
Comparison of Extracellular Vesicle Isolation Methods for miRNA Sequencing

 , ,
, ,  , , , ,
, , , ,  , , and
, , and  add
Show full author list
add
Show full author list
Abstract
1. Introduction
2. Results
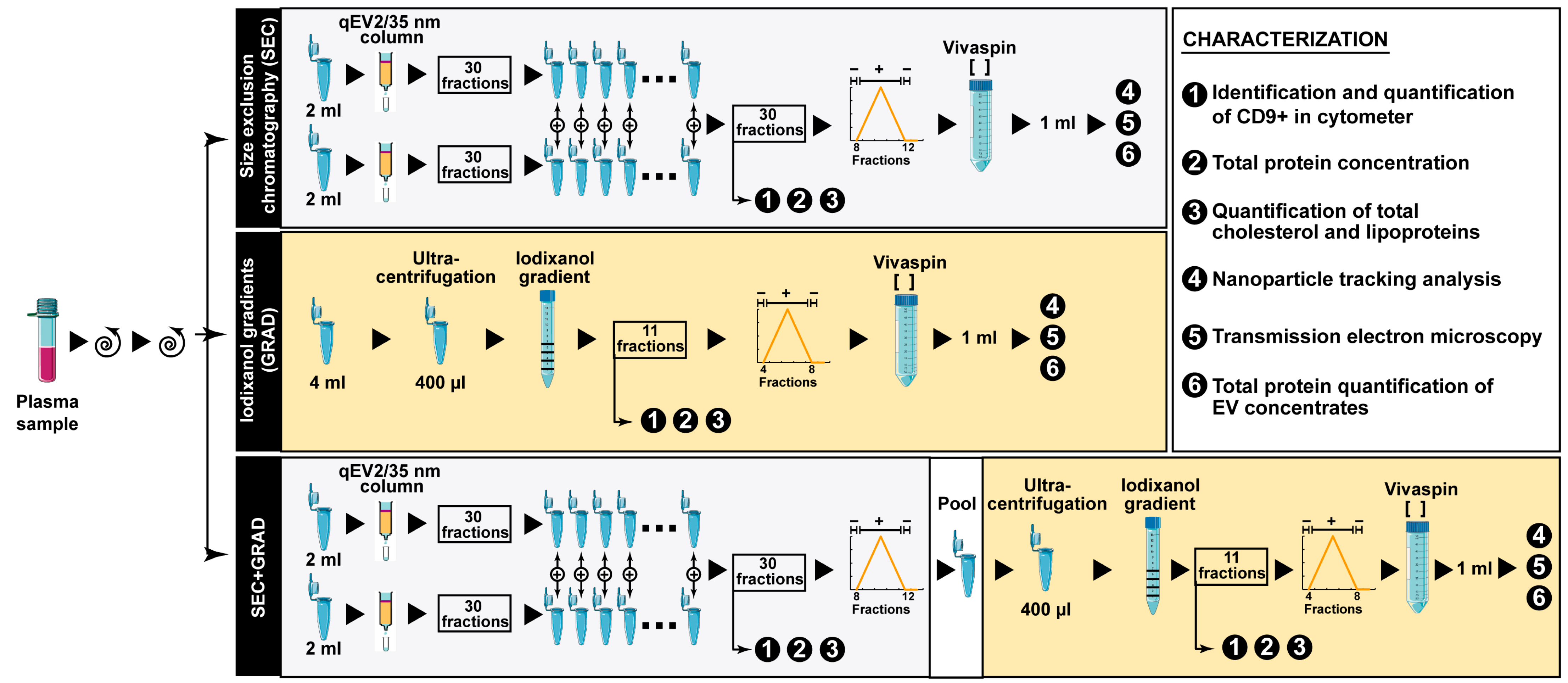
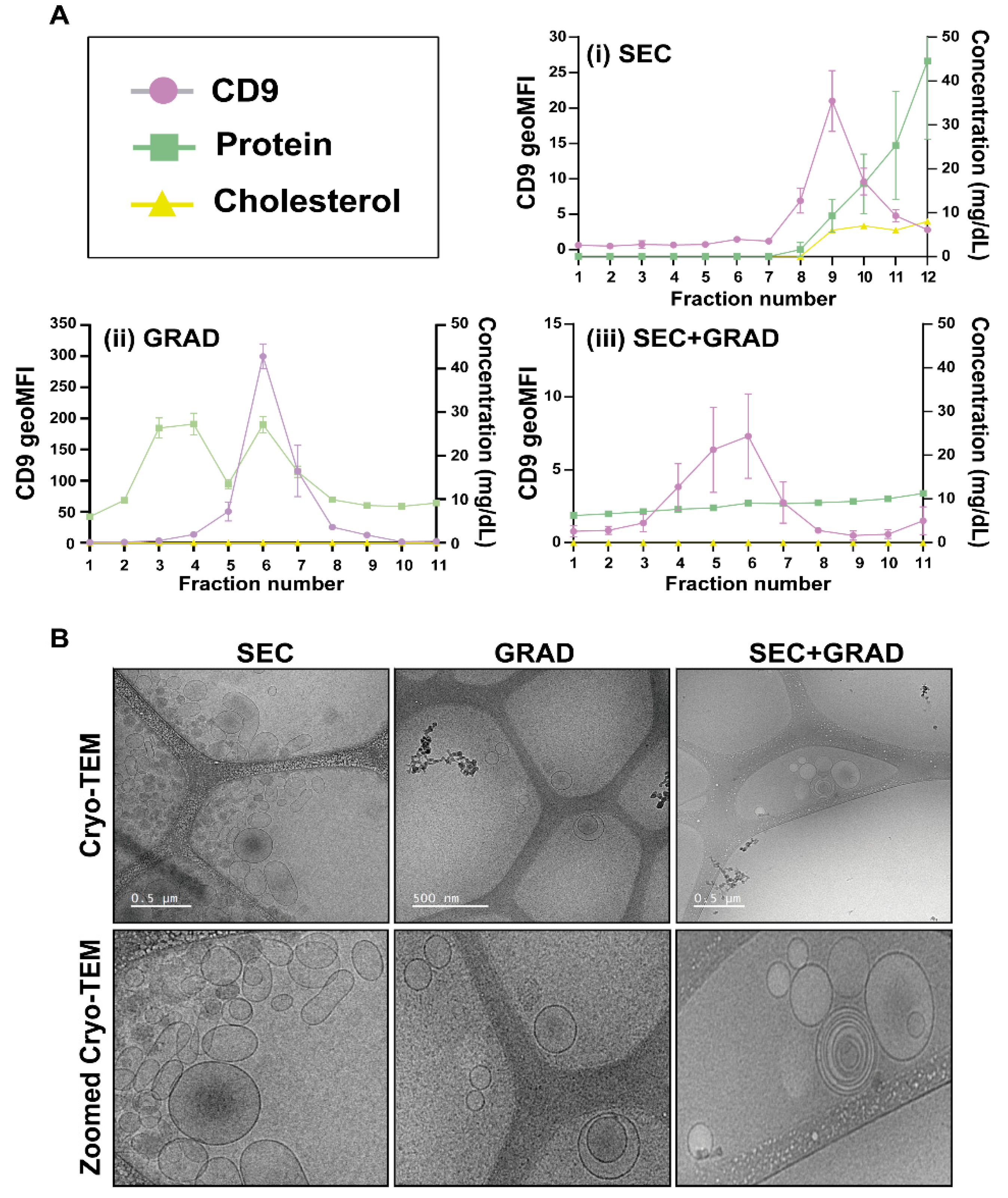
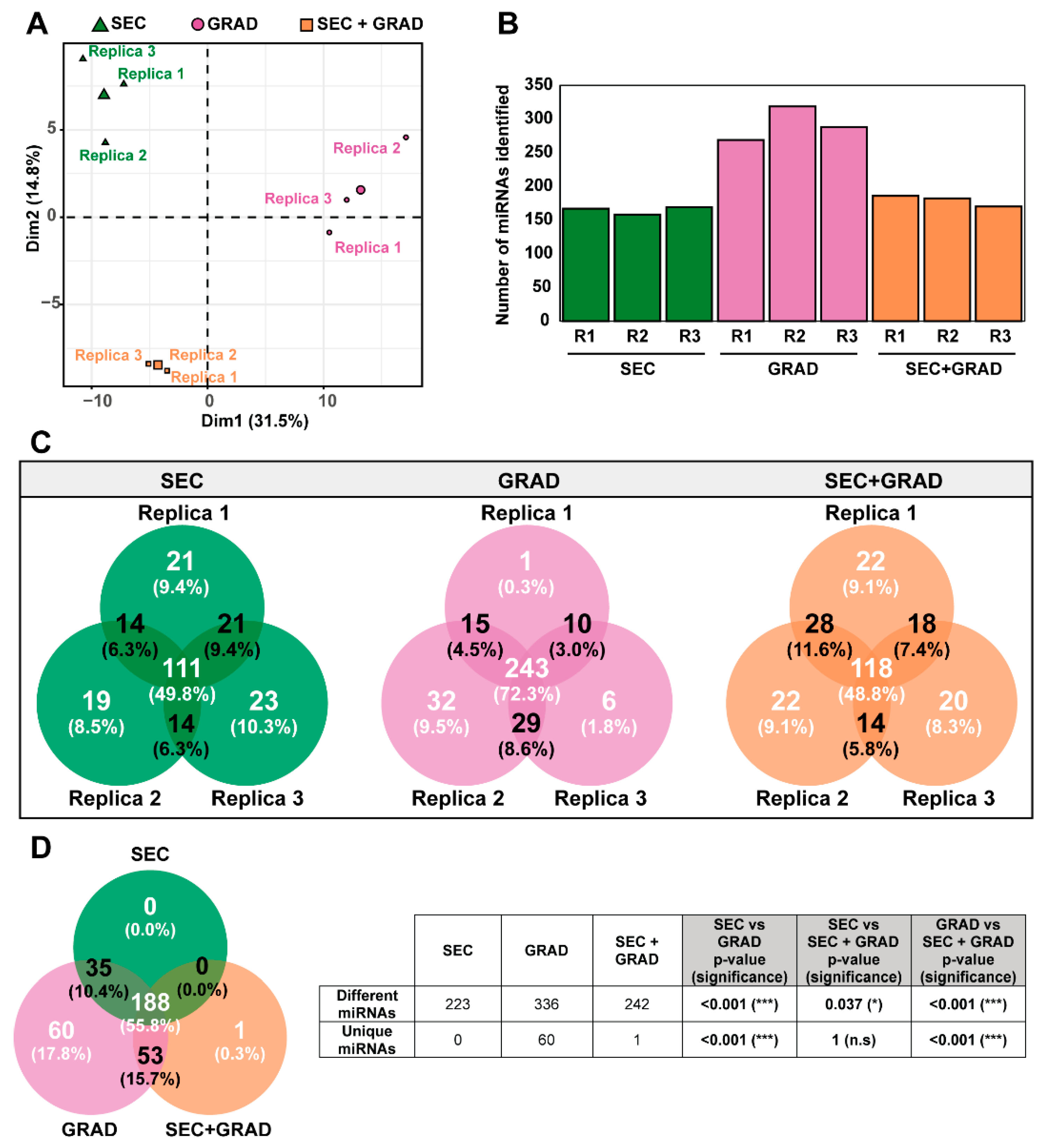
2.1. Comparative Characterization of Three EV Isolation Methods
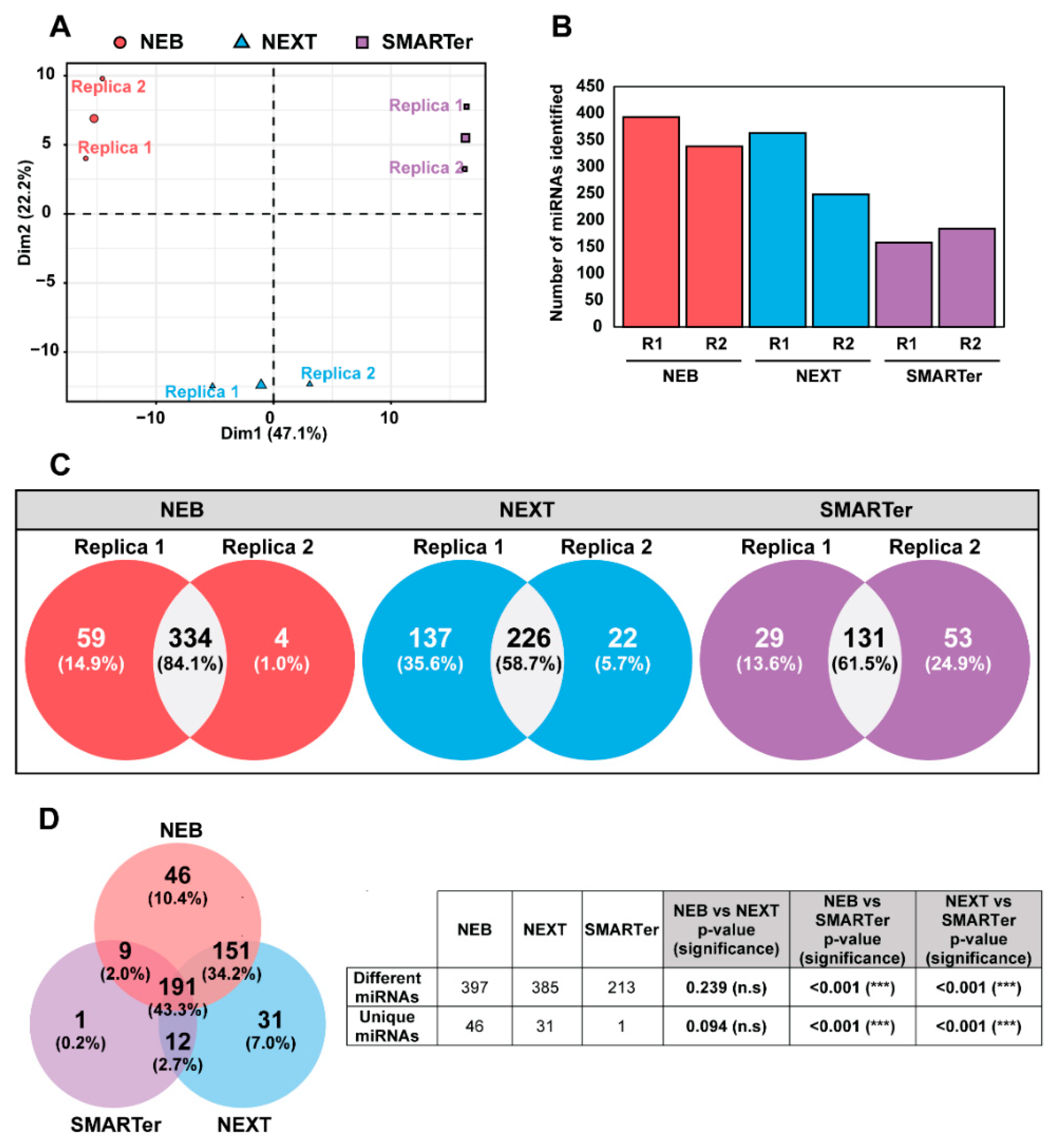
2.2. miRNA Library Evaluation for Next-Generation Sequencing
2.3. Selection of an EV Isolation Method According to miRNA Sequence Recovery
3. Discussion
4. Materials and Methods
4.1. Plasma Isolation
4.2. Cell Debris, Apoptotic Bodies, and Microvesicle Removal
4.3. Extracellular Vesicle Isolation Methods
4.4. EV Characterization
4.5. RNA Extraction
4.6. Library Preparation, Size Selection, and Sequencing
4.7. Bioinformatics Analyses
4.8. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bhaskaran, M.; Mohan, M. MicroRNAs: History, Biogenesis, and Their Evolving Role in Animal Development and Disease. Vet. Pathol. 2014, 51, 759–774. [Google Scholar] [CrossRef] [PubMed]
- Huntzinger, E.; Izaurralde, E. Gene silencing by microRNAs: Contributions of translational repression and mRNA decay. Nat. Rev. Genet. 2011, 12, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Steinbichler, T.B.; Dudás, J.; Riechelmann, H.; Skvortsova, I.I. The role of exosomes in cancer metastasis. Semin. Cancer Biol. 2017, 44, 170–181. [Google Scholar] [CrossRef]
- Vickers, K.C.; Palmisano, B.T.; Shoucri, B.M.; Shamburek, R.D.; Remaley, A.T. MicroRNAs are transported in plasma and delivered to recipient cells by high-density lipoproteins. Nat. Cell Biol. 2011, 13, 423–433. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, P.S.; Parkin, R.K.; Kroh, E.M.; Fritz, B.R.; Wyman, S.K.; Pogosova-Agadjanyan, E.L.; Peterson, A.; Noteboom, J.; O’Briant, K.C.; Allen, A.; et al. Circulating microRNAs as stable blood-based markers for cancer detection. Proc. Natl. Acad. Sci. USA 2008, 105, 10513–10518. [Google Scholar] [CrossRef] [PubMed]
- Winter, J.; Diederichs, S. Argonaute proteins regulate microRNA stability: Increased microRNA abundance by Argonaute proteins is due to microRNA stabilization. RNA Biol. 2011, 8, 1149–1157. [Google Scholar] [CrossRef]
- Simons, M.; Raposo, G. Exosomes—Vesicular carriers for intercellular communication. Curr. Opin. Cell Biol. 2009, 21, 575–581. [Google Scholar] [CrossRef]
- Gallo, A.; Tandon, M.; Alevizos, I.; Illei, G.G. The majority of microRNAs detectable in serum and saliva is concentrated in exosomes. PLoS ONE 2012, 7, e30679. [Google Scholar] [CrossRef]
- Sun, Z.; Shi, K.; Yang, S.; Liu, J.; Zhou, Q.; Wang, G.; Song, J.; Li, Z.; Zhang, Z.; Yuan, W. Effect of exosomal miRNA on cancer biology and clinical applications. Mol. Cancer 2018, 17, 147. [Google Scholar] [CrossRef]
- Colombo, M.; Raposo, G.; Théry, C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu. Rev. Cell Dev. Biol. 2014, 30, 255–289. [Google Scholar] [CrossRef]
- Doyle, L.M.; Wang, M.Z. Overview of Extracellular Vesicles, Their Origin, Composition, Purpose, and Methods for Exosome Isolation and Analysis. Cells 2019, 8, 727. [Google Scholar] [CrossRef] [PubMed]
- Raposo, G.; Stoorvogel, W. Extracellular vesicles: Exosomes, microvesicles, and friends. J. Cell Biol. 2013, 200, 373–383. [Google Scholar] [CrossRef] [PubMed]
- Zaborowski, M.P.; Balaj, L.; Breakefield, X.O.; Lai, C.P. Extracellular Vesicles: Composition, Biological Relevance, and Methods of Study. Bioscience 2015, 65, 783–797. [Google Scholar] [CrossRef]
- O’Grady, T.; Njock, M.S.; Lion, M.; Bruyr, J.; Mariavelle, E.; Galvan, B.; Boeckx, A.; Struman, I.; Dequiedt, F. Sorting and packaging of RNA into extracellular vesicles shape intracellular transcript levels. BMC Biol. 2022, 20, 72. [Google Scholar] [CrossRef]
- Baglio, S.R.; Rooijers, K.; Koppers-Lalic, D.; Verweij, F.J.; Pérez Lanzón, M.; Zini, N.; Naaijkens, B.; Perut, F.; Niessen, H.W.M.; Baldini, N.; et al. Human bone marrow- and adipose-mesenchymal stem cells secrete exosomes enriched in distinctive miRNA and tRNA species. Stem Cell Res. Ther. 2015, 6, 127. [Google Scholar] [CrossRef]
- D’souza, R.F.; Woodhead, J.S.T.; Zeng, N.; Blenkiron, C.; Merry, T.L.; Cameron-Smith, D.; Mitchell, C.J. Circulatory exosomal miRNA following intense exercise is unrelated to muscle and plasma miRNA abundances. Am. J. Physiol.-Endocrinol. Metab. 2018, 315, E723–E733. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, J.; Ochiya, T. Extracellular microRNAs and oxidative stress in liver injury: A systematic mini review. J. Clin. Biochem. Nutr. 2018, 63, 6–11. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, Y.; Liu, H.; Tang, W.H. Exosomes: Biogenesis, biologic function and clinical potential. Cell Biosci. 2019, 9, 19. [Google Scholar] [CrossRef]
- Johan, S.; Wurdinger, T.; van Rijn, S.; Meijer, D.; Gainche, L.; Sena-Esteves, M.; Curry, W.T., Jr.; Carter, R.S.; Krichevsky, A.M.; Breakefield, X.O. Glioblastoma microvesicles transport RNA and protein that promote tumor growth and provide diagnostic biomarkers. Nat. Cell Biol. 2008, 10, 1470–1476. [Google Scholar] [CrossRef]
- Chaput, N.; Théry, C. Exosomes: Immune properties and potential clinical implementations. Semin. Immunopathol. 2011, 33, 419–440. [Google Scholar] [CrossRef]
- de Miguel Pérez, D.; Rodriguez Martínez, A.; Ortigosa Palomo, A.; Delgado Ureña, M.; Garcia Puche, J.L.; Robles Remacho, A.; Exposito Hernandez, J.; Lorente Acosta, J.A.; Ortega Sánchez, F.G.; Serrano, M.J. Extracellular vesicle-miRNAs as liquid biopsy biomarkers for disease identification and prognosis in metastatic colorectal cancer patients. Sci. Rep. 2020, 10, 3974. [Google Scholar] [CrossRef] [PubMed]
- Valadi, H.; Ekström, K.; Bossios, A.; Sjöstrand, M.; Lee, J.J.; Lötvall, J.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007, 9, 654–659. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.M.; Abdelmohsen, K.; Mustapic, M.; Kapogiannis, D. Myriam Gorospe RNA in extracellular vesicles. Wiley Interdiscip. Rev. RNA 2017, 8, e1413. [Google Scholar] [CrossRef] [PubMed]
- Buschmann, D.; Kirchner, B.; Hermann, S.; Märte, M.; Wurmser, C.; Brandes, F.; Kotschote, S.; Bonin, M.; Steinlein, O.K.; Pfaffl, M.W.; et al. Evaluation of serum extracellular vesicle isolation methods for profiling miRNAs by next-generation sequencing. J. Extracell. Vesicles 2018, 7, 1481321. [Google Scholar] [CrossRef]
- Coenen-Stass, A.M.L.; Magen, I.; Brooks, T.; Ben-Dov, I.Z.; Greensmith, L.; Hornstein, E.; Fratta, P. Evaluation of methodologies for microRNA biomarker detection by next generation sequencing. RNA Biol. 2018, 15, 1133–1145. [Google Scholar]
- Witwer, K.W.; Buzás, E.I.; Bemis, L.T.; Bora, A.; Lässer, C.; Lötvall, J.; Nolte-’t Hoen, E.N.; Piper, M.G.; Sivaraman, S.; Skog, J.; et al. Standardization of sample collection, isolation and analysis methods in extracellular vesicle research. J. Extracell. Vesicles 2013, 2, 20360. [Google Scholar] [CrossRef]
- Michell, D.L.; Vickers, K.C. Lipoprotein carriers of microRNAs. Biochim. Biophys. Acta-Mol. Cell Biol. Lipids 2016, 1861, 2069–2074. [Google Scholar] [CrossRef]
- Brennan, K.; Martin, K.; FitzGerald, S.P.; O’Sullivan, J.; Wu, Y.; Blanco, A.; Richardson, C.; Mc Gee, M.M. A comparison of methods for the isolation and separation of extracellular vesicles from protein and lipid particles in human serum. Sci. Rep. 2020, 10, 1039. [Google Scholar] [CrossRef]
- Andreu, Z.; Yáñez-mó, M. Tetraspanins in extracellular vesicle formation and function. Front. Immunol. 2014, 5, 422. [Google Scholar] [CrossRef]
- Phillips, M.C. New insights into the determination of HDL structure by apolipoproteins. J. Lipid Res. 2013, 54, 2034–2048. [Google Scholar] [CrossRef]
- Behbodikhah, J.; Ahmed, S.; Elyasi, A.; Kasselman, L.J.; De Leon, J.; Glass, A.D.; Reiss, A.B. Apolipoprotein b and cardiovascular disease: Biomarker and potential therapeutic target. Metabolites 2021, 11, 690. [Google Scholar] [CrossRef] [PubMed]
- Jung, M.K.; Mun, J.Y. Sample preparation and imaging of exosomes by transmission electron microscopy. J. Vis. Exp. 2018, 2018, 5–9. [Google Scholar] [CrossRef]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq-A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2013, 7, 2803–2809. [Google Scholar] [CrossRef]
- Brochado-Kith, Ó.; Sanz, A.G.; Real, L.M.; Crespo-García, J.; Ryan-Murúa, P.; Macías, J.; Cabezas-González, J.; Troya, J.; Pineda, J.A.; Arias-Loste, M.T.; et al. MicroRNA profile of HCV spontaneous clarified individuals, denotes previous HCV infection. J. Clin. Med. 2019, 8, 849. [Google Scholar] [CrossRef]
- Théry, C.; Amigorena, S.; Raposo, G.; Clayton, A. Isolation and Characterization of Exosomes from Cell Culture Supernatants. Curr. Protoc. Cell Biol. 2006, 30, 3.22.1–3.22.29. [Google Scholar] [CrossRef]
- Coughlan, C.; Bruce, K.D.; Burgy, O.; Boyd, T.D.; Michel, C.R.; Garcia-Perez, J.E.; Adame, V.; Anton, P.; Bettcher, B.M.; Chial, H.J.; et al. Exosome Isolation by Ultracentrifugation and Precipitation and Techniques for Downstream Analyses. Curr. Protoc. Cell Biol. 2020, 88, e110. [Google Scholar] [CrossRef]
- Crescitelli, R.; Lässer, C.; Lötvall, J. Isolation and characterization of extracellular vesicle subpopulations from tissues. Nat. Protoc. 2021, 16, 1548–1580. [Google Scholar] [CrossRef]
- Böing, A.N.; van der Pol, E.; Grootemaat, A.E.; Coumans, F.A.W.; Sturk, A.; Nieuwland, R. Single-step isolation of extracellular vesicles by size-exclusion chromatography. J. Extracell. Vesicles 2014, 3, 23430. [Google Scholar] [CrossRef]
- Huang, X.; Yuan, T.; Tschannen, M.; Sun, Z.; Jacob, H.; Du, M.; Liang, M.; Dittmar, R.L.; Liu, Y.; Liang, M.; et al. Characterization of human plasma-derived exosomal RNAs by deep sequencing. BMC Genom. 2013, 14, 319. [Google Scholar] [CrossRef]
- Baldrich, P.; Tamim, S.; Mathioni, S.; Meyers, B. Ligation bias is a major contributor to nonstoichiometric abundances of secondary siRNAs and impacts analyses of microRNAs. bioRxiv 2020, 2020-09. [Google Scholar] [CrossRef]
- Shtratnikova, V.; Naumov, V.; Bezuglov, V.; Zheludkevich, A.; Smigulina, L.; Dikov, Y.; Denisova, T.; Suvorov, A.; Pilsner, J.R.; Hauser, R.; et al. Optimization of small RNA extraction and comparative study of NGS library preparation from low count sperm samples. Syst. Biol. Reprod. Med. 2021, 67, 230–243. [Google Scholar] [CrossRef] [PubMed]
- Dard-Dascot, C.; Naquin, D.; d’Aubenton-Carafa, Y.; Alix, K.; Thermes, C.; van Dijk, E. Systematic comparison of small RNA library preparation protocols for next-generation sequencing. BMC Genom. 2018, 19, 118. [Google Scholar] [CrossRef]
- Yagi, Y.; Ohkubo, T.; Kawaji, H.; Machida, A.; Miyata, H.; Goda, S.; Roy, S.; Hayashizaki, Y.; Suzuki, H.; Yokota, T. Next-generation sequencing-based small RNA profiling of cerebrospinal fluid exosomes. Neurosci. Lett. 2017, 636, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Prieto-Fernández, E.; Aransay, A.M.; Royo, F.; González, E.; Lozano, J.J.; Santos-Zorrozua, B.; Macias-Camara, N.; González, M.; Garay, R.P.; Benito, J.; et al. A comprehensive study of vesicular and non-vesicular miRNAs from a volume of cerebrospinal fluid compatible with clinical practice. Theranostics 2019, 9, 4567–4579. [Google Scholar] [CrossRef]
- Martínez-González, E.; Brochado-Kith, Ó.; Gómez-Sanz, A.; Martín-Carbonero, L.; Jimenez-Sousa, M.Á.; Martínez-Román, P.; Resino, S.; Briz, V.; Fernández-Rodríguez, A. Comparison of methods and characterization of small RNAs from plasma extracellular vesicles of HIV/HCV coinfected patients. Sci. Rep. 2020, 10, 11140. [Google Scholar] [CrossRef]
- Monguió-Tortajada, M.; Morón-Font, M.; Gámez-Valero, A.; Carreras-Planella, L.; Borràs, F.E.; Franquesa, M. Extracellular-Vesicle Isolation from Different Biological Fluids by Size-Exclusion Chromatography. Curr. Protoc. Stem Cell Biol. 2019, 49, e82. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| EV Isolation Methods | Concentration (Particles/mL) | Mean (nm) | Mode (nm) | D10 (nm) | D50 (nm) | D90 (nm) |
|---|---|---|---|---|---|---|
| SEC1 | 1.29 × 1012 ± 1.24 × 1011 | 128.4 ± 1.5 | 96.4 ± 5.4 | 80.9 ± 2.1 | 116.2 ± 1.3 | 194.1 ± 2.1 |
| SEC2 | 5.63 × 1011 ± 3.89 × 1010 | 134.5 ± 2.4 | 108.4 ± 4.1 | 87.6 ± 0.8 | 121.2 ± 1.6 | 206.7 ± 8.5 |
| SEC3 | 3.93 × 1011 ± 2.22 × 109 | 137.2 ± 1.9 | 114.8 ± 7.7 | 78.3 ± 2.3 | 124.2 ± 1.8 | 215.7 ± 3.4 |
| GRAD1 | 7.29 × 109 ± 8.16 × 107 | 134.7 ± 1.2 | 95.5 ± 1.1 | 86.1 ± 2.8 | 120.6 ± 1.2 | 200.9 ± 4.7 |
| GRAD2 | 9.55 × 109 ± 1.95 × 108 | 133.7 ± 1.6 | 94.3 ± 2.5 | 82.8 ± 1.4 | 120.2 ± 1.8 | 203.7 ± 3.3 |
| GRAD3 | 8.68 × 109 ± 3.18 × 108 | 141.1 ± 9.2 | 95.1 ± 4.0 | 85.3 ± 5.0 | 123.7 ± 6.7 | 215.9 ± 14.0 |
| SEC+GRAD1 | 4.90 × 109 ± 2.23 × 108 | 164.2 ± 2.7 | 129.1 ± 8.1 | 94.1 ± 2.9 | 146.2 ± 3.2 | 259.4 ± 11.2 |
| SEC+GRAD2 | 4.30 × 109 ± 1.31 × 109 | 179.7 ± 7.3 | 140.4 ± 28.7 | 96.1 ± 3.9 | 161.0 ± 14.4 | 277.6 ± 12.5 |
| SEC+GRAD3 | 2.29 × 109 ± 1.25 × 108 | 167.6 ± 3.0 | 111.5 ± 2.5 | 94.9 ± 9.0 | 148.3 ± 2.9 | 278.9 ± 16.8 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Llorens-Revull, M.; Martínez-González, B.; Quer, J.; Esteban, J.I.; Núñez-Moreno, G.; Mínguez, P.; Burgui, I.; Ramos-Ruíz, R.; Soria, M.E.; Rico, A.; et al. Comparison of Extracellular Vesicle Isolation Methods for miRNA Sequencing. Int. J. Mol. Sci. 2023, 24, 12183. https://doi.org/10.3390/ijms241512183
Llorens-Revull M, Martínez-González B, Quer J, Esteban JI, Núñez-Moreno G, Mínguez P, Burgui I, Ramos-Ruíz R, Soria ME, Rico A, et al. Comparison of Extracellular Vesicle Isolation Methods for miRNA Sequencing. International Journal of Molecular Sciences. 2023; 24(15):12183. https://doi.org/10.3390/ijms241512183
Chicago/Turabian StyleLlorens-Revull, Meritxell, Brenda Martínez-González, Josep Quer, Juan Ignacio Esteban, Gonzalo Núñez-Moreno, Pablo Mínguez, Idoia Burgui, Ricardo Ramos-Ruíz, María Eugenia Soria, Angie Rico, and et al. 2023. "Comparison of Extracellular Vesicle Isolation Methods for miRNA Sequencing" International Journal of Molecular Sciences 24, no. 15: 12183. https://doi.org/10.3390/ijms241512183
APA StyleLlorens-Revull, M., Martínez-González, B., Quer, J., Esteban, J. I., Núñez-Moreno, G., Mínguez, P., Burgui, I., Ramos-Ruíz, R., Soria, M. E., Rico, A., Riveiro-Barciela, M., Sauleda, S., Piron, M., Corrales, I., Borràs, F. E., Rodríguez-Frías, F., Rando, A., Ramírez-Serra, C., Camós, S., ... Costafreda, M. I. (2023). Comparison of Extracellular Vesicle Isolation Methods for miRNA Sequencing. International Journal of Molecular Sciences, 24(15), 12183. https://doi.org/10.3390/ijms241512183