
Effect of Vitamin E on Transcriptomic Alterations in Alzheimer’s Disease

 ,
, 
Abstract
1. Introduction
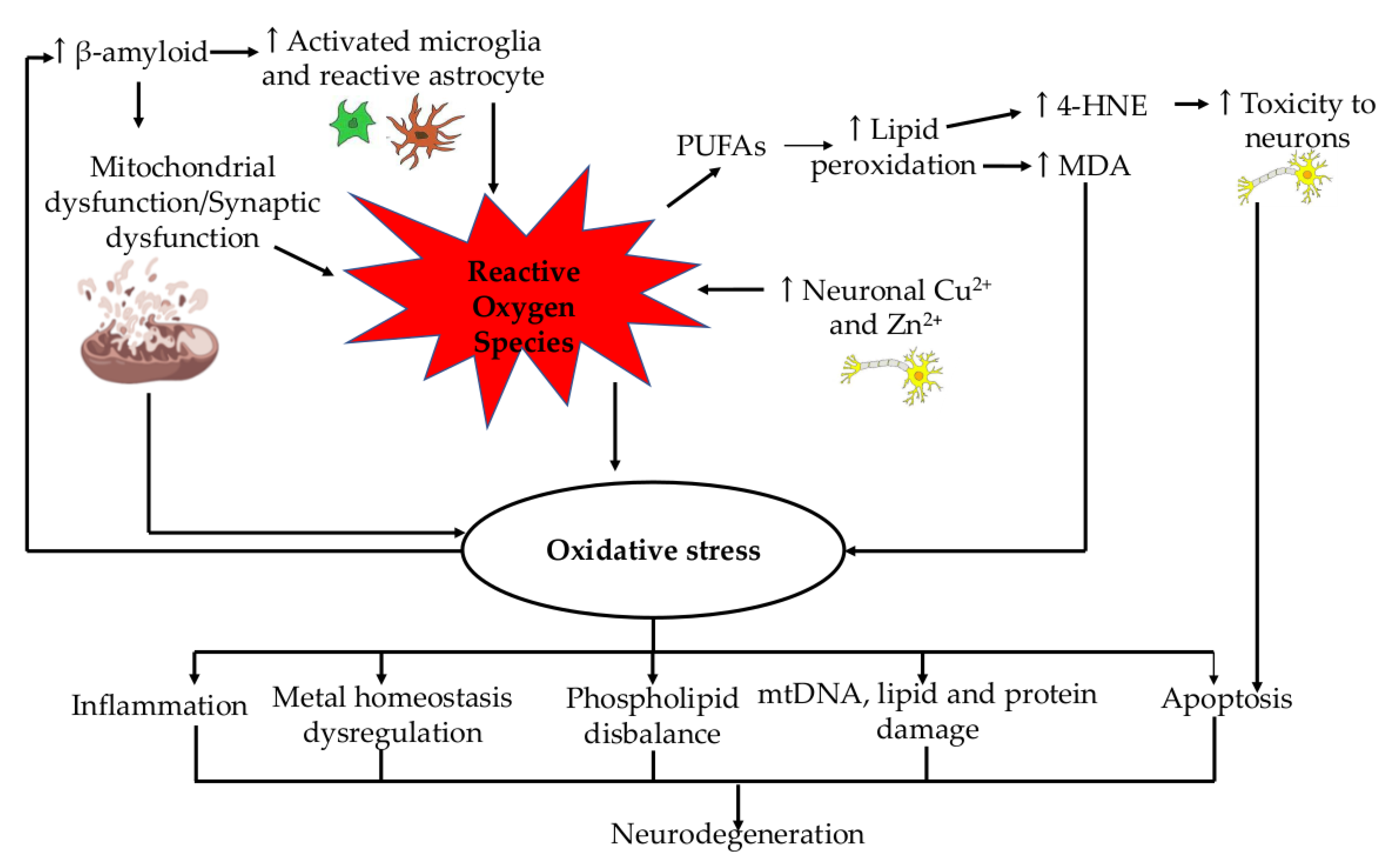
2. Role of Oxidative Stress in Alzheimer’s Disease
3. Transcriptomic Alterations in Alzheimer’s Disease
4. Vitamin E
5. Effect of Vitamin E’s Transcriptomic Alterations in Alzheimer’s Disease
6. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rowe, J.W.; Kahn, R.L. Human aging: Usual and successful. Science 1987, 237, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Tesch-Römer, C.; Wahl, H.-W. Toward a More Comprehensive Concept of Successful Aging: Disability and Care Needs. J. Gerontol. B Psychol. Sci. Soc. Sci. 2017, 72, 310–318. [Google Scholar] [CrossRef] [PubMed]
- Tomasetti, C.; Poling, J.; Roberts, N.J.; London, N.R.; Pittman, M.E.; Haffner, M.C.; Rizzo, A.; Baras, A.; Karim, B.; Kim, A.; et al. Cell division rates decrease with age, providing a potential explanation for the age-dependent deceleration in cancer incidence. Proc. Natl. Acad. Sci. USA 2019, 116, 20482–20488. [Google Scholar] [CrossRef] [PubMed]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Dan, X.; Babbar, M.; Wei, Y.; Hasselbalch, S.G.; Croteau, D.L.; Bohr, V.A. Ageing as a risk factor for neurodegenerative disease. Nat. Rev. Neurol. 2019, 15, 565–581. [Google Scholar] [CrossRef] [PubMed]
- Cheignon, C.; Tomas, M.; Bonnefont-Rousselot, D.; Faller, P.; Hureau, C.; Collin, F. Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox Biol. 2018, 14, 450–464. [Google Scholar] [CrossRef]
- Pecorini, S.; Gibellini, L.; Biasi, S.; Bianchini, E.; Milena, N.; Cossarizza, A.; Pinti, M. Mitochondria, Oxidative Stress, Cancer, and Aging. In Geriatric Oncology; Springer: Cham, Switzerland, 2020; pp. 183–204. ISBN 978-3-319-57414-1. [Google Scholar]
- Luo, J.; Mills, K.; le Cessie, S.; Noordam, R.; van Heemst, D. Ageing, age-related diseases and oxidative stress: What to do next? Ageing Res. Rev. 2020, 57, 100982. [Google Scholar] [CrossRef]
- Fan, L.; Mao, C.; Hu, X.; Zhang, S.; Yang, Z.; Hu, Z.; Sun, H.; Fan, Y.; Dong, Y.; Yang, J.; et al. New Insights Into the Pathogenesis of Alzheimer’s Disease. Front. Neurol. 2020, 10, 1312. [Google Scholar] [CrossRef]
- Magistri, M.; Velmeshev, D.; Makhmutova, M.; Faghihi, M.A. Transcriptomics Profiling of Alzheimer’s Disease Reveal Neurovascular Defects, Altered Amyloid-β Homeostasis, and Deregulated Expression of Long Noncoding RNAs. J. Alzheimers Dis. 2015, 48, 647–665. [Google Scholar] [CrossRef]
- Association, A. 2018 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2018, 14, 367–429. [Google Scholar] [CrossRef]
- Hebert, L.E.; Weuve, J.; Scherr, P.A.; Evans, D.A. Alzheimer disease in the United States (2010–2050) estimated using the 2010 census. Neurology 2013, 80, 1778–1783. [Google Scholar] [CrossRef] [PubMed]
- Barnes, D.E.; Yaffe, K. The projected effect of risk factor reduction on Alzheimer’s disease prevalence. Lancet. Neurol. 2011, 10, 819–828. [Google Scholar] [CrossRef] [PubMed]
- Kritsilis, M.; Rizou, S.V.; Koutsoudaki, P.N.; Evangelou, K.; Gorgoulis, V.G.; Papadopoulos, D. Ageing, Cellular Senescence and Neurodegenerative Disease. Int. J. Mol. Sci. 2018, 19, 2937. [Google Scholar] [CrossRef] [PubMed]
- Ma, M.W.; Wang, J.; Zhang, Q.; Wang, R.; Dhandapani, K.M.; Vadlamudi, R.K.; Brann, D.W. NADPH oxidase in brain injury and neurodegenerative disorders. Mol. Neurodegener. 2017, 12, 7. [Google Scholar] [CrossRef] [PubMed]
- Shea, D.; Hsu, C.-C.; Bi, T.M.; Paranjapye, N.; Childers, M.C.; Cochran, J.; Tomberlin, C.P.; Wang, L.; Paris, D.; Zonderman, J.; et al. α-Sheet secondary structure in amyloid β-peptide drives aggregation and toxicity in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2019, 116, 8895–8900. [Google Scholar] [CrossRef]
- Jara, C.; Aránguiz, A.; Cerpa, W.; Tapia-Rojas, C.; Quintanilla, R.A. Genetic ablation of tau improves mitochondrial function and cognitive abilities in the hippocampus. Redox Biol. 2018, 18, 279–294. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.S.; Bloom, G.S. Tau: The Center of a Signaling Nexus in Alzheimer’s Disease. Front. Neurosci. 2016, 10, 31. [Google Scholar] [CrossRef]
- Busche, M.A.; Wegmann, S.; Dujardin, S.; Commins, C.; Schiantarelli, J.; Klickstein, N.; Kamath, T.V.; Carlson, G.A.; Nelken, I.; Hyman, B.T. Tau impairs neural circuits, dominating amyloid-β effects, in Alzheimer models in vivo. Nat. Neurosci. 2019, 22, 57–64. [Google Scholar] [CrossRef]
- Barten, D.M.; Fanara, P.; Andorfer, C.; Hoque, N.; Wong, P.Y.A.; Husted, K.H.; Cadelina, G.W.; DeCarr, L.B.; Yang, L.; Liu, V.; et al. Hyperdynamic Microtubules, Cognitive Deficits, and Pathology Are Improved in Tau Transgenic Mice with Low Doses of the Microtubule-Stabilizing Agent BMS-241027. J. Neurosci. 2012, 32, 7137–7145. [Google Scholar] [CrossRef]
- Gauthier, S.; Feldman, H.H.; Schneider, L.S.; Wilcock, G.K.; Frisoni, G.B.; Hardlund, J.H.; Moebius, H.J.; Bentham, P.; Kook, K.A.; Wischik, D.J.; et al. Efficacy and safety of tau-aggregation inhibitor therapy in patients with mild or moderate Alzheimer’s disease: A randomised, controlled, double-blind, parallel-arm, phase 3 trial. Lancet 2016, 388, 2873–2884. [Google Scholar] [CrossRef]
- Novak, P.; Zilka, N.; Zilkova, M.; Kovacech, B.; Skrabana, R.; Ondrus, M.; Fialova, L.; Kontsekova, E.; Otto, M.; Novak, M. AADvac1, an Active Immunotherapy for Alzheimer’s Disease and Non Alzheimer Tauopathies: An Overview of Preclinical and Clinical Development. J. Prev. Alzheimer’s Dis. 2019, 6, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Cassidy, L.; Fernandez, F.; Johnson, J.B.; Naiker, M.; Owoola, A.G.; Broszczak, D.A. Oxidative stress in alzheimer’s disease: A review on emergent natural polyphenolic therapeutics. Complement. Ther. Med. 2020, 49, 102294. [Google Scholar] [CrossRef] [PubMed]
- Stefanatos, R.; Sanz, A. The role of mitochondrial ROS in the aging brain. FEBS Lett. 2018, 592, 743–758. [Google Scholar] [CrossRef] [PubMed]
- Tadokoro, K.; Ohta, Y.; Inufusa, H.; Loon, A.F.N.; Abe, K. Prevention of Cognitive Decline in Alzheimer’s Disease by Novel Antioxidative Supplements. Int. J. Mol. Sci. 2020, 21, 1974. [Google Scholar] [CrossRef] [PubMed]
- Cenini, G.; Lloret, A.; Cascella, R. Oxidative Stress in Neurodegenerative Diseases: From a Mitochondrial Point of View. Oxid. Med. Cell. Longev. 2019, 2019, 2105607. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Oxidative Stress: A Key Modulator in Neurodegenerative Diseases. Molecules 2019, 24, 1583. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Halliwell, B. Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nat. Rev. Neurosci. 2019, 20, 148–160. [Google Scholar] [CrossRef]
- Simunkova, M.; Alwasel, S.H.; Alhazza, I.M.; Jomova, K.; Kollar, V.; Rusko, M.; Valko, M. Management of oxidative stress and other pathologies in Alzheimer’s disease. Arch. Toxicol. 2019, 93, 2491–2513. [Google Scholar] [CrossRef]
- Buccellato, F.R.; D’Anca, M.; Fenoglio, C.; Scarpini, E.; Galimberti, D. Role of Oxidative Damage in Alzheimer’s Disease and Neurodegeneration: From Pathogenic Mechanisms to Biomarker Discovery. Antioxidants 2021, 10, 1353. [Google Scholar] [CrossRef]
- Teleanu, D.M.; Niculescu, A.-G.; Lungu, I.I.; Radu, C.I.; Vladâcenco, O.; Roza, E.; Costăchescu, B.; Grumezescu, A.M.; Teleanu, R.I. An Overview of Oxidative Stress, Neuroinflammation, and Neurodegenerative Diseases. Int. J. Mol. Sci. 2022, 23, 5938. [Google Scholar] [CrossRef]
- Kausar, S.; Wang, F.; Cui, H. The Role of Mitochondria in Reactive Oxygen Species Generation and Its Implications for Neurodegenerative Diseases. Cells 2018, 7, 274. [Google Scholar] [CrossRef] [PubMed]
- Larosa, V.; Remacle, C. Insights into the respiratory chain and oxidative stress. Biosci. Rep. 2018, 38, BSR20171492. [Google Scholar] [CrossRef] [PubMed]
- Collin, F. Chemical Basis of Reactive Oxygen Species Reactivity and Involvement in Neurodegenerative Diseases. Int. J. Mol. Sci. 2019, 20, 2407. [Google Scholar] [CrossRef]
- Tabassum, R.; Jeong, N.Y.; Jung, J. Therapeutic importance of hydrogen sulfide in age-associated neurodegenerative diseases. Neural Regen. Res. 2020, 15, 653–662. [Google Scholar] [CrossRef] [PubMed]
- Aslebagh, R.; Pfeffer, B.A.; Fliesler, S.J.; Darie, C.C. Mass spectrometry-based proteomics of oxidative stress: Identification of 4-hydroxy-2-nonenal (HNE) adducts of amino acids using lysozyme and bovine serum albumin as model proteins. Electrophoresis 2016, 37, 2615–2623. [Google Scholar] [CrossRef] [PubMed]
- Moldogazieva, N.T.; Mokhosoev, I.M.; Mel’nikova, T.I.; Porozov, Y.B.; Terentiev, A.A. Oxidative Stress and Advanced Lipoxidation and Glycation End Products (ALEs and AGEs) in Aging and Age-Related Diseases. Oxid. Med. Cell. Longev. 2019, 2019, 3085756. [Google Scholar] [CrossRef]
- Khalil, B.; Morderer, D.; Price, P.L.; Liu, F.; Rossoll, W. mRNP assembly, axonal transport, and local translation in neurodegenerative diseases. Brain Res. 2018, 1693, 75–91. [Google Scholar] [CrossRef]
- Mathys, H.; Davila-Velderrain, J.; Peng, Z.; Gao, F.; Mohammadi, S.; Young, J.Z.; Menon, M.; He, L.; Abdurrob, F.; Jiang, X.; et al. Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 2019, 570, 332–337. [Google Scholar] [CrossRef]
- Sinha, S.; Jones, B.M.; Traniello, I.M.; Bukhari, S.A.; Halfon, M.S.; Hofmann, H.A.; Huang, S.; Katz, P.S.; Keagy, J.; Lynch, V.J.; et al. Behavior-related gene regulatory networks: A new level of organization in the brain. Proc. Natl. Acad. Sci. USA 2020, 117, 23270–23279. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, J.; Huang, B.O.; Xu, Y.-M.; Li, J.; Huang, L.-F.; Lin, J.; Zhang, J.; Min, Q.-H.; Yang, W.-M.; et al. Mechanism of alternative splicing and its regulation. Biomed. Rep. 2015, 3, 152–158. [Google Scholar] [CrossRef]
- Ishii, T. Chapter 11—Transcriptome Analysis of Adrenocortical Cells in Health and Disease. In Cellular Endocrinology in Health and Disease; Ulloa-Aguirre, A., Conn, P.M., Eds.; Academic Press: Boston, MA, USA, 2014; pp. 169–192. ISBN 978-0-12-408134-5. [Google Scholar]
- Bagyinszky, E.; Van Giau, V.; An, S.A. Transcriptomics in Alzheimer’s Disease: Aspects and Challenges. Int. J. Mol. Sci. 2020, 21, 3517. [Google Scholar] [CrossRef]
- Patel, H.; Dobson, R.J.B.; Newhouse, S.J. A Meta-Analysis of Alzheimer’s Disease Brain Transcriptomic Data. J. Alzheimers Dis. 2019, 68, 1635–1656. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, H.; Long, J.; Pan, G.; He, T.; Anichtchik, O.; Belshaw, R.; Albani, D.; Edison, P.; Green, E.K.; et al. Systematic Analysis and Biomarker Study for Alzheimer’s Disease. Sci. Rep. 2018, 8, 17394. [Google Scholar] [CrossRef] [PubMed]
- Stopa, E.G.; Tanis, K.Q.; Miller, M.C.; Nikonova, E.V.; Podtelezhnikov, A.A.; Finney, E.M.; Stone, D.J.; Camargo, L.M.; Parker, L.; Verma, A.; et al. Comparative transcriptomics of choroid plexus in Alzheimer’s disease, frontotemporal dementia and Huntington’s disease: Implications for CSF homeostasis. Fluids Barriers CNS 2018, 15, 18. [Google Scholar] [CrossRef]
- Lowe, R.; Shirley, N.; Bleackley, M.; Dolan, S.; Shafee, T. Transcriptomics technologies. PLoS Comput. Biol. 2017, 13, e1005457. [Google Scholar] [CrossRef] [PubMed]
- Chu, Y.; Corey, D.R. RNA sequencing: Platform selection, experimental design, and data interpretation. Nucleic Acid Ther. 2012, 22, 271–274. [Google Scholar] [CrossRef] [PubMed]
- Annese, A.; Manzari, C.; Lionetti, C.; Picardi, E.; Horner, D.S.; Chiara, M.; Caratozzolo, M.F.; Tullo, A.; Fosso, B.; Pesole, G.; et al. Whole transcriptome profiling of Late-Onset Alzheimer’s Disease patients provides insights into the molecular changes involved in the disease. Sci. Rep. 2018, 8, 4282. [Google Scholar] [CrossRef] [PubMed]
- Van Rooij, J.G.J.; Meeter, L.H.H.; Melhem, S.; Nijholt, D.A.T.; Wong, T.H.; Rozemuller, A.; Uitterlinden, A.G.; van Meurs, J.G.; van Swieten, J.C. Hippocampal transcriptome profiling combined with protein-protein interaction analysis elucidates Alzheimer’s disease pathways and genes. Neurobiol. Aging 2019, 74, 225–233. [Google Scholar] [CrossRef]
- Chen, G.; Ning, B.; Shi, T. Single-Cell RNA-Seq Technologies and Related Computational Data Analysis. Front. Genet. 2019, 10, 317. [Google Scholar] [CrossRef]
- Long, J.M.; Ray, B.; Lahiri, D.K. MicroRNA-153 physiologically inhibits expression of amyloid-β precursor protein in cultured human fetal brain cells and is dysregulated in a subset of Alzheimer disease patients. J. Biol. Chem. 2012, 287, 31298–31310. [Google Scholar] [CrossRef]
- Angelucci, F.; Cechova, K.; Valis, M.; Kuca, K.; Zhang, B.; Hort, J. MicroRNAs in Alzheimer’s Disease: Diagnostic Markers or Therapeutic Agents? Front. Pharmacol. 2019, 10, 665. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.K.; Kamisah, Y.; Mohamed, N.; Muhammad, N.; Masbah, N.; Mohd Fahami, N.A.; Mohamed, I.N.; Shuid, A.N.; Mohd Saad, Q.; Abdullah, A.; et al. Potential Role of Tocotrienols on Non-Communicable Diseases: A Review of Current Evidence. Nutrients 2020, 12, 259. [Google Scholar] [CrossRef] [PubMed]
- Browne, D.; McGuinness, B.; Woodside, J.V.; McKay, G.J. Vitamin E and Alzheimer’s disease: What do we know so far? Clin. Interv. Aging 2019, 14, 1303–1317. [Google Scholar] [CrossRef] [PubMed]
- Lloret, A.; Esteve, D.; Monllor, P.; Cervera-Ferri, A.; Lloret, A. The Effectiveness of Vitamin E Treatment in Alzheimer’s Disease. Int. J. Mol. Sci. 2019, 20, 879. [Google Scholar] [CrossRef]
- Khadangi, F.; Azzi, A. Vitamin E—The Next 100 Years. IUBMB Life 2019, 71, 411–415. [Google Scholar] [CrossRef]
- Fischer, A.; Rimbach, G. Gene Regulatory Activity of Vitamin E BT. In Vitamin E in Human Health; Weber, P., Birringer, M., Blumberg, J.B., Eggersdorfer, M., Frank, J., Eds.; Springer International Publishing: Cham, Switzerland, 2019; pp. 81–98. ISBN 978-3-030-05315-4. [Google Scholar]
- Ismail, M.; Alsalahi, A.; Imam, M.U.; Ooi, D.J.; Khaza’ai, H.; Aljaberi, M.A.; Shamsudin, M.N.; Idrus, Z. Safety and Neuroprotective Efficacy of Palm Oil and Tocotrienol-Rich Fraction from Palm Oil: A Systematic Review. Nutrients 2020, 12, 521. [Google Scholar] [CrossRef]
- Boccardi, V.; Baroni, M.; Mangialasche, F.; Mecocci, P. Vitamin E family: Role in the pathogenesis and treatment of Alzheimer’s disease. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2016, 2, 182–191. [Google Scholar] [CrossRef]
- İçer, M.; Arslan, N.; Karadağ, G. Effects of vitamin E on neurodegenerative diseases: An update. Acta Neurobiol. Exp. 2021, 81, 21–33. [Google Scholar] [CrossRef]
- Manosso, L.M.; Camargo, A.; Dafre, A.L.; Rodrigues, A.L.S. Vitamin E for the management of major depressive disorder: Possible role of the anti-inflammatory and antioxidant systems. Nutr. Neurosci. 2022, 25, 1310–1324. [Google Scholar] [CrossRef]
- Mehrabadi, S.; Sadr, S.S. Administration of Vitamin D3 and E supplements reduces neuronal loss and oxidative stress in a model of rats with Alzheimer’s disease. Neurol. Res. 2020, 42, 862–868. [Google Scholar] [CrossRef]
- Giraldo, E.; Lloret, A.; Fuchsberger, T.; Viña, J. Aβ and tau toxicities in Alzheimer’s are linked via oxidative stress-induced p38 activation: Protective role of vitamin E. Redox Biol. 2014, 2, 873–877. [Google Scholar] [CrossRef] [PubMed]
- Bozaykut, P.; Karademir, B.; Yazgan, B.; Sozen, E.; Siow, R.C.M.; Mann, G.E.; Ozer, N.K. Effects of vitamin E on peroxisome proliferator-activated receptor γ and nuclear factor-erythroid 2-related factor 2 in hypercholesterolemia-induced atherosclerosis. Free Radic. Biol. Med. 2014, 70, 174–181. [Google Scholar] [CrossRef] [PubMed]
- Testa, G.; Gamba, P.; Di Scipio, F.; Sprio, A.E.; Salamone, P.; Gargiulo, S.; Sottero, B.; Biasi, F.; Berta, G.N.; Poli, G.; et al. Potentiation of amyloid-β peptide neurotoxicity in human dental-pulp neuron-like cells by the membrane lipid peroxidation product 4-hydroxynonenal. Free Radic. Biol. Med. 2012, 53, 1708–1717. [Google Scholar] [CrossRef] [PubMed]
- Arrozi, A.P.; Shukri, S.N.S.; Murshid, N.M.; Shahzalli, A.B.A.; Ngah, W.Z.W.; Damanhuri, H.A.; Makpol, S. Alpha- and Gamma-Tocopherol Modulates the Amyloidogenic Pathway of Amyloid Precursor Protein in an in vitro Model of Alzheimer’s Disease: A Transcriptional Study. Front. Cell. Neurosci. 2022, 16, 846459. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, N.F.; Yanagisawa, D.; Durani, L.W.; Hamezah, H.S.; Damanhuri, H.A.; Wan Ngah, W.Z.; Tsuji, M.; Kiuchi, Y.; Ono, K.; Tooyama, I. Tocotrienol-Rich Fraction Modulates Amyloid Pathology and Improves Cognitive Function in AβPP/PS1 Mice. J. Alzheimers Dis. 2017, 55, 597–612. [Google Scholar] [CrossRef] [PubMed]
- Dysken, M.W.; Sano, M.; Asthana, S.; Vertrees, J.E.; Pallaki, M.; Llorente, M.; Love, S.; Schellenberg, G.D.; McCarten, J.R.; Malphurs, J.; et al. Effect of vitamin E and memantine on functional decline in Alzheimer disease: The TEAM-AD VA cooperative randomized trial. JAMA 2014, 311, 33–44. [Google Scholar] [CrossRef]
- Kryscio, R.J.; Abner, E.L.; Caban-Holt, A.; Lovell, M.; Goodman, P.; Darke, A.K.; Yee, M.; Crowley, J.; Schmitt, F.A. Association of Antioxidant Supplement Use and Dementia in the Prevention of Alzheimer’s Disease by Vitamin E and Selenium Trial (PREADViSE). JAMA Neurol. 2017, 74, 567–573. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Author | Study Design | Brain Region | Result |
|---|---|---|---|
| Patel et al. [44] | Study type: Meta-analysis with 1501 (746 AD, 755 controls) male and female subjects using microarray (Affymetrix and Illumina) and scRNA-seq technology. Number of patients: 1501 (746 AD, 755 controls) Sex: Male and Female Technology: microarray (Affymetrix and Illumina); scRNA-seq | Frontal Lobe Temporal lobe Parietal lobe Cerebellum |
|
| Li et al. [45] | Systematic analysis with 245 AD, 143 MCI and 182 control male and female subjects using microarray (Illumina) technology. |
Occipital Visual Cortex Dorsolateral Prefrontal Cortex Superior Temporal Gyrus Anterior Cingulate Inferior Temporal Gyrus Superior Parietal Lobule Putamen Posterior Cingulate Cortex Nucleus Accumbens Inferior Frontal Gyrus Caudate Nucleus Precentral Gyrus Amygdala Para hippocampal Gyrus Temporal Pole Middle Temporal Gyrus Frontal Pole Hippocampus |
|
| Stopa et al. [46] | Comparative study with 6 Ctrl, 7 AD, 4 FTD and 3 HD male and Female subjects using microarray (Affymetrix) technology. | Choroid plexus |
|
| Mathys et al. [39] | Longitudinal cohort study with 48 AD (24 elevated Aβ and 24 individuals with no or very low Aβ) male and female subjects using scRNA-seq technology | Prefrontal cortex |
|
| Annese et al. [49] | Cohort study with 18 (6 controls, 6 AD and 6 PD) males subjects using scRNA-seq (Illumina HiSeq2000) technology | Hippocampus Temporal gyrus Frontal gyrus |
|
| Van Rooij et al. [50] | Cohort study with 18 (10 controls and 8 AD) male and female subjects using scRNA-seq technology | Hippocampus |
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ekeuku, S.O.; Mohd Murshid, N.; Shukri, S.N.; Mohd Sahardi, N.F.N.; Makpol, S. Effect of Vitamin E on Transcriptomic Alterations in Alzheimer’s Disease. Int. J. Mol. Sci. 2023, 24, 12372. https://doi.org/10.3390/ijms241512372
Ekeuku SO, Mohd Murshid N, Shukri SN, Mohd Sahardi NFN, Makpol S. Effect of Vitamin E on Transcriptomic Alterations in Alzheimer’s Disease. International Journal of Molecular Sciences. 2023; 24(15):12372. https://doi.org/10.3390/ijms241512372
Chicago/Turabian StyleEkeuku, Sophia Ogechi, Nuraqila Mohd Murshid, Siti Nursyazwani Shukri, Nur Fatin Nabilah Mohd Sahardi, and Suzana Makpol. 2023. "Effect of Vitamin E on Transcriptomic Alterations in Alzheimer’s Disease" International Journal of Molecular Sciences 24, no. 15: 12372. https://doi.org/10.3390/ijms241512372
APA StyleEkeuku, S. O., Mohd Murshid, N., Shukri, S. N., Mohd Sahardi, N. F. N., & Makpol, S. (2023). Effect of Vitamin E on Transcriptomic Alterations in Alzheimer’s Disease. International Journal of Molecular Sciences, 24(15), 12372. https://doi.org/10.3390/ijms241512372