guide to appropriate use of correlation coefficient in medical research. Malawi Med. J. 2012, 24, 69–71. [Google Scholar]
- Schober, P.; Boer, C.; Schwarte, L.A. Correlation coefficients: Appropriate use and interpretation. Anesth. Analg. 2018, 126, 1763–1768. [Google Scholar] [CrossRef]
- Zhang, Z.; Yuan, K.H. Practical Statistical Power Analysis Using Webpower and R; Zhang, Z., Yuan, K.H., Eds.; ISDSA Press: Granger, IN, USA, 2018. [Google Scholar]
- Cohen, J. A power primer. Psychol. Bull. 1992, 112, 155–159. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinical Course | Inoculum | Adjuvant | Mouse Strain | Model | Reference |
|---|---|---|---|---|---|
| RR | PLP139–151 | CFA | SJL/J | EAE | [18] |
| hyperacute | PLP139–151 | CFA/curdlan | SJL/J | EAE | [18] |
| PP, SP | MOG92–106 | CFA/curdlan | SJL/J | EAE | [19] |
| PP | MOG92–106 | CFA | A.SW | EAE | [20] |
| monophasic/chronic | MOG35–55 | CFA, PT | C57BL/6 | EAE | [23] |
| PP | TMEV | – | SJL/J | virus | [18] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moriguchi, K.; Nakamura, Y.; Park, A.-M.; Sato, F.; Kuwahara, M.; Khadka, S.; Omura, S.; Ahmad, I.; Kusunoki, S.; Tsunoda, I. Anti-Glycolipid Antibody Examination in Five EAE Models and Theiler’s Virus Model of Multiple Sclerosis: Detection of Anti-GM1, GM3, GM4, and Sulfatide Antibodies in Relapsing-Remitting EAE. Int. J. Mol. Sci. 2023, 24, 12937. https://doi.org/10.3390/ijms241612937
Moriguchi K, Nakamura Y, Park A-M, Sato F, Kuwahara M, Khadka S, Omura S, Ahmad I, Kusunoki S, Tsunoda I. Anti-Glycolipid Antibody Examination in Five EAE Models and Theiler’s Virus Model of Multiple Sclerosis: Detection of Anti-GM1, GM3, GM4, and Sulfatide Antibodies in Relapsing-Remitting EAE. International Journal of Molecular Sciences. 2023; 24(16):12937. https://doi.org/10.3390/ijms241612937
Chicago/Turabian StyleMoriguchi, Kota, Yumina Nakamura, Ah-Mee Park, Fumitaka Sato, Motoi Kuwahara, Sundar Khadka, Seiichi Omura, Ijaz Ahmad, Susumu Kusunoki, and Ikuo Tsunoda. 2023. "Anti-Glycolipid Antibody Examination in Five EAE Models and Theiler’s Virus Model of Multiple Sclerosis: Detection of Anti-GM1, GM3, GM4, and Sulfatide Antibodies in Relapsing-Remitting EAE" International Journal of Molecular Sciences 24, no. 16: 12937. https://doi.org/10.3390/ijms241612937
APA StyleMoriguchi, K., Nakamura, Y., Park, A.-M., Sato, F., Kuwahara, M., Khadka, S., Omura, S., Ahmad, I., Kusunoki, S., & Tsunoda, I. (2023). Anti-Glycolipid Antibody Examination in Five EAE Models and Theiler’s Virus Model of Multiple Sclerosis: Detection of Anti-GM1, GM3, GM4, and Sulfatide Antibodies in Relapsing-Remitting EAE. International Journal of Molecular Sciences, 24(16), 12937. https://doi.org/10.3390/ijms241612937