

A Novel Genetic Variant in MBD5 Associated with Severe Epilepsy and Intellectual Disability: Potential Implications on Neural Primary Cilia
 , ,
, ,  ,
,  ,
,  ,
,  ,
,  and
and 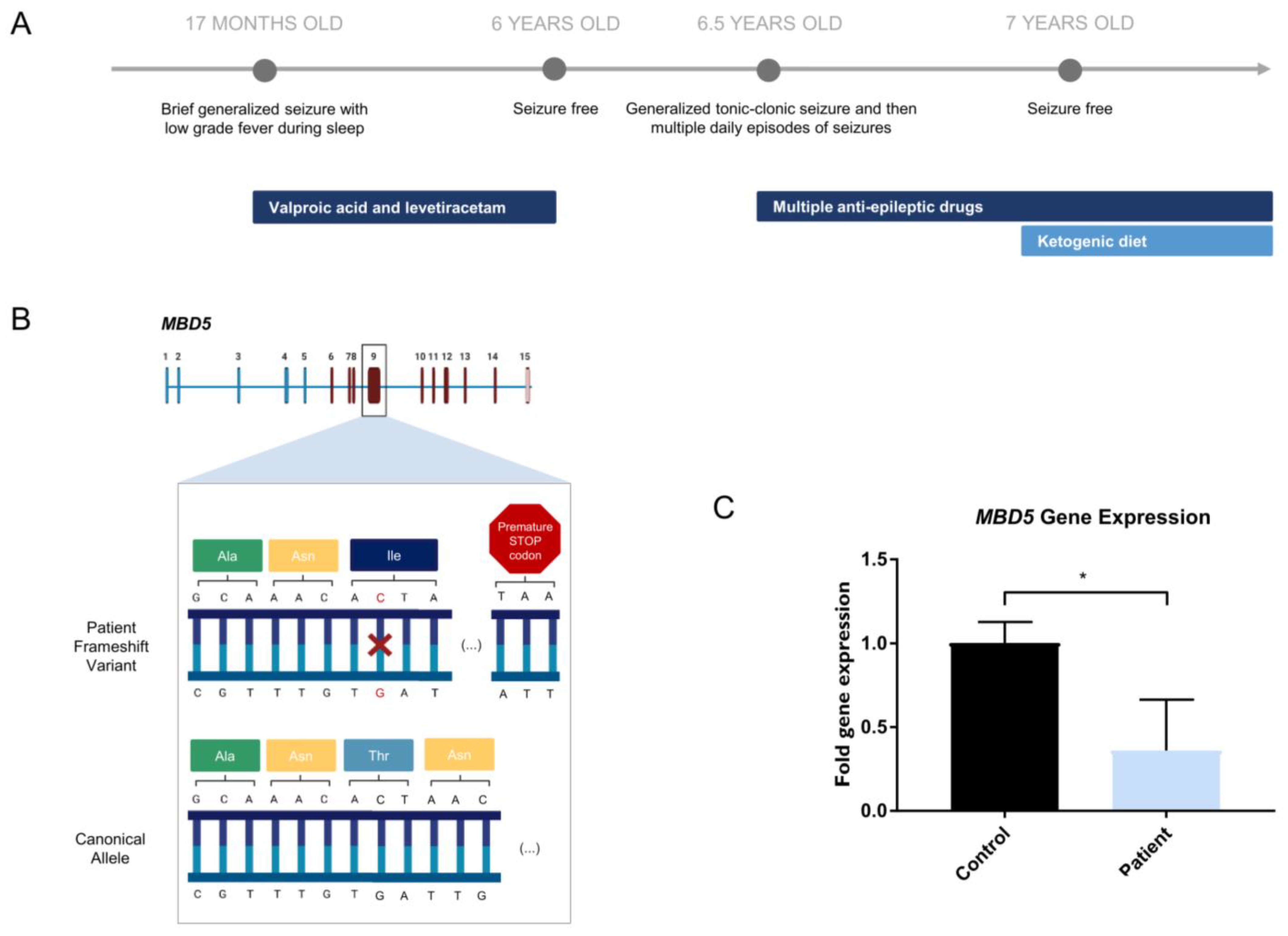{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Clinical Report of a Patient with Severe Epilepsy and Psychomotor Developmental Delay
2.2. Novel MBD5 Genetic Variant Shows Haploinsufficiency
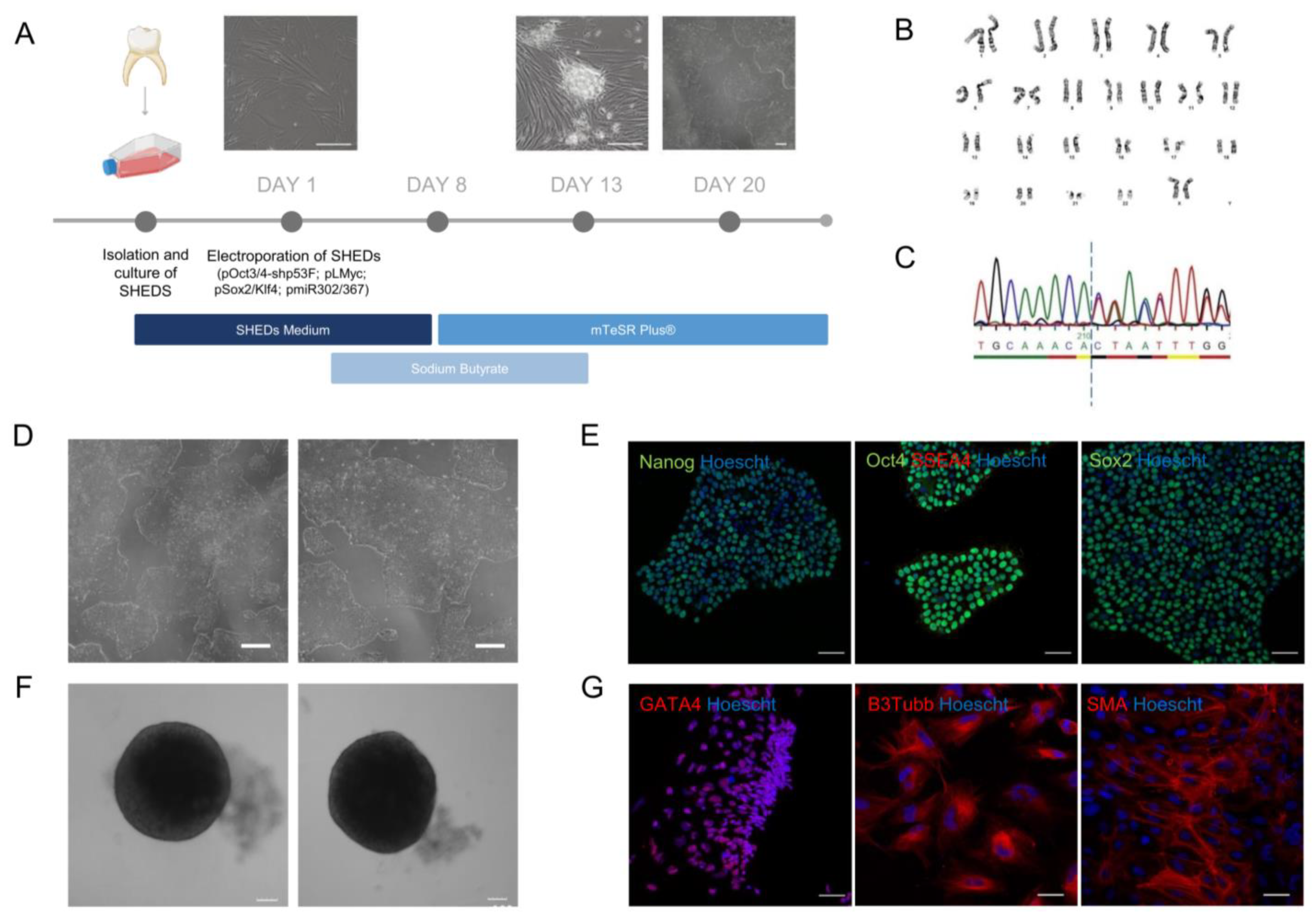
2.3. Establishment of an iPSC Cell Line from Patient-Derived Dental Stem Cells
2.4. CRISPR-Cas9 HDR-Mediated Correction the MBD5 Variant in the Patient-Derived iPSCs
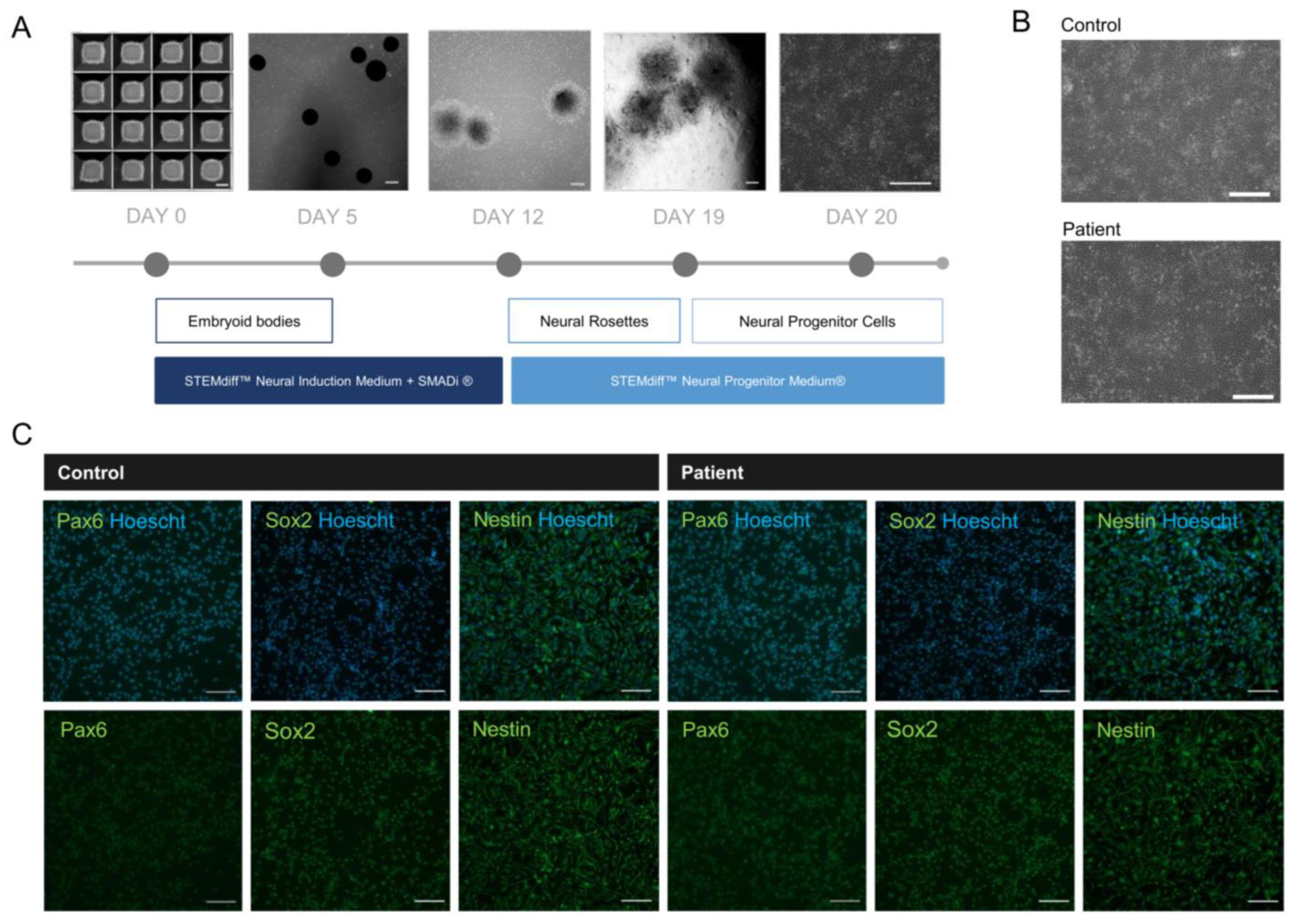
2.5. NPCs Reveal Neural Identity Markers
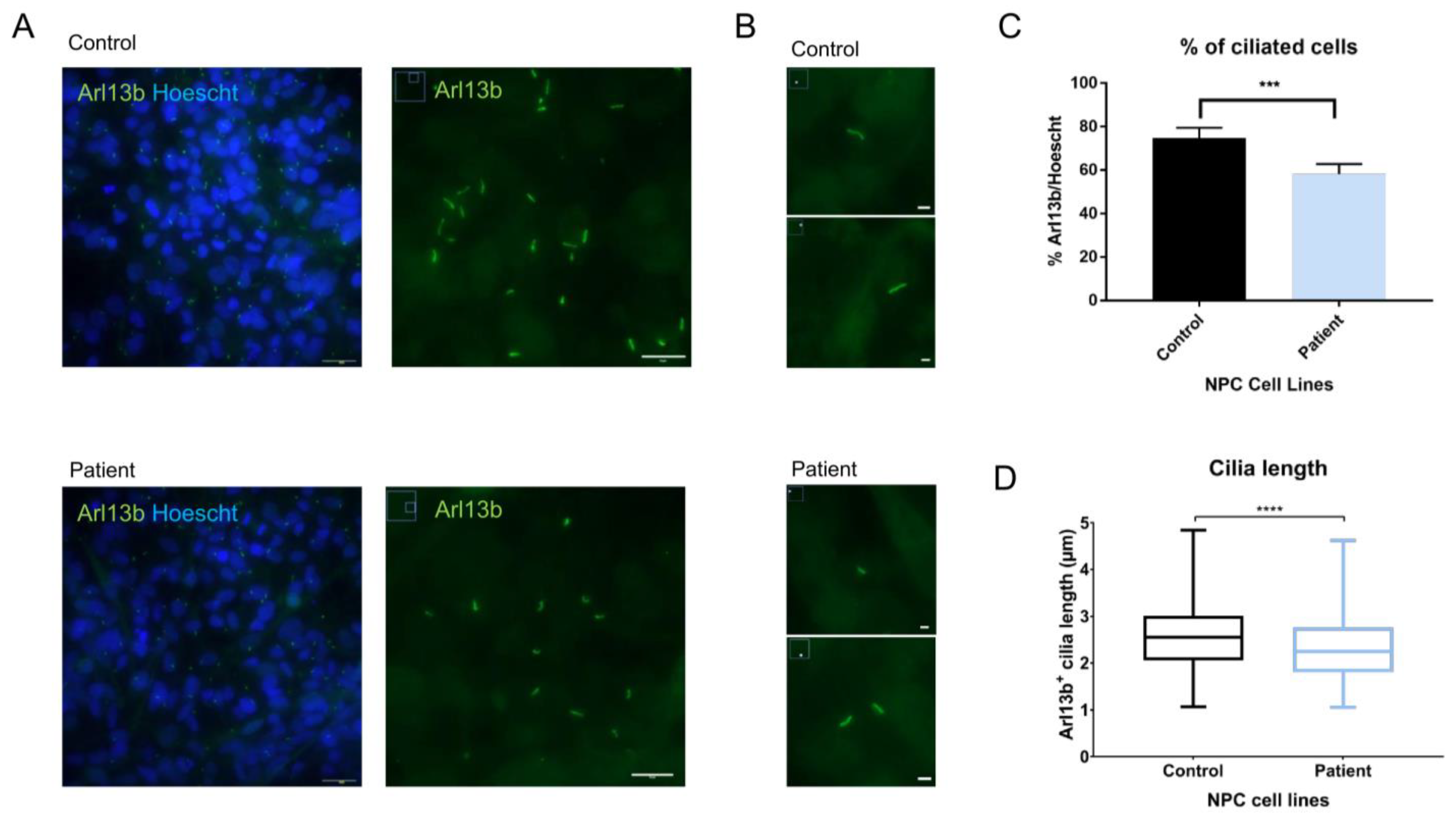
2.6. Evaluation of Primary Cilia Markers and Length in NPCs Shows Alterations in Arl13b+ Cilia Length in Patient Cells
3. Discussion
4. Materials and Methods
4.1. Culture and Cryopreservation of SHEDs
4.2. Reprogramming Patient-Derived SHEDs into iPSCs
4.3. Immunostaining of iPSCs
4.4. Trilineage Differentiation of iPSCs
4.5. Sanger Sequencing
4.6. Karyotyping
4.7. Correction of MBD5 c.2297delC in iPSCs via CRISPR/Cas9
4.8. Neural Progenitor Cell Differentiation
4.9. Immunostaining of NPCs
4.10. Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
4.11. Primary Cilia Count and Length Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Francés, L.; Quintero, J.; Fernández, A.; Ruiz, A.; Caules, J.; Fillon, G.; Hervás, A.; Soler, C.V. Current state of knowledge on the prevalence of neurodevelopmental disorders in childhood according to the DSM-5: A systematic review in accordance with the PRISMA criteria. Child Adolesc. Psychiatry Ment. Health 2022, 16, 1–15. [Google Scholar] [CrossRef]
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, 5th ed.; American Psychiatric Association: Washington, DC, USA, 2013. [Google Scholar] [CrossRef]
- Cardoso, A.R.; Lopes-Marques, M.; Silva, R.M.; Serrano, C.; Amorim, A.; Prata, M.J.; Azevedo, L. Essential genetic findings in neurodevelopmental disorders. Hum. Genom. 2019, 13, 31. [Google Scholar] [CrossRef] [PubMed]
- De Rubeis, S.; He, X.; Goldberg, A.P.; Poultney, C.S.; Samocha, K.; Cicek, A.E.; Kou, Y.; Liu, L.; Fromer, M.; Walker, S.; et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 2014, 515, 209–215. [Google Scholar] [CrossRef]
- Peça, J.; Feng, G. Cellular and synaptic network defects in autism. Curr. Opin. Neurobiol. 2012, 22, 866–872. [Google Scholar] [CrossRef] [PubMed]
- Talkowski, M.E.; Mullegama, S.V.; Rosenfeld, J.A.; van Bon, B.W.M.M.; Shen, Y.; Repnikova, E.A.; Gastier-Foster, J.; Thrush, D.L.; Kathiresan, S.; Ruderfer, D.M.; et al. Assessment of 2q23.1 microdeletion syndrome implicates MBD5 as a single causal locus of intellectual disability, epilepsy, and autism spectrum disorder. Am. J. Hum. Genet. 2011, 89, 551–563. [Google Scholar] [CrossRef] [PubMed]
- Mullegama, S.V.; Elsea, S.H. Clinical and Molecular Aspects of MBD5-Associated Neurodevelopmental Disorder (MAND). Eur. J. Hum. Genet. 2016, 24, 1235–1243. [Google Scholar] [CrossRef] [PubMed]
- Laget, S.; Joulie, M.; Le Masson, F.; Sasai, N.; Christians, E.; Pradhan, S.; Roberts, R.J.; Defossez, P.-A.A. The human proteins MBD5 and MBD6 associate with heterochromatin but they do not bind methylated DNA. PLoS ONE 2010, 5, e11982. [Google Scholar] [CrossRef]
- Klose, R.J.; Bird, A.P. Genomic DNA methylation: The mark and its mediators. Trends Biochem. Sci. 2006, 31, 89–97. [Google Scholar] [CrossRef]
- Roloff, T.C.; Ropers, H.H.; Nuber, U.A. Comparative study of methyl-CpG-binding domain proteins. BMC Genom. 2003, 4, 1. [Google Scholar] [CrossRef]
- Muñoz, E.M.; de Souza, F.S.J.; Rath, M.F.; Martínez Cerdeño, V. Editorial: Transcription Regulation—Brain Development and Homeostasis—A Finely Tuned and Orchestrated Scenario in Physiology and Pathology. Front. Mol. Neurosci. 2022, 14, 834607. [Google Scholar] [CrossRef]
- Qin, S.; Min, J. Structure and function of the nucleosome-binding PWWP domain. Trends Biochem. Sci. 2014, 39, 536–547. [Google Scholar] [CrossRef] [PubMed]
- Seabra, C.M.; Aneichyk, T.; Erdin, S.; Tai, D.J.C.; De Esch, C.E.F.; Razaz, P.; An, Y.; Manavalan, P.; Ragavendran, A.; Stortchevoi, A.; et al. Transcriptional consequences of MBD5 disruption in mouse brain and CRISPR-derived neurons. Mol. Autism 2020, 11, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Frasca, A.; Spiombi, E.; Palmieri, M.; Albizzati, E.; Valente, M.M.; Bergo, A.; Leva, B.; Kilstrup-nielsen, C.; Bianchi, F.; Di Carlo, V.; et al. MECP 2 mutations affect ciliogenesis: A novel perspective for Rett syndrome and related disorders. EMBO Mol. Med. 2020, 12, e10270. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.; Panda, S.; Lee, H.Y. Primary Ciliary Deficits in the Dentate Gyrus of Fragile X Syndrome. Stem Cell Rep. 2020, 15, 454–466. [Google Scholar] [CrossRef]
- Tereshko, L.; Turrigiano, G.G.; Sengupta, P. Primary cilia in the postnatal brain: Subcellular compartments for organizing neuromodulatory signaling. Curr. Opin. Neurobiol. 2022, 74, 102533. [Google Scholar] [CrossRef]
- Truong, M.E.; Bilekova, S.; Choksi, S.P.; Li, W.; Bugaj, L.J.; Xu, K.; Reiter, J.F.; Truong, M.E.; Bilekova, S.; Choksi, S.P.; et al. Article Vertebrate cells differentially interpret ciliary and extraciliary cAMP ll ll Article Vertebrate cells differentially interpret ciliary and extraciliary cAMP. Cell 2021, 184, 2911–2926. [Google Scholar] [CrossRef]
- Sheu, S.-H.; Upadhyayula, S.; Dupuy, V.; Pang, S.; Deng, F.; Wan, J.; Walpita, D.; Pasolli, H.A.; Houser, J.; Sanchez-Martinez, S.; et al. A serotonergic axon-cilium synapse drives nuclear signaling to alter chromatin accessibility. Cell 2022, 185, 3390–3407.e18. [Google Scholar] [CrossRef]
- Guo, J.; Otis, J.M.; Higginbotham, H.; Monckton, C.; Cheng, J.G.; Asokan, A.; Mykytyn, K.; Caspary, T.; Stuber, G.D.; Anton, E.S. Primary Cilia Signaling Shapes the Development of Interneuronal Connectivity. Dev. Cell 2017, 42, 286–300.e4. [Google Scholar] [CrossRef]
- Valente, E.M.; Rosti, R.O.; Gibbs, E.; Gleeson, J.G. Primary cilia in neurodevelopmental disorders. Nat. Rev. Neurol. 2013, 10, 27–36. [Google Scholar] [CrossRef]
- Youn, Y.H.; Han, Y.G. Primary Cilia in Brain Development and Diseases. Am. J. Pathol. 2018, 188, 11–22. [Google Scholar] [CrossRef]
- Hasenpusch-Theil, K.; Theil, T. The Multifaceted Roles of Primary Cilia in the Development of the Cerebral Cortex. Front. Cell Dev. Biol. 2021, 9, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Andreu-Cervera, A.; Catala, M.; Schneider-Maunoury, S. Cilia, ciliopathies and hedgehog-related forebrain developmental disorders. Neurobiol. Dis. 2021, 150, 105236. [Google Scholar] [CrossRef]
- Olstad, E.W.; Ringers, C.; Hansen, J.N.; Wens, A.; Brandt, C.; Wachten, D.; Yaksi, E.; Jurisch-Yaksi, N. Ciliary Beating Compartmentalizes Cerebrospinal Fluid Flow in the Brain and Regulates Ventricular Development. Curr. Biol. 2019, 29, 229–241.e6. [Google Scholar] [CrossRef] [PubMed]
- Higginbotham, H.; Guo, J.; Yokota, Y.; Umberger, N.L.; Su, C.Y.; Li, J.; Verma, N.; Hirt, J.; Ghukasyan, V.; Caspary, T.; et al. Arl13b-regulated cilia activities are essential for polarized radial glial scaffold formation. Nat. Neurosci. 2013, 16, 1000–1007. [Google Scholar] [CrossRef] [PubMed]
- Baudoin, J.P.; Viou, L.; Launay, P.S.; Luccardini, C.; Espeso Gil, S.; Kiyasova, V.; Irinopoulou, T.; Alvarez, C.; Rio, J.P.; Boudier, T.; et al. Tangentially Migrating Neurons Assemble a Primary Cilium that Promotes Their Reorientation to the Cortical Plate. Neuron 2012, 76, 1108–1122. [Google Scholar] [CrossRef]
- Guo, J.; Otis, J.M.; Suciu, S.K.; Stuber, G.D.; Caspary, T.; Anton, E.S.; Guo, J.; Otis, J.M.; Suciu, S.K.; Catalano, C.; et al. Primary Cilia Signaling Promotes Axonal Tract Development and Is Disrupted in Joubert Syndrome- Article Primary Cilia Signaling Promotes Axonal Tract Development and Is Disrupted in Joubert Syndrome-Related Disorders Models. Dev. Cell 2019, 51, 759–773.e6. [Google Scholar] [CrossRef] [PubMed]
- Lancaster, M.A.; Gleeson, J.G. The primary cilium as a cellular signaling center: Lessons from disease. Curr. Opin. Genet. Dev. 2009, 19, 220–229. [Google Scholar] [CrossRef]
- Louvi, A.; Grove, E.A. Cilia in the CNS: The quiet organelle claims center stage. Neuron 2011, 69, 1046–1060. [Google Scholar] [CrossRef]
- Guo, J.; Higginbotham, H.; Li, J.; Nichols, J.; Hirt, J.; Ghukasyan, V.; Anton, E.S. Developmental disruptions underlying brain abnormalities in ciliopathies. Nat. Commun. 2015, 6, 7857. [Google Scholar] [CrossRef]
- Shi, M.; Zhang, H.; Wang, L.; Zhu, C.; Sheng, K.; Du, Y.; Wang, K.; Dias, A.; Chen, S.; Whitman, M.; et al. Premature termination codons are recognized in the nucleus in a reading-frame-dependent manner. Cell Discov. 2015, 1, 15001. [Google Scholar] [CrossRef]
- Han, X.; Wei, Y.; Wang, H.; Wang, F.; Ju, Z.; Li, T. Nonsense-mediated mRNA decay: A “nonsense” pathway makes sense in stem cell biology. Nucleic Acids Res. 2018, 46, 1038–1051. [Google Scholar] [CrossRef]
- Howden, S.E.; Maufort, J.P.; Duffin, B.M.; Elefanty, A.G.; Stanley, E.G.; Thomson, J.A. Simultaneous Reprogramming and Gene Correction of Patient Fibroblasts. Stem Cell Rep. 2015, 5, 1109–1118. [Google Scholar] [CrossRef]
- Zhang, X.; Li, Z.; Liu, Y.; Gai, Z. Great expectations: Induced pluripotent stem cell technologies in neurodevelopmental impairments. Int. J. Med. Sci. 2020, 18, 459–473. [Google Scholar] [CrossRef] [PubMed]
- Taapken, S.M.; Nisler, B.S.; Newton, M.A.; Sampsell-Barron, T.L.; Leonhard, K.A.; McIntire, E.M.; Montgomery, K.D. Karotypic abnormalities in human induced pluripotent stem cells and embryonic stem cells. Nat. Biotechnol. 2011, 29, 313–314. [Google Scholar] [CrossRef] [PubMed]
- Walker, T.; Huang, J.; Young, K. Neural Stem and Progenitor Cells in Nervous System Function and Therapy. Stem Cells Int. 2016, 2016, 1890568. [Google Scholar] [CrossRef]
- Park, S.M.; Lim, J.S.; Ramakrishina, S.; Kim, S.H.; Kim, W.K.; Lee, J.; Kang, H.C.; Reiter, J.F.; Kim, D.S.; Kim, H.; et al. Brain Somatic Mutations in MTOR Disrupt Neuronal Ciliogenesis, Leading to Focal Cortical Dyslamination. Neuron 2018, 99, 83–97.e7. [Google Scholar] [CrossRef] [PubMed]
- Kossoff, E.H.; Thiele, E.A.; Pfeifer, H.H.; McGrogan, J.R.; Freeman, J.M. Tuberous Sclerosis Complex and the Ketogenic Diet. Epilepsia 2005, 46, 1684–1686. [Google Scholar] [CrossRef]
- Coppola, G.; Klepper, J.; Ammendola, E.; Fiorillo, M.; della Corte, R.; Capano, G.; Pascotto, A. The effects of the ketogenic diet in refractory partial seizures with reference to tuberous sclerosis. Eur. J. Paediatr. Neurol. 2006, 10, 148–151. [Google Scholar] [CrossRef]
- Grocott, O.R.; Herrington, K.S.; Pfeifer, H.H.; Thiele, E.A.; Thibert, R.L. Low glycemic index treatment for seizure control in Angelman syndrome: A case series from the Center for Dietary Therapy of Epilepsy at the Massachusetts General Hospital. Epilepsy Behav. 2017, 68, 45–50. [Google Scholar] [CrossRef]
- Liebhaber, G.M.; Riemann, E.; Matthias Baumeister, F.A. Ketogenic Diet in Rett Syndrome. J. Child Neurol. 2003, 18, 74–75. [Google Scholar] [CrossRef] [PubMed]
- Rho, J.M. How does the ketogenic diet induce anti-seizure effects? Neurosci. Lett. 2017, 637, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Olson, C.A.; Vuong, H.E.; Yano, J.M.; Liang, Q.Y.; Nusbaum, D.J.; Hsiao, E.Y. The Gut Microbiota Mediates the Anti-Seizure Effects of the Ketogenic Diet. Cell 2018, 173, 1728–1741.e13. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhou, S.; Zhou, Y.; Yu, L.; Zhang, L.; Wang, Y. Altered gut microbiome composition in children with refractory epilepsy after ketogenic diet. Epilepsy Res. 2018, 145, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Calderón, N.; Betancourt, L.; Hernández, L.; Rada, P. A ketogenic diet modifies glutamate, gamma-aminobutyric acid and agmatine levels in the hippocampus of rats: A microdialysis study. Neurosci. Lett. 2017, 642, 158–162. [Google Scholar] [CrossRef] [PubMed]
- Bough, K.J.; Wetherington, J.; Hassel, B.; Pare, J.F.; Gawryluk, J.W.; Greene, J.G.; Shaw, R.; Smith, Y.; Geiger, J.D.; Dingledine, R.J. Mitochondrial biogenesis in the anticonvulsant mechanism of the ketogenic diet. Ann. Neurol. 2006, 60, 223–235. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.X.-Q.; Abdian, N.; Maussion, G.; Thomas, R.A.; Demirova, I.; Cai, E.; Tabatabaei, M.; Beitel, L.K.; Karamchandani, J.; Fon, E.A.; et al. A Multistep Workflow to Evaluate Newly Generated iPSCs and Their Ability to Generate Different Cell Types. Methods Protoc. 2021, 4, 50. [Google Scholar] [CrossRef]
- Namba, T.; Huttner, W.B. Neural progenitor cells and their role in the development and evolutionary expansion of the neocortex. Wiley Interdiscip. Rev. Dev. Biol. 2017, 6, e256. [Google Scholar] [CrossRef]
- Martínez-Cerdeño, V.; Noctor, S.C. Neural progenitor cell terminology. Front. Neuroanat. 2018, 12, 104. [Google Scholar] [CrossRef]
- Suzuki, H.; Imajo, Y.; Funaba, M.; Nishida, N.; Sakamoto, T.; Sakai, T. Current Concepts of Neural Stem/Progenitor Cell Therapy for Chronic Spinal Cord Injury. Front. Cell. Neurosci. 2022, 15, 794692. [Google Scholar] [CrossRef]
- Mullegama, S.V.; Klein, S.D.; Williams, S.R.; Innis, J.W.; Probst, F.J.; Haldeman-Englert, C.; Martinez-Agosto, J.A.; Yang, Y.; Tian, Y.; Elsea, S.H.; et al. Transcriptome analysis of MBD5-associated neurodevelopmental disorder (MAND) neural progenitor cells reveals dysregulation of autism-associated genes. Sci. Rep. 2021, 11, 11295. [Google Scholar] [CrossRef]
- Tang, F.; Zhang, X.; Ke, P.; Liu, J.; Zhang, Z.; Hu, D.; Gu, J.; Zhang, H.; Guo, H.; Zang, Q.; et al. MBD5 regulates NMDA receptor expression and seizures by inhibiting Stat1 transcription. Neurobiol. Dis. 2023, 181, 106103. [Google Scholar] [CrossRef]
- Shiwaku, H.; Umino, A.; Umino, M.; Nishikawa, T. Phencyclidine-induced dysregulation of primary cilia in the rodent brain. Brain Res. 2017, 1674, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Varga, E.; Nemes, C.; Bock, I.; Táncos, Z.; Berzsenyi, S.; Lévay, G.; Román, V.; Kobolák, J.; Dinnyés, A. Establishment of an induced pluripotent stem cell ( iPSC ) line from a 9-year old male with autism spectrum disorder (ASD). Stem Cell Res. 2017, 21, 19–22. [Google Scholar] [CrossRef] [PubMed]
- Caspary, T.; Larkins, C.E.; Anderson, K.V. The Graded Response to Sonic Hedgehog Depends on Cilia Architecture. Dev. Cell 2007, 12, 767–778. [Google Scholar] [CrossRef] [PubMed]
- Mishra, P.; Singh, U.; Pandey, C.; Mishra, P.; Pandey, G. Application of student’s t-test, analysis of variance, and covariance. Ann. Card. Anaesth. 2019, 22, 407. [Google Scholar] [CrossRef]
- Almuzzaini, B.; Alatwi, N.S.; Alsaif, S.; Al Balwi, M.A. A novel interstitial deletion of chromosome 2q21.1-q23.3: Case report and literature review. Mol. Genet. Genom. Med. 2020, 8, e1135. [Google Scholar] [CrossRef]
- Bonnet, C.; Ali Khan, A.; Bresso, E.; Vigouroux, C.; Béri, M.; Lejczak, S.; Deemer, B.; Andrieux, J.; Philippe, C.; Moncla, A.; et al. Extended spectrum of MBD5 mutations in neurodevelopmental disorders. Eur. J. Hum. Genet. 2013, 21, 1457–1461. [Google Scholar] [CrossRef]
- Bravo-Oro, A.; Lurie, I.W.; Elizondo-Cárdenas, G.; Peña-Zepeda, C.; Salazar-Martínez, A.; Correa-González, C.; Castrillo, J.L.; Avila, S.; Esmer, C. A novel interstitial deletion of 2q22.3 q23.3 in a patient with dysmorphic features, epilepsy, aganglionosis, pure red cell aplasia, and skeletal malformations. Am. J. Med. Genet. A 2015, 167A, 1865–1871. [Google Scholar] [CrossRef]
- Castro-Gago, M.; Gómez-Lado, C.; Barros-Angueira, F.; Trujillo-Ariza, M.V.; Fuentes-Pita, P.; López-Vázquez, A.M.; Eirís-Puñal, J. De novo heterozygous mutation in the MBD5 gene associated with bilateral band heterotopia and polymicrogyria. Rev. Neurol. 2019, 69, 492–496. [Google Scholar]
- Chung, B.H.Y.; Stavropoulos, J.; Marshall, C.R.; Weksberg, R.; Scherer, S.W.; Yoon, G. 2q23 de novo microdeletion involving the MBD5 gene in a patient with developmental delay, postnatal microcephaly and distinct facial features. Am. J. Med. Genet. Part A 2011, 155, 424–429. [Google Scholar] [CrossRef]
- Chung, B.H.Y.; Mullegama, S.; Marshall, C.R.; Lionel, A.C.; Weksberg, R.; Dupuis, L.; Brick, L.; Li, C.; Scherer, S.W.; Aradhya, S.; et al. Severe intellectual disability and autistic features associated with microduplication 2q23.1. Eur. J. Hum. Genet. 2012, 20, 398–403. [Google Scholar] [CrossRef] [PubMed]
- Cukier, H.N.; Lee, J.M.; Ma, D.; Young, J.I.; Mayo, V.; Butler, B.L.; Ramsook, S.S.; Rantus, J.A.; Abrams, A.J.; Whitehead, P.L.; et al. The Expanding Role of MBD Genes in Autism: Identification of a MECP2 Duplication and Novel Alterations in MBD5, MBD6, and SETDB1. Autism Res. 2012, 5, 385–397. [Google Scholar] [CrossRef]
- De Vries, B.B.A.; Pfundt, R.; Leisink, M.; Koolen, D.A.; Vissers, L.E.L.M.; Janssen, I.M.; Van Reijmersdal, S.; Nillesen, W.M.; Huys, E.H.L.P.G.; De Leeuw, N.; et al. Diagnostic genome profiling in mental retardation. Am. J. Hum. Genet. 2005, 77, 606–616. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; An, Y.; Yu, L.; Liu, R.; Qin, Y.; Guo, X.; Sun, D.; Zhou, S.; Wu, B.; Jiang, Y.; et al. A genomic copy number variant analysis implicates the MBD5 and HNRNPU genes in Chinese children with infantile spasms and expands the clinical spectrum of 2q23.1 deletion. BMC Med. Genet. 2014, 15, 62. [Google Scholar] [CrossRef]
- Fry, A.E.; Rees, E.; Thompson, R.; Mantripragada, K.; Blake, P.; Jones, G.; Morgan, S.; Jose, S.; Mugalaasi, H.; Archer, H.; et al. Pathogenic copy number variants and SCN1A mutations in patients with intellectual disability and childhood-onset epilepsy. BMC Med. Genet. 2016, 17, 34. [Google Scholar] [CrossRef]
- Gokben, S.; Onay, H.; Yilmaz, S.; Atik, T.; Serdaroglu, G.; Tekin, H.; Ozkinay, F. Targeted next generation sequencing: The diagnostic value in early-onset epileptic encephalopathy. Acta Neurol. Belg. 2017, 117, 131–138. [Google Scholar] [CrossRef]
- Hamdan, F.F.; Srour, M.; Capo-Chichi, J.-M.; Daoud, H.; Nassif, C.; Patry, L.; Massicotte, C.; Ambalavanan, A.; Spiegelman, D.; Diallo, O.; et al. De novo mutations in moderate or severe intellectual disability. PLoS Genet. 2014, 10, e1004772. [Google Scholar] [CrossRef] [PubMed]
- Han, J.Y.; Lee, I.G.; Jang, W.; Kim, M.; Kim, Y.; Jang, J.H.; Park, J. Diagnostic exome sequencing identifies a heterozygous MBD5 frameshift mutation in a family with intellectual disability and epilepsy. Eur. J. Med. Genet. 2017, 60, 559–564. [Google Scholar] [CrossRef]
- Ishizuka, K.; Kimura, H.; Yoshimi, A.; Banno, M.; Kushima, I.; Uno, Y.; Okada, T.; Mori, D.; Aleksic, B.; Ozaki, N. Investigation of single-nucleotide variants in MBD5 associated with autism spectrum disorders and schizophrenia phenotypes. Nagoya J. Med. Sci. 2016, 78, 465–474. [Google Scholar]
- Jaillard, S.; Dubourg, C.; Gérard-Blanluet, M.; Delahaye, A.; Pasquier, L.; Dupont, C.; Henry, C.; Tabet, A.C.; Lucas, J.; Aboura, A.; et al. 2q23.1 microdeletion identified by array comparative genomic hybridisation: An emerging phenotype with Angelman-like features? J. Med. Genet. 2009, 46, 847–855. [Google Scholar] [CrossRef]
- Kushima, I.; Aleksic, B.; Nakatochi, M.; Shimamura, T.; Shiino, T.; Yoshimi, A.; Kimura, H.; Takasaki, Y.; Wang, C.; Xing, J.; et al. High-resolution copy number variation analysis of schizophrenia in Japan. Mol. Psychiatry 2017, 22, 430–440. [Google Scholar] [CrossRef]
- Le, T.N.U.; Ha, T.M.T. MBD5-related intellectual disability in a Vietnamese child. Am. J. Med. Genet. Part A 2021, 185, 1321–1323. [Google Scholar] [CrossRef]
- Motobayashi, M.; Nishimura-Tadaki, A.; Inaba, Y.; Kosho, T.; Miyatake, S.; Niimi, T.; Nishimura, T.; Wakui, K.; Fukushima, Y.; Matsumoto, N.; et al. Neurodevelopmental features in 2q23.1 microdeletion syndrome: Report of a new patient with intractable seizures and review of literature. Am. J. Med. Genet. Part A 2012, 158 A, 861–868. [Google Scholar] [CrossRef]
- Mullegama, S.V.; Rosenfeld, J.A.; Orellana, C.; Van Bon, B.W.M.M.; Halbach, S.; Repnikova, E.A.; Brick, L.; Li, C.; Dupuis, L.; Rosello, M.; et al. Reciprocal deletion and duplication at 2q23.1 indicates a role for MBD5 in autism spectrum disorder. Eur. J. Hum. Genet. 2014, 22, 57–63. [Google Scholar] [CrossRef]
- Mullegama, S.V.; Elsea, S.H. Intragenic MBD5 familial deletion variant does not negatively impact MBD5 mRNA expression. Mol. Cytogenet. 2014, 7, 80. [Google Scholar] [CrossRef][Green Version]
- Myers, K.A.; Marini, C.; Carvill, G.L.; McTague, A.; Panetta, J.; Stutterd, C.; Stanley, T.; Marin, S.; Nguyen, J.; Barba, C.; et al. Phenotypic spectrum of seizure disorders in MBD5-associated neurodevelopmental disorder. Neurol. Genet. 2021, 7, e579. [Google Scholar] [CrossRef]
- Noh, G.J.; Graham, J.M.J. 2q23.1 microdeletion of the MBD5 gene in a female with seizures, developmental delay and distinct dysmorphic features. Eur. J. Med. Genet. 2012, 55, 59–62. [Google Scholar] [CrossRef]
- Ohori, S.; Tsuburaya, R.S.; Kinoshita, M.; Miyagi, E.; Mizuguchi, T.; Mitsuhashi, S.; Frith, M.C.; Matsumoto, N. Long-read whole-genome sequencing identified a partial MBD5 deletion in an exome-negative patient with neurodevelopmental disorder. J. Hum. Genet. 2021, 66, 697–705. [Google Scholar] [CrossRef]
- Orrico, A.; Galli, L.; Rossi, M.; Cortesi, A.; Mazzi, M.; Caterino, E. The Variable Expression of a Novel MBD5 Gene Frameshift Mutation in an Italian Family. Neuropediatrics 2021, 52, 138–141. [Google Scholar] [CrossRef]
- Shichiji, M.; Ito, Y.; Shimojima, K.; Nakamu, H.; Oguni, H.; Osawa, M.; Yamamoto, T. A cryptic microdeletion including MBD5 occurring within the breakpoint of a reciprocal translocation between chromosomes 2 and 5 in a patient with developmental delay and obesity. Am. J. Med. Genet. Part A 2013, 161, 850–855. [Google Scholar] [CrossRef]
- Tadros, S.; Wang, R.; Waters, J.J.; Waterman, C.; Collins, A.L.; Collinson, M.N.; Ahn, J.W.; Josifova, D.; Chetan, R.; Kumar, A. Inherited 2q23.1 microdeletions involving the MBD5 locus. Mol. Genet. Genom. Med. 2017, 5, 608–613. [Google Scholar] [CrossRef]
- Turner, T.N.; Hormozdiari, F.; Duyzend, M.H.; McClymont, S.A.; Hook, P.W.; Iossifov, I.; Raja, A.; Baker, C.; Hoekzema, K.; Stessman, H.A.; et al. Genome Sequencing of Autism-Affected Families Reveals Disruption of Putative Noncoding Regulatory DNA. Am. J. Hum. Genet. 2016, 98, 58–74. [Google Scholar] [CrossRef]
- Van Bon, B.W.M.; Koolen, D.A.; Brueton, L.; McMullan, D.; Lichtenbelt, K.D.; Adès, L.C.; Peters, G.; Gibson, K.; Novara, F.; Pramparo, T.; et al. The 2q23.1 microdeletion syndrome: Clinical and behavioural phenotype. Eur. J. Hum. Genet. 2010, 18, 163–170. [Google Scholar] [CrossRef]
- Verhoeven, W.; Egger, J.; Kipp, J.; Verheul-Aan de Wiel, J.; Ockeloen, C.; Kleefstra, T.; Pfundt, R. A novel MBD5 mutation in an intellectually disabled adult female patient with epilepsy: Suggestive of early onset dementia? Mol. Genet. Genom. Med. 2019, 7, e849. [Google Scholar] [CrossRef]
- Wagenstaller, J.; Spranger, S.; Lorenz-Depiereux, B.; Kazmierczak, B.; Nathrath, M.; Wahl, D.; Heye, B.; Glaser, D.; Liebscher, V.; Meitinger, T.; et al. Copy-number variations measured by single-nucleotide-polymorphism oligonucleotide arrays in patients with mental retardation. Am. J. Hum. Genet. 2007, 81, 768–779. [Google Scholar] [CrossRef]
- Wang, Y.; Du, X.; Bin, R.; Yu, S.; Xia, Z.; Zheng, G.; Zhong, J.; Zhang, Y.; Jiang, Y.-H.; Wang, Y. Genetic Variants Identified from Epilepsy of Unknown Etiology in Chinese Children by Targeted Exome Sequencing. Sci. Rep. 2017, 7, 40319. [Google Scholar] [CrossRef]
- Williams, S.R.; Mullegama, S.V.; Rosenfeld, J.A.; Dagli, A.I.; Hatchwell, E.; Allen, W.P.; Williams, C.A.; Elsea, S.H. Haploinsufficiency of MBD5 associated with a syndrome involving microcephaly, intellectual disabilities, severe speech impairment, and seizures. Eur. J. Hum. Genet. 2010, 18, 436–441. [Google Scholar] [CrossRef]
- Woodbury-Smith, M.; Nicolson, R.; Zarrei, M.; Yuen, R.K.C.; Walker, S.; Howe, J.; Uddin, M.; Hoang, N.; Buchanan, J.A.; Chrysler, C.; et al. Variable phenotype expression in a family segregating microdeletions of the NRXN1 and MBD5 autism spectrum disorder susceptibility genes. NPJ Genom. Med. 2017, 2, 17. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martins, M.; Oliveira, A.R.; Martins, S.; Vieira, J.P.; Perdigão, P.; Fernandes, A.R.; de Almeida, L.P.; Palma, P.J.; Sequeira, D.B.; Santos, J.M.M.; et al. A Novel Genetic Variant in MBD5 Associated with Severe Epilepsy and Intellectual Disability: Potential Implications on Neural Primary Cilia. Int. J. Mol. Sci. 2023, 24, 12603. https://doi.org/10.3390/ijms241612603
Martins M, Oliveira AR, Martins S, Vieira JP, Perdigão P, Fernandes AR, de Almeida LP, Palma PJ, Sequeira DB, Santos JMM, et al. A Novel Genetic Variant in MBD5 Associated with Severe Epilepsy and Intellectual Disability: Potential Implications on Neural Primary Cilia. International Journal of Molecular Sciences. 2023; 24(16):12603. https://doi.org/10.3390/ijms241612603
Chicago/Turabian StyleMartins, Mariana, Ana Rafaela Oliveira, Solange Martins, José Pedro Vieira, Pedro Perdigão, Ana Rita Fernandes, Luís Pereira de Almeida, Paulo Jorge Palma, Diana Bela Sequeira, João Miguel Marques Santos, and et al. 2023. "A Novel Genetic Variant in MBD5 Associated with Severe Epilepsy and Intellectual Disability: Potential Implications on Neural Primary Cilia" International Journal of Molecular Sciences 24, no. 16: 12603. https://doi.org/10.3390/ijms241612603
APA StyleMartins, M., Oliveira, A. R., Martins, S., Vieira, J. P., Perdigão, P., Fernandes, A. R., de Almeida, L. P., Palma, P. J., Sequeira, D. B., Santos, J. M. M., Duque, F., Oliveira, G., Cardoso, A. L., Peça, J., & Seabra, C. M. (2023). A Novel Genetic Variant in MBD5 Associated with Severe Epilepsy and Intellectual Disability: Potential Implications on Neural Primary Cilia. International Journal of Molecular Sciences, 24(16), 12603. https://doi.org/10.3390/ijms241612603