1. Introduction
Mixed-lineage leukemia 1 (MLL1), also known as MLL, KMT2A, HRX, HTRX and ALL1, is one of the six mixed-lineage leukemia (MLL) family histone methyltransferases (HMT) in mammals [
1,
2,
3]. It mainly introduces 1-, 2- and 3-methylation into histone H3K4 through the evolutionarily conserved set domain. MLL1 and H3K4 methylation (H3K4me) take place in the promoter, transcription initiation site and 5′ transcription region of target genes and promote transcription initiation, so they play an important role in transcriptional regulation, especially in the early development of zygotic gene activation (ZGA) and hematopoiesis [
4,
5].
The H3K4 HMT activity of MLL1 is controlled by the core complex, which is composed of MLL1 and WDR5. The activity of MLL1 alone is weak, but its H3K4 HMT activity is greatly enhanced with the formation of the core complex [
6]. The MLL1 core complex consists of WDR5 and MLL1 proteins [
6,
7]. By disrupting the protein interaction between WDR5 and MLL1, the MLL1 core complex can be effectively dissociated and MLL1 activity can be inhibited [
8]. MM-102 is one of the compounds that can prevent the interaction between MLL1 and WDR5, inhibiting MLL1 activity. When mouse bone marrow cells transfected with the
MLL1-AF9 fusion gene were cultured with MM-102, the expression of
MLL1 target genes
HoxA9 and
Meis-1 was greatly reduced [
9]. Furthermore, down-regulating H3K4me3 through MM-102 can improve ZGA and abnormal expression patterns of epigenetic chromatin-modifying enzymes, as well as pluripotency and apoptosis genes in the blastocyst stage, and greatly improve efficiency and embryo quality in porcine somatic cell nuclear transfer, making it closer to that of in in vivo embryos [
10]. Our latest research found that adding appropriate concentrations of MM-102 and 3i (MM-102, PD0325901 and CHIR99021) during the ZGA stage of mouse and bovine in vitro fertilization (IVF) embryo development can greatly improve the development of IVF blastocysts without affecting their quality [
11]. MM-401 is another inhibitor of MLL1, and can transform mouse EpiSCs into a naïve pluripotent state [
12]. MLL1 inhibition causes the redistribution of H3K4me1 in enhancers, germline determinants and EpiSC markers to regulate the pluripotency regulatory network and restore the ability of EpiSCs to participate in embryonic development and germline chimerism [
12]. However, there is still a lack of relevant experimental studies on whether MLL1 inhibition by MM-102 alone or combined with other small-molecule inhibitors can transform human or livestock prime ESCs into a naïve state.
Cattle provide humans with high-quality meat products and nutritious dairy products, which have important economic and research value. The establishment of bovine pluripotent stem cells (bPSCs) has great significance for germplasm conservation, gene editing, breeding and understanding the developmental specificity of ruminants. In 2018, Bogliotti et al. obtained bovine prime ESCs, known as CTFR-bESCs, in a culture system supplemented with fibroblast growth factor 2 (FGF2) and WNT signaling pathway inhibitor IWR1, which exhibited characteristics of prime PSCs [
13]. Recently, bovine expanded pluripotent stem cells were established from blastocysts or via the reprogramming of bovine embryonic fibroblasts, which possess embryonic and extraembryonic development potential [
14]. However, whether we can transform bovine prime ESCs into a naïve state remains undetermined.
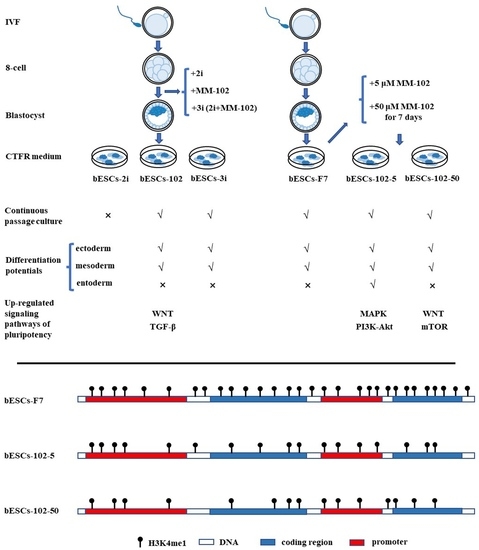
In this study, bovine prime ESCs were established from normal cultured IVF embryos, or those treated with MM-102 or 3i (MM-102, PD0325901 and CHIR99021)-. MM-102 improved the pluripotency and enhanced the capacity of the differentiation of bESCs. At the same time, after the addition of MM-102, bESCs could differentiate the endoderm in teratoma in vivo, and the signaling pathways of pluripotency were activated by MM-102 treatment. The effects of MLL1 inhibition on H3K4 methylation were determined via RNA-sequencing, Western blotting and ChIP-Seq analysis of H3K4me1. The modification pattern of H3K4me1 was altered by MM-102. In particular, the quantity of H3K4me1 in the promoter region of the genes was decreased, but the total H3K4me1 modification was significantly increased in the treated cells. Moreover, DNA methylation was comprehensively investigated via genome-wide DNA methylation sequencing. The pattern of DNA methylation modification was also significantly altered after MM-102 treatment, the methylation level of the promoter was reduced, and the expression of DNMT3B was significantly increased. This study will provide new insight into stem cell state transformation through the regulation of epigenetic modification.
3. Discussion
Given the highly accessible and hyperactive chromatin structures in pluripotent stem cells (PSCs), it is generally assumed that H3K4me plays an important ‘‘housekeeping’’ role in PSCs and is necessary for PSCs to maintain self-renewal and unlimited differentiation potential [
15]. Zhang et al. found that H3K4me1 was significantly different between mouse ESCs and EpiSCs, but H3K4me3 was not. Interestingly, the inhibition of MLL1 led to a genome-wide change in H3K4me1 in EpiSCs and the global redistribution of H3K4me1 at the enhancers, and repressed lineage determinant factors and EpiSC markers, which indirectly regulate the transcription of mouse ESCs [
12]. Naïve PSCs are distinguished from primed PSCs by their capacity to form teratomas and chimeric animals following their introduction into pre-implantation embryos, and exhibit unique transcription makers and DNA methylation levels [
15]. In recent years, significant advances have been made in the conversion of the primed state of hPSCs into naïve pluripotency. Human naïve pluripotency has shown better differentiation than the primed state, which is associated with the accumulation of DNA methylation and epigenetic-repressive marks in the primed state [
16]. Previous studies showed that MLL1 inhibition transformed mouse EpiSCs into a naïve pluripotent state [
12], and the combined treatment of the MLL1 inhibitor and 1α,25-dihydroxyvitamin D3 (1,25-(OH)2D3) enhanced the functionality of expanded PSCs, triggering an extended 2C-like state in vitro and robust totipotent-like properties in vivo [
17]; this indicated the MLL1 inhibition increases the differentiation capacity of PSCs. In this study, we found that MLL1 inhibition improved the pluripotency and differentiation potential of bESCs by decreasing H3K4me1 modification at the promoters and altered the distribution of DNA methylation in bESCs. However, MLL1 inhibition increased some naïve and primed markers simultaneously, but did not transform bESCs from a primed to a naïve state as in the mouse EpiSCs. We also found that MLL1 inhibition by itself cannot as effectively transform bovine ESCs to a naïve state. This may be due to the following: firstly, the MLL1 inhibitor used in our study is different from that used at the Dou Lab [
12], which may lead to differences in some effects; secondly, MM-102 only up-regulated some of the stem cell signaling pathways, such as PI3K-Akt and WNT, which may not be enough to transform bESCs from a primed to a naïve state; finally, the characteristics of PSCs from domestic animals are quite different from those from mice and humans, and further investigation is needed to thoroughly define the PSCs of domestic animals.
MLL1 epigenetically regulates gene expression patterns that specify cellular identity in both embryonic development and adult stem cell populations.
Mll1 is required for the expression of neurogenic—but not gliogenic—transcriptional modules in multipotent neural stem cell (NSC) populations, which further indicates that specific
Mll1-dependent genes may be useful for direct reprogramming strategies [
18]. MLL1 induced the activation of a Rac/Rho/Integrin signaling axis, to enhance hematopoiesis from murine embryonic stem cells [
19]. In this study, MLL1 inhibition in bESCs increased the expression of PRDM14, which is a histone lysine methyltransferase and a common marker of primordial germ cells and pluripotent embryonic stem cells [
20]. Moreover, the inhibition of MLL1 down-regulated the expression level of
DNMT3A and up-regulated the expression level of
DNMT3B. DNMT3B is the primary driver of de novo DNA methylation in actively transcribed genes, while DNMT3A plays a minimal role in ESCs [
21]. These results indicate the effects of MLL1 inhibition in bESCs, mainly on an epigenetic level. MLL1 inhibition changed the distribution of DNA methylation, which was mainly reflected in the increase in the overall CpG methylation level, the decrease in mCG in the upstream and downstream regions of the genes, and the decrease in mCG in the promoter region. The increased expression of DNMT3B in bESCs is associated with increased DNA methylation (CpG). Meanwhile, mCG in the promoter region of pluripotent genes such as
OCT4 and NANOG was decreased upon MLL1 inhibition, following the bisulfite genomic sequencing of the promoter regions. These changes may be one of the reasons why MM-102 changes the pluripotency of bESCs. Although the distribution of DNA methylation was changed following MLL1 inhibition, the global DNA hypomethylation observed in naïve PSCs was not achieved. To obtain real naïve PSCs for bovines, a thorough understanding of pluripotency control in ruminants is needed.
In conclusion, MLL1 inhibition enhanced the pluripotency of bovine embryonic stem cells, which enriched the promoter of pluripotency genes with H3K4me1 to increase the expression of pluripotency genes, and MM-102 altered the global DNA methylation distribution of bESCs.
4. Materials and Methods
4.1. Animal Care and Use
All experiments with mice (generation of embryonic fibroblasts and teratoma formation) were conducted in accordance with the Guide for Care and Use of Laboratory Research Involving Animals and approved by the Inner Mongolia University’s Animal Care and Use Committee (approval code: IMU-mouse-2021-037, approval date: 2021-2-26). The C57 mice used in the production of mouse fibroblasts were obtained from the animal breeding room of our laboratory. The teratoma nod-SCID mice used in the differentiation test were purchased from Beijing Wetong Lihua Co., LTD., Beijing, China, and tested in our laboratory’s temporary animal breeding room.
4.2. Bovine Embryo Culture
The bovine in vitro fertilization and embryo culture are described in our previous report [
11]. In short, bovine ovaries were obtained from a local abattoir, and oocytes were cultured for 22–24 h after maturation and fertilized with frozen cattle sperm. Six hours after fertilization, the eggs were transferred to the embryonic culture medium. When the embryos had developed to the 8-cell stage at 48 h, MM-102, PD0325901 and CHIR99021 (2i), or MM-102 plus PD0325901 and CHIR99021 (3i), was added to the medium. The embryos were then cultured for 7–8 days to develop into blastocysts.
4.3. Derivation of bESC Cell Lines
The bovine blastocysts obtained via in vitro fertilization were removed from the zona pellucida and plated in 4-well plates, and mTeSR-E6 (05946, STEMCELL Vancouver, Technologies, Canada) was added with 20 ng/mL FGF-2 (100-18B, STEMCELL Technologies, Vancouver, BC, Canada), 2.5 μM IWR-1 (I0161, Sigma-Aldrich, Shanghai, China) and 0.1328 g/mL low fatty acid BSA (219989925, MP Biomedicals, Auckland, NZ, USA), and were incubated at 37 °C and 5% CO2. After 48–72 h, ICMs adhered to the feeder layer, and the medium was changed daily. Outgrowths (after 8–10 days in culture) were dissociated and passaged using TrypLE (12563011, Gibco, CA, USA) and were reseeded in the presence of 10 μM Rho kinase (ROCK) inhibitor Y-27632 (SCM075, Sigma, Shanghai, China). The established bESC lines were grown in 12-well plates and were passaged every 5 days at a 1:4–1:6 split ratio. To increase cell survival, the ROCK inhibitor Y-27632 (10 μM) was added to the wells 1 h before passaging and was also added to the newly prepared wells containing MEFs treated with mitomycin and fresh culture medium during the first 24 h of cultivation.
4.4. Alkaline Phosphatase (AP) Staining
Prior to AP staining, the bESCs were cultured on feeder cells for 4 days. Following the removal of the culture medium, the cells were washed with DPBS and fixed for 10 min at room temperature using 4% paraformaldehyde. The fixed cells were washed three times with DPBS and stained for 1–3 h at room temperature in the dark, using the BCIP/NBT Color Development Substrate Kit (C3206, Beyotime Biotechnology, Shanghai, China). The cells were washed with DPBS to terminate the staining reaction and were subsequently maintained in DPBS.
4.5. Karyotype Analysis
During the flourishing period of cell division, which is usually on the 4th day of culture, 2 mg/mL colchicine (64-86-8, Sigma, Shanghai, China) was added to the culture medium and incubated at 37 °C for 2.5 h. Then, the cells were treated with 8 mL 0.075 mol KCl solution in a 37 °C water bath for 30 min. We collected the cells and added 1mL fixing solution (acetic acid: methanol = 1:3), and the solution was mixed. After centrifugation at 1500 rpm for 5 min, the supernatant was discarded, 10 mL fixing solution was added and mixed, and the cells stood at room temperature for 15min. After carrying out these fixing procedures three times, 50–200 μL fixing solution was added to the cells and mixed well, according to the amount of precipitation. Then, the cells were dropped from a height of approximately 1 m onto a pre-cooled slide at −20 °C, and left to dry overnight. The prepared sample was immersed in 0.025% trypsin solution preheated at 37 °C, and digested for 10 s; then, it was quickly washed twice in 0.85% NaCl solution, and dried. The sample was dyed in Jimsa solution (48,900, Sigma, Shanghai, China) for 15 min; then, we slowly washed off the excess with water, and dried it. Photographs were taken and analyzed using a cytogenetics workstation.
4.6. Quantitative Real-Time PCR (qRT-PCR)
The quantification of mRNAs was conducted via real-time PCR using specific primers. Real-time PCR was performed using an Applied Biosystems 7500 sequence detection system (Thermo Fischer Scientific, Shanghai, China) and KAPA SYBR® FAST Universal qPCR master mix (Kapa Biosystems Pty, Boston, MA, USA). The PCR samples were analyzed in 96-well plates. Each reaction (20 mL) contained forward and reverse primers at 0.2 mM and 10 mL SYBR Green PCR master mix. The PCR steps included incubation for 5 min at 95 °C, followed by 40 cycles of 95°C for 10 s, 60 °C for 20 s, and 72 °C for 30 s. All reactions were performed at least in triplicate, and product identity was confirmed via melting curve analysis. Relative expression levels were determined using the 2−ΔΔCT method, and normalized against GAPDH levels.
4.7. Statistical Analysis
Statistical analyses of qPCR results were performed using GraphPad Prism 8.3.0, and statistically significant differences between groups were identified using a paired t-test, with bESCs-102, bESCs-3i, bESCs-102-5, and bESCs-102-50 as the treatment groups and bESCs-F7 as the control group. The blastocyst rates, TE, ICM and total cells of the blastocysts subjected to different treatments were analyzed via a chi-squared test with Yates’ correction. The analyses were performed using the statistical software GraphPad PRISM 6.0 (GraphPad Software, Inc., La Jolla, CA, USA), and the results are presented as the means ± SDs. p-values less than 0.05 were considered statistically significant.
4.8. RNA-seq Analysis
Transcriptome (RNA) sequencing was performed by Tianjin Nuohe Zhiyuan Biotechnology Co., LTD, Tianjin, China. The PCA plot and volcano plot in this paper were generated using ggplot2 (version: 3.3.5) in R software 4.1.0, and the heatmap was generated using pheatmap (version:1.0.12); all track data (BS-seq, Chip-seq) were transformed to bigwig format used deeptools (version:3.4.3) [
22], and then visualized using the Integrative Genomics Viewer (IGV, 2.14.1) [
23].
4.9. Immunofluorescence Staining (IF)
Cells were cultured on coverslips on a 4-well plate. When grown to a suitable density, the cells were fixed with 4% paraformaldehyde for 10 min, and then treated with Triton X-100 to penetrate the cell membrane. After three washes with PBS, the cells were incubated with the primary antibody at 4 °C overnight. Then, the cells were incubated with the secondary antibody at room temperature for 1 h. After the removal of antibodies, the cells were incubated at room temperature with DAPI for 5 min. The slide was sealed after a microscopic examination. The antibodies used in this experiment included anti-OCT4 (sc-5279, Santa Cruz Biotechnology, Shanghai, China), anti-SOX2 (L1D6A2, Cell Signaling, Boston, MA, USA), anti-NANOG (500-P236, Peprotech, Shanghai, China), anti-SSEA-1 (MAB4301, Sigma, Shanghai, China), anti-SSEA-4 (MAB4304, Millipore, Shanghai, China), anti-TRA-1-60 (MAB4360, Shanghai, China), anti-TRA-1-81 (MAB4381, Shanghai, China), anti-AFP (AF5369, R&D Systems, Minneapolis, MN, USA), anti-SMA (ab5694, Abcam, Cambridge, MU, USA) and anti-GFAP (I1044, DAKO, Copenhagen, Denmark).
4.10. Flow Cytometry Analysis
The cells were fixed with 4% paraformaldehyde for 10 min and permeabilized with 0.8% Triton X-100 for 10 min. Then, the cells were incubated with antibodies in 10% goat serum for 1h at room temperature. The primary antibody was incubated at 4 °C overnight and the secondary antibody was incubated at room temperature for 1 h. The DPBS was used to wash off the antibodies between each step. After incubation, the cells were screened and analyzed via flow cytometry (Cytoflex LX), according to the corresponding fluorescence intensity. The antibodies used in this experiment included anti-SSEA-1 (MAB4301, Sigma) and anti-SSEA-4 (MAB4304, Millipore).
4.11. Western Blotting (WB)
Cells were collected and protein was extracted using a Mammalian Protein Extraction Reagent (CWBIO, Taizhou, Jiangsu, China), according to the manufacturer’s procedure. A cracking product was added to the loading buffer and boiled in boiling water for 5 min for denaturation. We added the same amount of protein sample into the hole of the SDS-PAGE gel, and maintained a constant pressure of 90–120 V for 30 min–3 h. After the marker was separated, the gel was cut according to the marker position to obtain the corresponding protein sizes, and transferred to a nitrocellulose membrane with a constant current of 200 mA for 30 min–1 h. After the transfer of the required strip was completed, we placed it in 5% sealing solution and placed it at room temperature for 1 h. We transferred the membrane to the diluent of the primary antibody at 4 °C overnight, and washed the membrane 3 times with TBST solution. The secondary antibody was incubated at room temperature for 1 h, and TBST was used to wash the membrane 3 times; it was exposed to an indicator after the treatment. The antibodies used in this experiment included anti-H3 (4620S, Cell Signaling Technology, Boston, MA, USA), anti-H3K4me1 (ab8895, Abcam, Cambridge, MU, USA), anti-H3K4me2 (ab32356, Abcam), anti-H3K27me3 (ab6002, Abcam), anti-GAPDH (10494-1-AP, Proteintech, Franklin Lakes, NJ, USA), anti-PRDM14 (ab187881, Abcam), anti-P300 (ab54984, Abcam), anti-G9A (ab18894, Abcam), anti-DNMT3A (D23G1, Cell Signaling Technology, Boston, MA, USA) and anti-DNMT3B (bs-0301R, Bioss, Beijing, China). Images were obtained using a Tanon 5200 Multi Automatic Fluorescence and Chemiluminescence Imaging System (Tanon, Shanghai, China).
4.12. Formation of Embryoid Bodies (EB) In Vitro
When the density of the cells reached 80%, the cells were digested and collected. The differentiation medium from the first stage (90% IMDM + 10% FBS) was used to suspend the cells in low-adherent 35 mm culture dishes. The embryoid body formed at 6–7 days. The embryoid bodies were collected and centrifuged. Cells were resuspended in the second-stage differentiation medium (90%DMEM + 10% FBS) and plated into four-well plates for adherent growth. After 3 weeks of culture, the differentiated cells were identified via immunofluorescence staining, with the specific antibodies of three germ layers. The antibodies used in this experiment included anti-α-SMA (ab244177, Abcam, Cambridge, MU, USA), anti-AFP (MAB1368, R&D Systems, Minneapolis, MN, USA) and anti-GFAP (Z0334, DAKO, Copenhagen, Denmark).
4.13. Teratoma Formation In Vivo
The cells were collected and suspended in an appropriate amount of DPBS, and approximately 1 × 107 cells were injected subcutaneously into Nod scid mice at each site. When the tumors were visible, the teratomas were removed from the mice. The resulting teratomas were paraffin-embedded, sectioned, and HE stained for analysis.
4.14. ChIP-seq Analysis
The Simple ChIP
® Plus Enzymatic Chromatin IP Kit (Magnetic Beads) (9005, Cell Signaling) was utilized to extract DNA fragments from the cells. The sequencing was performed by Tianjin Nuohe Zhiyuan Biotechnology Co., LTD, Tianjin, China. Illumina reads were first mapped to the UCSC bosTau9 reference using bwa-mem (version:0.7.17) [
24] with default parameters. Next, Picard (
http://broadinstitute.github.io/picard/, accessed on 12 June 2023, version 2.23.1) was used to mark up the PCR duplicates. Then, we used macs2 (v2.2.7.1, -nomodel-broad-broad-cutoff 0.1-shift 0-gsize 2.7e9-keep-dup auto) [
25] to call the peaks. H3K4me1 signals were normalized using the MA norm (version: 1.1.4) [
26] method for the quantitative comparison of ChIP-seq data, and the significant differential peaks were determined as log10(
p-value) < 0 and M-value > 1. Subsequently, the differential peaks were annotated to UCSC bosTau9 using the R package ChIP seeker (version: 1.28.3) [
27] and TxDb.Btaurus.UCSC.bosTau9.refGene (version: 3.10.0).
4.15. Bisulfite Genomic Sequencing Analysis
All cell lines in this experiment were used to analyze the promoter methylation of OCT4 and NANOG via bisulfite sequencing PCR. DNA treatment and methylation-specific PCR were executed using the ZYMO EZ DNA Methylation-Gold Kit (ZYMO RESEARCH) and Takara Ex Taq (Hot Start Version), according to the manufacturers’ protocols. The PCR products were ligated into a pEASY-T1 Cloning Vector (Trans Gen Biotech, Beijing, China) for methylation sequencing. At least 10 clones per gene were sequenced and analyzed for each sample.
4.16. Whole-Genome Methylation Sequencing
DNA methylation sequencing was performed by Tianjin Nuohe Zhiyuan Biotechnology Co., LTD. The R package edge R (version: 3.34.1) [
28] was used to analyze the differences between the treatments and the control. The read counts were tested for differential expression using the ‘exact test’. The differentially expressed genes (DEGs) in the data set with |log2 (fold change)|≥ 1.5 and adjusted
p ≤ 0.05 were selected for the subsequent analyses. Next, the cluster profiler (version: 4.0.5) [
29] package was used to annotate and enrich the GO and KEGG pathways for DEGs.
p ≤ 0.05 was determined as a cut-off criterion for significant enrichment.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}








