
Transcriptional Regulators Controlling Virulence in Pseudomonas aeruginosa


Abstract
1. Introduction
1.1. Motility and Attachment Factors
1.2. Biofilm Formation Factors

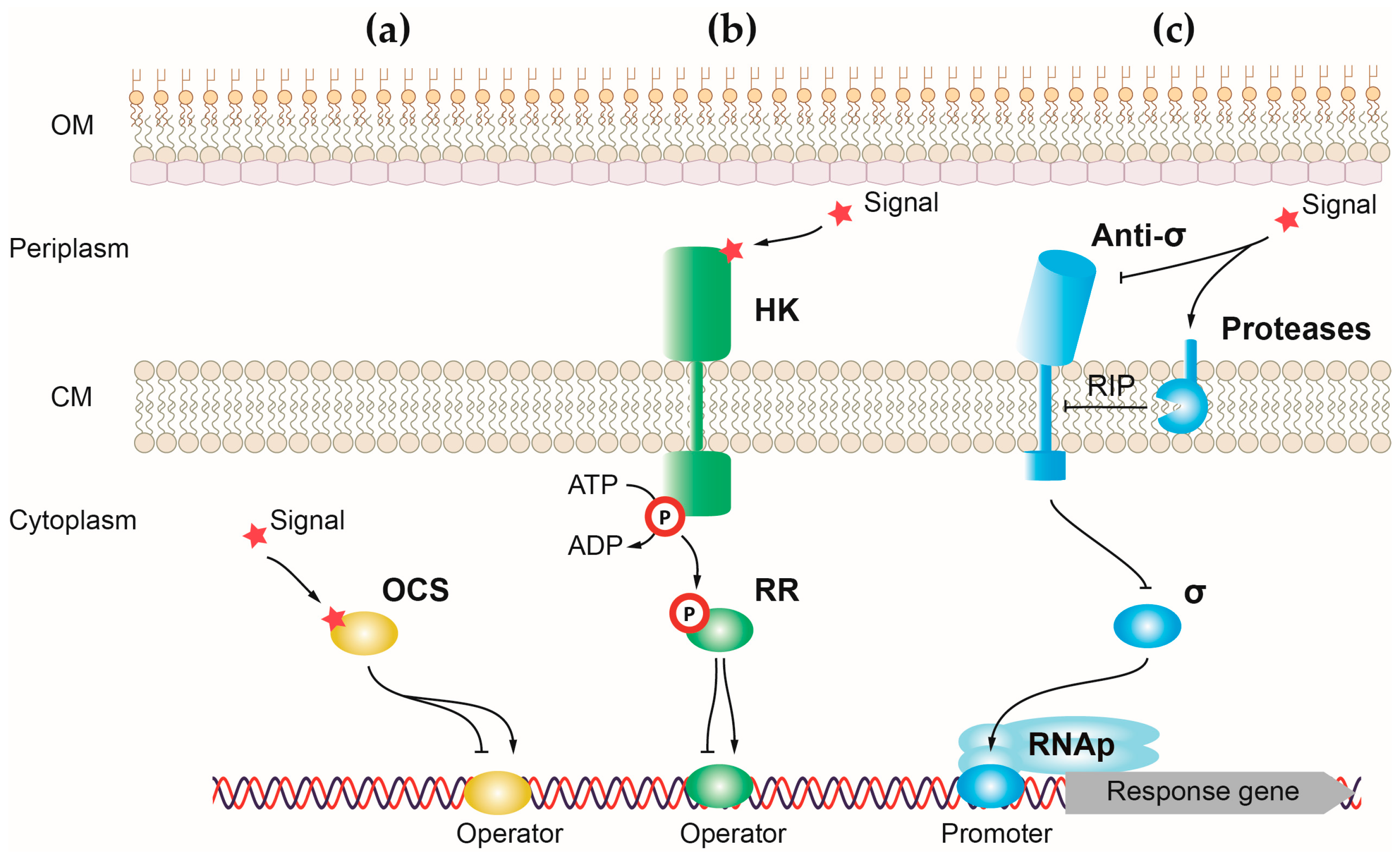{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Virulence Hallmark | Virulence Factors | Genes Involved | Function in Virulence | Main Transcriptional Regulator(s) | References |
|---|---|---|---|---|---|
| Bacterial motility and attachment | Flagellum | flg, fli, and flh gene clusters | Swimming and swarming motility | AmrZ, PilS-PilR, FleS-FleR, σFliA | [20,21,22] |
| Type IV pili | flp, tad, afimU fimV, and pilABCD | Twitching motility and attachment to solid surfaces | AmrZ, PchR, PilS-PilR, PprA-PprB, GacS-GacA, BfiS-BfiR, GtrS-GltR, σSigX | [12,22,23,24,25,26,27,28] | |
| Adhesins | bapA, cupA, cupB, cupC, cupD, and cupE gene clusters | Attachment to host cells and solid surfaces | RocS-RocR-RocA1, RcsC-RcsB, PvrS-PvrR, PprA-PprB, GacS-GacA, BfiS-BfiR, GtrS-GltR, σSigX, σRpoS | [11,23,24,26,27,29,30,31] | |
| Lectins | lecA and lecB | Adherence to epithelial cells | PqsR, Fur, PchR, σAlgT | [28,32,33,34] | |
| Biofilm formation | Esopolysaccharide alginate | alg gene cluster | Component of the extracellular matrix | AmrZ, PchR, FimS-AlgR, KinB-AlgB, σAlgT, σRpoH, σRpoS, σSbrI | [28,35,36,37,38,39,40] |
| Exopolysaccharides Pel and Psl | pel and psl gene clusters | Component of the extracellular matrix | GacS-GacA, BfiS-BfiR, GtrS-GltR, σAlgT, σRpoS | [24,26,27,33,36,37,41] | |
| eDNA | phdA | Component of the extracellular matrix | BfmS-BfmR | [42] | |
| Rhamnolipids | rhlAB and rhlC | Biosurfactant | RhlR, PtxR | [43,44] | |
| Glycine betaine | betA and betB | Biosurfactant | BetI, GbdR | [45] | |
| Extracellular invasive enzymes and toxins | Elastases | lasA and lasB | Degradation of host elastin and collagen | LasR, RhlR, PtxR, PchR, FleS-FleR, σAlgT | [28,43,44,46,47,48,49] |
| Protease IV | piv/prpL | Degradation of host fibrinogen | Fur, σPvdS, σFpvI | [50,51] | |
| Alkaline protease | aprA | Degradation of host transferrin and matrix-associated proteins | LasR, PvrA, σSigX, σAlgT | [23,46,47,52] | |
| Phospholipases | plcH, exoU, pldA, pldB, and tplE | Degradation of host cell membranes and lung surfactants | ExsA, GbdR, PvrA, SphR, GacS-GacA BfiS-BfiR, GtrS-GltR, σFliA | [45,52,53,54] | |
| Exotoxin S and T | exoS and exoT | Cytotoxicity and cytoskeleton disruption | ExsA, GacS-GacA, BfiS-BfiR, GtrS-GltR, σFliA | [24,26,27,53,55] | |
| ExoY adenylate cyclase | exoY | Disruption of actin cytoskeleton and increased host cell permeability | ExsA, GacS-GacA, BfiS-BfiR, GtrS-GltR, σFliA | [24,26,27,53,55] | |
| Exotoxin A | exoA/toxA | Inhibition of host protein synthesis | PtxR, PtxS, GtrS-GltR, FleS-FleR, σPvdS | [48,51,56,57] | |
| Pro-inflammatory toxins | lptA, lptE, and osmE | Induction of host inflammatory response | σAlgT | [58] | |
| Toxic secondary metabolites | Hydrogen cyanide | hcnABC | Arrest respiration in host cells | LasR, RhlR, PchR, AmpR | [28,47,59] |
| Phenazine and pyocyanin | phzABCDEFG phzM | Cause oxidative stress and cytotoxicity | RhlR, PqsR, CdpR, PvrA, PchR, AmpR | [28,43,44,52,59,60] | |
| Acquisition and homeostasis of iron | Pyoverdine | pvd gene cluster | Siderophore production and iron acquisition | Fur, AmpR, σPvdS, σFpvI | [59,61,62] |
| Pyochelin | pchDCBA, pchEF, and fptABCX gene clusters | Siderophore production, iron acquisition, and tissue damage | Fur, PchR | [61] | |
| Heme and hemophore | phu, has, and hxu gene clusters | Iron acquisition from heme | Fur, AmpR, σHasI, σHxuI | [59,61,62] | |
| Iron storage and detoxification | prrF1, prrF2, brfB, sodB, and katA genes | Prevent iron accumulation and toxicity | Fur, PchR | [28,61] |
1.3. Extracellular Invasive Enzymes and Toxins
1.4. Toxic Secondary Metabolites
1.5. Iron Acquisition Systems and Factors Controlling Iron Homeostasis
1.6. Regulation of Virulence
2. Control of P. aeruginosa Virulence by One-Component Systems
2.1. OCSs Responding to Quorum Sensing (QS)
2.1.1. LasR
2.1.2. RhlR
2.1.3. PqsR
2.2. OCSs Regulating Motility, Attachment, and Biofilm Formation
AmrZ
2.3. OCSs Regulating the Production and Secretion of Extracellular Enzymes, Toxins, and Toxic Secondary Metabolites
2.3.1. ExsA
2.3.2. Sfa2
2.3.3. GbdR
2.3.4. PvrA
2.3.5. SphR
2.3.6. PtxR
2.3.7. SoxR
2.4. OCSs Regulating the Acquisition and Homeostasis of Iron
2.4.1. Fur
2.4.2. PchR
2.4.3. AmpR
3. Control of P. aeruginosa Virulence by Two-Component Systems
3.1. TCSs Regulating Motility, Attachment, and Biofilm Formation
3.1.1. PilS-PilR
3.1.2. FleS-FleR
3.1.3. RocS-RocR-RocA1
3.1.4. RcsC-RcsB and PvrS-PvrR
3.1.5. PprA-PprB
3.1.6. FimS-AlgR
3.1.7. KinB-AlgB
3.1.8. GacS-GacA
3.1.9. BfiS-BfiR, BfmS-BfmR, and MifS-MifR
3.1.10. GtrS-GltR
3.2. TCSs Regulating the Production and Secretion of Extracellular Enzymes, Toxins, and Toxic Secondary Metabolites
PhoR-PhoB
3.3. TCSs Regulating the Acquisition and Homeostasis of Iron
BqsS-BqsR
4. Control of P. aeruginosa Virulence by Sigma (σ) Factors
4.1. σ Factors Regulating Motility, Attachment, and Biofilm Formation
4.1.1. σFliA (σ28)
4.1.2. σSigX
4.1.3. σAlgT
4.1.4. σRpoH (σ32)
4.1.5. σRpoS (σ38)
4.1.6. σSbrI
4.2. σ Factors Regulating the Production and Secretion of Extracellular Enzymes, Toxins, and Toxic Secondary Metabolites
σVreI
4.3. σ Factors Regulating the Acquisition and Homeostasis of Iron
4.3.1. σPvdS and σFpvI
4.3.2. σHasI and σHxuI
5. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Morin, C.D.; Déziel, E.; Gauthier, J.; Levesque, R.C.; Lau, G.W. An Organ System-Based Synopsis of Pseudomonas aeruginosa Virulence. Virulence 2021, 12, 1469–1507. [Google Scholar] [CrossRef]
- Muggeo, A.; Coraux, C.; Guillard, T. Current Concepts on Pseudomonas aeruginosa Interaction with Human Airway Epithelium. PLoS Pathog. 2023, 19, e1011221. [Google Scholar] [CrossRef]
- Buhl, M.; Peter, S.; Willmann, M. Prevalence and Risk Factors Associated with Colonization and Infection of Extensively Drug-Resistant Pseudomonas aeruginosa: A Systematic Review. Expert Rev. Anti-Infect. Ther. 2015, 13, 1159–1170. [Google Scholar] [CrossRef] [PubMed]
- Hauser, A.R.; Jain, M.; Bar-Meir, M.; McColley, S.A. Clinical Significance of Microbial Infection and Adaptation in Cystic Fibrosis. Clin. Microbiol. Rev. 2011, 24, 29–70. [Google Scholar] [CrossRef] [PubMed]
- Livermore, D.M. Multiple Mechanisms of Antimicrobial Resistance in Pseudomonas aeruginosa: Our Worst Nightmare? Clin. Infect. Dis. 2002, 34, 634–640. [Google Scholar] [CrossRef]
- Karakonstantis, S.; Kritsotakis, E.I.; Gikas, A. Pandrug-Resistant Gram-Negative Bacteria: A Systematic Review of Current Epidemiology, Prognosis and Treatment Options. J. Antimicrob. Chemother. 2020, 75, 271–282. [Google Scholar] [CrossRef]
- Chadha, J.; Harjai, K.; Chhibber, S. Revisiting the Virulence Hallmarks of Pseudomonas aeruginosa: A Chronicle through the Perspective of Quorum Sensing. Environ. Microbiol. 2021, 24, 2630–2656. [Google Scholar] [CrossRef]
- Khan, F.; Pham, D.T.N.; Oloketuyi, S.F.; Kim, Y.M. Regulation and Controlling the Motility Properties of Pseudomonas aeruginosa. Appl. Microbiol. Biotechnol. 2020, 104, 33–49. [Google Scholar] [CrossRef]
- Burrows, L.L. Pseudomonas aeruginosa Twitching Motility: Type IV Pili in Action. Annu. Rev. Microbiol. 2012, 66, 493–520. [Google Scholar] [CrossRef] [PubMed]
- Webster, S.S.; Wong, G.C.L.; O’Toole, G.A. The Power of Touch: Type 4 Pili, the von Willebrand A Domain, and Surface Sensing by Pseudomonas aeruginosa. J. Bacteriol. 2022, 204, e00084-22. [Google Scholar] [CrossRef]
- Giraud, C.; Bernard, C.S.; Calderon, V.; Yang, L.; Filloux, A.; Molin, S.; Fichant, G.; Bordi, C.; De Bentzmann, S. The PprA-PprB Two-Component System Activates CupE, the First Non-Archetypal Pseudomonas aeruginosa Chaperone-Usher Pathway System Assembling Fimbriae. Environ. Microbiol. 2011, 13, 666–683. [Google Scholar] [CrossRef]
- De Bentzmann, S.; Giraud, C.; Bernard, C.S.; Calderon, V.; Ewald, F.; Plésiat, P.; Nguyen, C.; Grunwald, D.; Attree, I.; Jeannot, K.; et al. Unique Biofilm Signature, Drug Susceptibility and Decreased Virulence in Drosophila through the Pseudomonas aeruginosa Two-Component System PprAB. PLoS Pathog. 2012, 8, 1003052. [Google Scholar] [CrossRef]
- Chemani, C.; Imberty, A.; De Bentzmann, S.; Pierre, M.; Wimmerová, M.; Guery, B.P.; Faure, K. Role of LecA and LecB Lectins in Pseudomonas aeruginosa-Induced Lung Injury and Effect of Carbohydrate Ligands. Infect. Immun. 2009, 77, 2065–2075. [Google Scholar] [CrossRef] [PubMed]
- Petrova, O.E.; Schurr, J.R.; Schurr, M.J.; Sauer, K. The Novel Pseudomonas aeruginosa Two-Component Regulator BfmR Controls Bacteriophage-Mediated Lysis and DNA Release during Biofilm Development through PhdA. Mol. Microbiol. 2011, 81, 767–783. [Google Scholar] [CrossRef] [PubMed]
- Ramsey, D.M.; Wozniak, D.J. Understanding the Control of Pseudomonas aeruginosa Alginate Synthesis and the Prospects for Management of Chronic Infections in Cystic Fibrosis. Mol. Microbiol. 2005, 56, 309–322. [Google Scholar] [CrossRef] [PubMed]
- Davey, M.E.; Caiazza, N.C.; O’Toole, G.A. Rhamnolipid Surfactant Production Affects Biofilm Architecture in Pseudomonas aeruginosa PAO1. J. Bacteriol. 2003, 185, 1027. [Google Scholar] [CrossRef]
- Van Gennip, M.; Christensen, L.D.; Alhede, M.; Phipps, R.; Jensen, P.Ø.; Christophersen, L.; Pamp, S.J.; Moser, C.; Mikkelsen, P.J.; Koh, A.Y.; et al. Inactivation of the RhlA Gene in Pseudomonas aeruginosa Prevents Rhamnolipid Production, Disabling the Protection against Polymorphonuclear Leukocytes. APMIS 2009, 117, 537–546. [Google Scholar] [CrossRef]
- Zulianello, L.; Canard, C.; Köhler, T.; Caille, D.; Lacroix, J.S.; Meda, P. Rhamnolipids are Virulence Factors That Promote Early Infiltration of Primary Human Airway Epithelia by Pseudomonas aeruginosa. Infect. Immun. 2006, 74, 3134–3147. [Google Scholar] [CrossRef]
- Wargo, M.J. Choline Catabolism to Glycine Betaine Contributes to Pseudomonas aeruginosa Survival during Murine Lung Infection. PLoS ONE 2013, 8, e56850. [Google Scholar] [CrossRef]
- Potvin, E.; Sanschagrin, F.; Levesque, R.C. Sigma Factors in Pseudomonas aeruginosa. FEMS Microbiol. Rev. 2008, 32, 38–55. [Google Scholar] [CrossRef]
- Kilmury, S.L.N.; Burrows, L.L. The Pseudomonas aeruginosa PilSR Two-Component System Regulates Both Twitching and Swimming Motilities. MBio 2018, 9, 1–13. [Google Scholar] [CrossRef]
- Tart, A.H.; Blanks, M.J.; Wozniak, D.J. The AlgT-Dependent Transcriptional Regulator AmrZ (AlgZ) Inhibits Flagellum Biosynthesis in Mucoid, Nonmotile Pseudomonas aeruginosa Cystic Fibrosis Isolates. J. Bacteriol. 2006, 188, 6483–6489. [Google Scholar] [CrossRef] [PubMed]
- Gicquel, G.; Bouffartigues, E.; Bains, M.; Oxaran, V.; Rosay, T.; Lesouhaitier, O.; Connil, N.; Bazire, A.; Maillot, O.; Bénard, M.; et al. The Extra-Cytoplasmic Function Sigma Factor SigX Modulates Biofilm and Virulence-Related Properties in Pseudomonas aeruginosa. PLoS ONE 2013, 8, e80407. [Google Scholar] [CrossRef] [PubMed]
- Brencic, A.; Lory, S. Determination of the Regulon and Identification of Novel MRNA Targets of Pseudomonas aeruginosa RsmA. Mol. Microbiol. 2009, 72, 612–632. [Google Scholar] [CrossRef]
- Hobbs, M.; Collie, E.S.R.; Free, P.D.; Livingston, S.P.; Mattick, J.S. PilS and PilR, a Two-component Transcriptional Regulatory System Controlling Expression of Type 4 Fimbriae in Pseudomonas aeruginosa. Mol. Microbiol. 1993, 7, 669–682. [Google Scholar] [CrossRef]
- O’Callaghan, J.; Jerry Reen, F.; Adams, C.; Casey, P.G.; Gahan, C.G.M.; O’Gara, F. A Novel Host-Responsive Sensor Mediates Virulence and Type III Secretion during Pseudomonas aeruginosa-Host Cell Interactions. Microbiology 2012, 158, 1057–1070. [Google Scholar] [CrossRef] [PubMed]
- Petrova, O.E.; Sauer, K. The Novel Two-Component Regulatory System BfiSR Regulates Biofilm Development by Controlling the Small RNA RsmZ through CafA. J. Bacteriol. 2010, 192, 5275–5288. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Shao, X.; Xie, Y.; Wang, T.; Zhang, Y.; Wang, X.; Deng, X. An Integrated Genomic Regulatory Network of Virulence-Related Transcriptional Factors in Pseudomonas aeruginosa. Nat. Commun. 2019, 10, 2931. [Google Scholar] [CrossRef]
- Kulasekara, H.D.; Ventre, I.; Kulasekara, B.R.; Lazdunski, A.; Filloux, A.; Lory, S. A Novel Two-Component System Controls the Expression of Pseudomonas aeruginosa Fimbrial Cup Genes. Mol. Microbiol. 2005, 55, 368–380. [Google Scholar] [CrossRef]
- Mikkelsen, H.; Hui, K.; Barraud, N.; Filloux, A. The Pathogenicity Island Encoded PvrSR/RcsCB Regulatory Network Controls Biofilm Formation and Dispersal in Pseudomonas aeruginosa PA14. Mol. Microbiol. 2013, 89, 450–463. [Google Scholar] [CrossRef]
- Wang, C.; Chen, W.; Xia, A.; Zhang, R.; Huang, Y.; Yang, S.; Ni, L.; Jin, F. Carbon Starvation Induces the Expression of Pprb-Regulated Genes in Pseudomonas aeruginosa. Appl. Environ. Microbiol. 2019, 85, e01705-19. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Yang, F.; Shao, X.; Yue, Z.; Li, Z.; Song, Y.; Pan, X.; Jin, Y.; Cheng, Z.; Ha, U.-H.; et al. ECF Sigma Factor HxuI Is Critical for In Vivo Fitness of Pseudomonas aeruginosa during Infection. Microbiol. Spectr. 2022, 10, e01620-21. [Google Scholar] [CrossRef] [PubMed]
- Bazire, A.; Shioya, K.; Soum-Soutéra, E.; Bouffartigues, E.; Ryder, C.; Guentas-Dombrowsky, L.; Hémery, G.; Linossier, I.; Chevalier, S.; Wozniak, D.J.; et al. The Sigma Factor AlgU Plays a Key Role in Formation of Robust Biofilms by Nonmucoid Pseudomonas aeruginosa. J. Bacteriol. 2010, 192, 3001–3010. [Google Scholar] [CrossRef]
- Déziel, E.; Gopalan, S.; Tampakaki, A.P.; Lépine, F.; Padfield, K.E.; Saucier, M.; Xiao, G.; Rahme, L.G. The Contribution of MvfR to Pseudomonas aeruginosa Pathogenesis and Quorum Sensing Circuitry Regulation: Multiple Quorum Sensing-Regulated Genes are Modulated without Affecting LasRI, RhlRI or the Production of N-Acyl-L-Homoserine Lactones. Mol. Microbiol. 2005, 55, 998–1014. [Google Scholar] [CrossRef] [PubMed]
- McGuffie, B.A.; Vallet-Gely, I.; Dove, S.L. σ Factor and Anti-σ Factor That Control Swarming Motility and Biofilm Formation in Pseudomonas aeruginosa. J. Bacteriol. 2016, 198, 755. [Google Scholar] [CrossRef]
- Suh, S.J.; Silo-Suh, L.; Woods, D.E.; Hassett, D.J.; West, S.E.H.; Ohman, D.E. Effect of RpoS Mutation on the Stress Response and Expression of Virulence Factors in Pseudomonas aeruginosa. J. Bacteriol. 1999, 181, 3890–3897. [Google Scholar] [CrossRef]
- Irie, Y.; Starkey, M.; Edwards, A.N.; Wozniak, D.J.; Romeo, T.; Parsek, M.R. Pseudomonas aeruginosa Biofilm Matrix Polysaccharide Psl Is Regulated Transcriptionally by RpoS and Post-Transcriptionally by RsmA. Mol. Microbiol. 2010, 78, 158–172. [Google Scholar] [CrossRef]
- Williamson, K.S.; Dlakić, M.; Akiyama, T.; Franklin, M.J. The Pseudomonas aeruginosa RpoH (Σ32) Regulon and Its Role in Essential Cellular Functions, Starvation Survival, and Antibiotic Tolerance. Int. J. Mol. Sci. 2023, 24, 1513. [Google Scholar] [CrossRef]
- Baynham, P.J.; Wozniak, D.J. Identification and Characterization of AlgZ, an AlgT-Dependent DNA-Binding Protein Required for Pseudomonas aeruginosa AlgD Transcription. Mol. Microbiol. 1996, 22, 97–108. [Google Scholar] [CrossRef]
- Chand, N.S.; Hung, D.T. The Two-Component Sensor Kinase KinB Acts as a Non-Canonical Switch between Acute and Chronic Infection. Virulence 2011, 2, 553–558. [Google Scholar] [CrossRef][Green Version]
- Stacey, S.D.; Pritchett, C.L. Pseudomonas aeruginosa AlgU Contributes to Posttranscriptional Activity by Increasing RsmA Expression in a MucA22 Strain. J. Bacteriol. 2016, 198, 1812–1826. [Google Scholar] [CrossRef]
- Petrova, O.E.; Schurr, J.R.; Schurr, M.J.; Sauer, K. Microcolony Formation by the Opportunistic Pathogen Pseudomonas aeruginosa Requires Pyruvate Andpyruvate Fermentation. Mol. Microbiol. 2012, 86, 819–835. [Google Scholar] [CrossRef]
- Carty, N.L.; Layland, N.; Colmer-Hamood, J.A.; Calfee, M.W.; Pesci, E.C.; Hamood, A.N. PtxR Modulates the Expression of QS-Controlled Virulence Factors in the Pseudomonas aeruginosa Strain PAO1. Mol. Microbiol. 2006, 61, 782–794. [Google Scholar] [CrossRef]
- Miranda, S.W.; Asfahl, K.L.; Dandekar, A.A.; Greenberg, E.P. Pseudomonas aeruginosa Quorum Sensing. Adv. Exp. Med. Biol. 2022, 1386, 95–115. [Google Scholar] [CrossRef]
- Bremer, E. Liberate and Grab It, Ingest and Digest It: The GbdR Regulon of the Pathogen Pseudomonas aeruginosa. J. Bacteriol. 2014, 196, 3–6. [Google Scholar] [CrossRef] [PubMed]
- Firoved, A.M.; Deretic, V. Microarray Analysis of Global Gene Expression in Mucoid Pseudomonas aeruginosa. J. Bacteriol. 2003, 185, 1071–1081. [Google Scholar] [CrossRef]
- Gilbert, K.B.; Kim, T.H.; Gupta, R.; Greenberg, E.P.; Schuster, M. Global Position Analysis of the Pseudomonas aeruginosa Quorum-Sensing Transcription Factor LasR. Mol. Microbiol. 2009, 73, 1072–1085. [Google Scholar] [CrossRef]
- Gellatly, S.L.; Bains, M.; Breidenstein, E.B.; Strehmel, J.; Reffuveille, F.; Taylor, P.K.; Yeung, A.T.; Overhage, J.; Hancock, R.E. Novel Roles for Two-Component Regulatory Systems in Cytotoxicity and Virulence-Related properties in Pseudomonas aeruginosa. AIMS Microbiol. 2018, 4, 173–191. [Google Scholar] [CrossRef] [PubMed]
- Rumbaugh, K.P.; Griswold, J.A.; Hamood, A.N. Contribution of the Regulatory Gene LasR to the Pathogenesis of Pseudomonas aeruginosa Infection of Burned Mice. J. Burn Care Rehabil. 1999, 20, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Ochsner, U.A.; Wilderman, P.J.; Vasil, A.I.; Vasil, M.L. GeneChip Expression Analysis of the Iron Starvation Response in Pseudomonas aeruginosa: Identification of Novel Pyoverdine Biosynthesis Genes. Mol. Microbiol. 2002, 45, 1277–1287. [Google Scholar] [CrossRef]
- Schulz, S.; Eckweiler, D.; Bielecka, A.; Nicolai, T.; Franke, R.; Dötsch, A.; Hornischer, K.; Bruchmann, S.; Düvel, J.; Häussler, S. Elucidation of Sigma Factor-Associated Networks in Pseudomonas aeruginosa Reveals a Modular Architecture with Limited and Function-Specific Crosstalk. PLoS Pathog. 2015, 11, e1004744. [Google Scholar] [CrossRef]
- Pan, X.; Liang, H.; Zhao, X.; Zhang, Q.; Chen, L.; Yue, Z.; Yin, L.; Jin, Y.; Bai, F.; Cheng, Z.; et al. Regulatory and Structural Mechanisms of PvrA-Mediated Regulation of the PQS Quorum-Sensing System and PHA Biosynthesis in Pseudomonas aeruginosa. Nucleic Acids Res. 2023, 51, 2691–2708. [Google Scholar] [CrossRef]
- Lo, Y.L.; Chen, C.L.; Shen, L.; Chen, Y.C.; Wang, Y.H.; Lee, C.C.; Wang, L.C.; Chuang, C.H.; Janapatla, R.P.; Chiu, C.H.; et al. Characterization of the Role of Global Regulator FliA in the Pathophysiology of Pseudomonas aeruginosa Infection. Res. Microbiol. 2018, 169, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Okino, N.; Ito, M. Molecular Mechanism for Sphingosine-Induced Pseudomonas Ceramidase Expression through the Transcriptional Regulator SphR. Sci. Rep. 2016, 6, 38797. [Google Scholar] [CrossRef] [PubMed]
- Urbanowski, M.L.; Lykken, G.L.; Yahr, T.L. A Secreted Regulatory Protein Couples Transcription to the Secretory Activity of the Pseudomonas aeruginosa Type III Secretion System. Proc. Natl. Acad. Sci. USA 2005, 102, 9930–9935. [Google Scholar] [CrossRef]
- Daddaoua, A.; Fillet, S.; Fernández, M.; Udaondo, Z.; Krell, T.; Ramos, J.L. Genes for Carbon Metabolism and the ToxA Virulence Factor in Pseudomonas aeruginosa are Regulated through Molecular Interactions of PtxR and PtxS. PLoS ONE 2012, 7, e39390. [Google Scholar] [CrossRef]
- Daddaoua, A.; Molina-Santiago, C.; De La Torre, J.; Krell, T.; Ramos, J.L. GtrS and GltR Form a Two-Component System: The Central Role of 2-Ketogluconate in the Expression of Exotoxin A and Glucose Catabolic Enzymes in Pseudomonas aeruginosa. Nucleic Acids Res. 2014, 42, 7654–7665. [Google Scholar] [CrossRef]
- Firoved, A.M.; Ornatowski, W.; Deretic, V. Microarray Analysis Reveals Induction of Lipoprotein Genes in Mucoid Pseudomonas aeruginosa: Implications for Inflammation in Cystic Fibrosis. Infect. Immun. 2004, 72, 5012. [Google Scholar] [CrossRef] [PubMed]
- Balasubramanian, D.; Kumari, H.; Jaric, M.; Fernandez, M.; Turner, K.H.; Dove, S.L.; Narasimhan, G.; Lory, S.; Mathee, K. Deep Sequencing Analyses Expands the Pseudomonas aeruginosa AmpR Regulon to Include Small RNA-Mediated Regulation of Iron Acquisition, Heat Shock and Oxidative Stress Response. Nucleic Acids Res. 2014, 42, 979. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Yu, X.; Zhu, M.; Kang, H.; Ma, J.; Wu, M.; Gan, J.; Deng, X.; Liang, H. Structural and Molecular Mechanism of CdpR Involved in Quorum-Sensing and Bacterial Virulence in Pseudomonas aeruginosa. PLoS Biol. 2016, 14, e1002449. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Jiménez, A.; Marcos-Torres, F.J.; Llamas, M.A. Mechanisms of Iron Homeostasis in Pseudomonas aeruginosa and Emerging Therapeutics Directed to Disrupt This Vital Process. Microb. Biotechnol. 2023, 16, 1475–1491. [Google Scholar] [CrossRef] [PubMed]
- Visca, P.; Imperi, F. An Essential Transcriptional Regulator: The Case of Pseudomonas aeruginosa Fur. Future Microbiol. 2018, 13, 853–856. [Google Scholar] [CrossRef]
- Galdino, A.C.M.; Branquinha, M.H.; Santos, A.L.S.; Viganor, L. Pseudomonas aeruginosa and Its Arsenal of Proteases: Weapons to Battle the Host. In Pathophysiological Aspects of Proteases; Springer: Berlin/Heidelberg, Germany, 2017; pp. 381–397. [Google Scholar] [CrossRef]
- López, D.J.; Collado, M.I.; Ibarguren, M.; Vasil, A.I.; Vasil, M.L.; Goñi, F.M.; Alonso, A. Multiple Phospholipid Substrates of Phospholipase C/Sphingomyelinase HR2 from Pseudomonas aeruginosa. Chem. Phys. Lipids 2011, 164, 78–82. [Google Scholar] [CrossRef] [PubMed]
- Okino, N.; Ito, M. Ceramidase Enhances Phospholipase C-Induced Hemolysis by Pseudomonas aeruginosa. J. Biol. Chem. 2007, 282, 6021–6030. [Google Scholar] [CrossRef] [PubMed]
- Vasil, M.L.; Stonehouse, M.J.; Vasil, A.I.; Wadsworth, S.J.; Goldfine, H.; Bolcome, R.E.; Chan, J. A Complex Extracellular Sphingomyelinase of Pseudomonas aeruginosa Inhibits Angiogenesis by Selective Cytotoxicity to Endothelial Cells. PLoS Pathog. 2009, 5, e1000420. [Google Scholar] [CrossRef]
- Wood, S.J.; Goldufsky, J.W.; Seu, M.Y.; Dorafshar, A.H.; Shafikhani, S.H. Pseudomonas aeruginosa Cytotoxins: Mechanisms of Cytotoxicity and Impact on Inflammatory Responses. Cells 2023, 12, 195. [Google Scholar] [CrossRef]
- Hauser, A.R. The Type III Secretion System of Pseudomonas aeruginosa: Infection by Injection. Nat. Rev. Microbiol. 2009, 7, 654–665. [Google Scholar] [CrossRef]
- Finck-Barbançon, V.; Goranson, J.; Zhu, L.; Sawa, T.; Wiener-Kronish, J.P.; Fleiszig, S.M.J.; Wu, C.; Mende-Mueller, L.; Frank, D.W. ExoU Expression by Pseudomonas aeruginosa Correlates with Acute Cytotoxicity and Epithelial Injury. Mol. Microbiol. 1997, 25, 547–557. [Google Scholar] [CrossRef]
- Roy-Burman, A.; Savel, R.H.; Racine, S.; Swanson, B.L.; Revadigar, N.S.; Fujimoto, J.; Sawa, T.; Frank, D.W.; Wiener-Kronish, J.P. Type III Protein Secretion Is Associated with Death in Lower Respiratory and Systemic Pseudomonas aeruginosa Infections. J. Infect. Dis. 2001, 183, 1767–1774. [Google Scholar] [CrossRef]
- Kroken, A.R.; Kumar, N.G.; Yahr, T.L.; Smith, B.E.; Nieto, V.; Horneman, H.; Evans, D.J.; Fleiszig, S.M.J. Exotoxin S Secreted by Internalized Pseudomonas aeruginosa Delays Lytic Host Cell Death. PLoS Pathog. 2022, 18, e1010306. [Google Scholar] [CrossRef]
- Wedekind, J.E.; Trame, C.B.; Dorywalska, M.; Koehl, P.; Raschke, T.M.; McKee, M.; FitzGerald, D.; Collier, R.J.; McKay, D.B. Refined Crystallographic Structure of Pseudomonas aeruginosa Exotoxin A and Its Implications for the Molecular Mechanism of Toxicity. J. Mol. Biol. 2001, 314, 823–837. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Youle, R.J.; FitzGerald, D.J.; Pastan, I. Pseudomonas Exotoxin A-Mediated Apoptosis Is Bak Dependent and Preceded by the Degradation of Mcl-1. Mol. Cell. Biol. 2010, 30, 3444–3452. [Google Scholar] [CrossRef] [PubMed]
- Jiang, F.; Waterfield, N.R.; Yang, J.; Yang, G.; Jin, Q. A Pseudomonas aeruginosa Type VI Secretion Phospholipase D Effector Targets Both Prokaryotic and Eukaryotic Cells. Cell Host Microbe 2014, 15, 600–610. [Google Scholar] [CrossRef]
- Sana, T.G.; Berni, B.; Bleves, S. The T6SSs of Pseudomonas aeruginosa Strain PAO1 and Their Effectors: Beyond Bacterial-Cell Targeting. Front. Cell. Infect. Microbiol. 2016, 6, 61. [Google Scholar] [CrossRef] [PubMed]
- Sana, T.G.; Baumann, C.; Merdes, A.; Soscia, C.; Rattei, T.; Hachani, A.; Jones, C.; Bennett, K.L.; Filloux, A.; Superti-Furga, G.; et al. Internalization of Pseudomonas aeruginosa Strain PAO1 into Epithelial Cells Is Promoted by Interaction of a T6SS Effector with the Microtubule Network. MBio 2015, 6, e00712-15. [Google Scholar] [CrossRef]
- Jiang, F.; Wang, X.; Wang, B.; Chen, L.; Zhao, Z.; Waterfield, N.R.; Yang, G.; Jin, Q. The Pseudomonas aeruginosa Type VI Secretion PGAP1-like Effector Induces Host Autophagy by Activating Endoplasmic Reticulum Stress. Cell Rep. 2016, 16, 1502–1509. [Google Scholar] [CrossRef]
- Usher, L.R.; Lawson, R.A.; Geary, I.; Taylor, C.J.; Bingle, C.D.; Taylor, G.W.; Whyte, M.K.B. Induction of Neutrophil Apoptosis by the Pseudomonas aeruginosa Exotoxin Pyocyanin: A Potential Mechanism of Persistent Infection. J. Immunol. 2002, 168, 1861–1868. [Google Scholar] [CrossRef]
- Lau, G.W.; Ran, H.; Kong, F.; Hassett, D.J.; Mavrodi, D. Pseudomonas aeruginosa Pyocyanin Is Critical for Lung Infection in Mice. Infect. Immun. 2004, 72, 4275–4278. [Google Scholar] [CrossRef]
- Anderson, R.D.; Roddam, L.F.; Bettiol, S.; Sanderson, K.; Reid, D.W. Biosignificance of Bacterial Cyanogenesis in the CF Lung. J. Cyst. Fibros. 2010, 9, 158–164. [Google Scholar] [CrossRef][Green Version]
- Otero-Asman, J.R.; García-García, A.I.; Civantos, C.; Quesada, J.M.; Llamas, M.A. Pseudomonas aeruginosa Possesses Three Distinct Systems for Sensing and Using the Host Molecule Haem. Environ. Microbiol. 2019, 21, 4629–4647. [Google Scholar] [CrossRef]
- Minandri, F.; Imperi, F.; Frangipani, E.; Bonchi, C.; Visaggio, D.; Facchini, M.; Pasquali, P.; Bragonzi, A.; Visca, P. Role of Iron Uptake Systems in Pseudomonas aeruginosa Virulence and Airway Infection. Infect. Immun. 2016, 84, 2324–2335. [Google Scholar] [CrossRef]
- Kirienko, N.V.; Cezairliyan, B.O.; Ausubel, F.M.; Powell, J.R. Pseudomonas aeruginosa PA14 Pathogenesis in Caenorhabditis Elegans. In Pseudomonas Methods and Protocols; Filloux, A., Ramos, J.-L., Eds.; Springer: New York, NY, USA, 2014; pp. 653–669. ISBN 978-1-4939-0473-0. [Google Scholar]
- Pasqua, M.; Visaggio, D.; Lo Sciuto, A.; Genah, S.; Banin, E.; Visca, P.; Imperi, F. Ferric Uptake Regulator Fur Is Conditionally Essential in Pseudomonas aeruginosa. J. Bacteriol. 2017, 199, e00472-17. [Google Scholar] [CrossRef] [PubMed]
- Reinhart, A.A.; Nguyen, A.T.; Brewer, L.K.; Bevere, J.; Jones, J.W.; Kane, M.A.; Damron, F.H.; Barbier, M.; Oglesby-Sherrouse, A.G. The Pseudomonas aeruginosa PrrF Small RNAs Regulate Iron Homeostasis during Acute Murine Lung Infection. Infect. Immun. 2017, 85, e76416. [Google Scholar] [CrossRef]
- Ishihama, A. Functional Modulation of Escherichia coli RNA Polymerase. Annu. Rev. Microbiol. 2000, 54, 499–518. [Google Scholar] [CrossRef] [PubMed]
- Wigneshweraraj, S.; Bose, D.; Burrows, P.C.; Joly, N.; Schumacher, J.; Rappas, M.; Pape, T.; Zhang, X.; Stockley, P.; Severinov, K.; et al. Modus Operandi of the Bacterial RNA Polymerase Containing the Σ54 Promoter-Specificity Factor. Mol. Microbiol. 2008, 68, 538–546. [Google Scholar] [CrossRef] [PubMed]
- Bastiaansen, K.C.; Bitter, W. ECF Sigma Factors: From Stress Management to Iron Uptake. In Bacterial Regulatory Networks; Caister Academic Press: Poole, UK, 2012. [Google Scholar]
- Paget, M.S. Bacterial Sigma Factors and Anti-Sigma Factors: Structure, Function and Distribution. Biomolecules 2015, 5, 1245–1265. [Google Scholar] [CrossRef]
- Browning, D.F.; Busby, S.J.W. Local and Global Regulation of Transcription Initiation in Bacteria. Nat. Rev. Microbiol. 2016, 14, 638–650. [Google Scholar] [CrossRef]
- Ulrich, L.E.; Koonin, E.V.; Zhulin, I.B. One-Component Systems Dominate Signal Transduction in Prokaryotes. Trends Microbiol. 2005, 13, 52–56. [Google Scholar] [CrossRef]
- Gumerov, V.M.; Ortega, D.R.; Adebali, O.; Ulrich, L.E.; Zhulin, I.B. MiST 3.0: An Updated Microbial Signal Transduction Database with an Emphasis on Chemosensory Systems. Nucleic Acids Res. 2020, 48, D459. [Google Scholar] [CrossRef]
- Cashin, P.; Goldsack, L.; Hall, D.; O’Toole, R. Contrasting Signal Transduction Mechanisms in Bacterial and Eukaryotic Gene Transcription. FEMS Microbiol. Lett. 2006, 261, 155–164. [Google Scholar] [CrossRef]
- Lee, J.; Zhang, L. The Hierarchy Quorum Sensing Network in Pseudomonas aeruginosa. Protein Cell 2015, 6, 26–41. [Google Scholar] [CrossRef]
- Rumbaugh, K.P.; Griswold, J.A.; Iglewski, B.H.; Hamood, A.N. Contribution of Quorum Sensing to the Virulence of Pseudomonas aeruginosa in Burn Wound Infections. Infect. Immun. 1999, 67, 5854–5862. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.; Gan, J.; Zhao, J.; Kong, W.; Zhang, J.; Zhu, M.; Li, F.; Song, Y.; Qin, J.; Liang, H. Crystal Structure of Pseudomonas aeruginosa RsaL Bound to Promoter DNA Reaffirms Its Role as a Global Regulator Involved in Quorum-Sensing. Nucleic Acids Res. 2017, 45, 699–710. [Google Scholar] [CrossRef] [PubMed]
- Lesprit, P.; Faurisson, F.; Join-Lambert, O.; Roudot-Thoraval, F.; Foglino, M.; Vissuzaine, C.; Carbon, C. Role of the Quorum-Sensing System in Experimental Pneumonia Due to Pseudomonas aeruginosa in Rats. Am. J. Respir. Crit. Care Med. 2003, 167, 1478–1482. [Google Scholar] [CrossRef]
- Ochsner, U.A.; Reiser, J. Autoinducer-Mediated Regulation of Rhamnolipid Biosurfactant Synthesis in Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA 1995, 92, 6424–6428. [Google Scholar] [CrossRef]
- Mukherjee, S.; Moustafa, D.; Smith, C.D.; Goldberg, J.B.; Bassler, B.L. The RhlR Quorum-Sensing Receptor Controls Pseudomonas aeruginosa Pathogenesis and Biofilm Development Independently of Its Canonical Homoserine Lactone Autoinducer. PLoS Pathog. 2017, 13, e1006504. [Google Scholar] [CrossRef] [PubMed]
- Pearson, J.P.; Feldman, M.; Iglewski, B.H.; Prince, A. Pseudomonas aeruginosa Cell-to-Cell Signaling Is Required for Virulence in a Model of Acute Pulmonary Infection. Infect. Immun. 2000, 68, 4331–4334. [Google Scholar] [CrossRef] [PubMed]
- O’Loughlin, C.T.; Miller, L.C.; Siryaporn, A.; Drescher, K.; Semmelhack, M.F.; Bassler, B.L. A Quorum-Sensing Inhibitor Blocks Pseudomonas aeruginosa Virulence and Biofilm Formation. Proc. Natl. Acad. Sci. USA 2013, 110, 17981–17986. [Google Scholar] [CrossRef]
- Diggle, S.P.; Matthijs, S.; Wright, V.J.; Fletcher, M.P.; Chhabra, S.R.; Lamont, I.L.; Kong, X.; Hider, R.C.; Cornelis, P.; Cámara, M.; et al. The Pseudomonas aeruginosa 4-Quinolone Signal Molecules HHQ and PQS Play Multifunctional Roles in Quorum Sensing and Iron Entrapment. Chem. Biol. 2007, 14, 87–96. [Google Scholar] [CrossRef]
- Baynham, P.J.; Ramsey, D.M.; Gvozdyev, B.V.; Cordonnier, E.M.; Wozniak, D.J. The Pseudomonas aeruginosa Ribbon-Helix-Helix DNA-Binding Protein AlgZ (AmrZ) Controls Twitching Motility and Biogenesis of Type IV Pili. J. Bacteriol. 2006, 188, 132–140. [Google Scholar] [CrossRef]
- Jones, C.J.; Newsom, D.; Kelly, B.; Irie, Y.; Jennings, L.K.; Xu, B.; Limoli, D.H.; Harrison, J.J.; Parsek, M.R.; White, P.; et al. ChIP-Seq and RNA-Seq Reveal an AmrZ-Mediated Mechanism for Cyclic Di-GMP Synthesis and Biofilm Development by Pseudomonas aeruginosa. PLoS Pathog. 2014, 10, e1003984. [Google Scholar] [CrossRef] [PubMed]
- Kalia, M.; Resch, M.D.; Cherny, K.E.; Sauer, K. The Alginate and Motility Regulator AmrZ Is Essential for the Regulation of the Dispersion Response by Pseudomonas aeruginosa Biofilms. mSphere 2022, 7, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Gallegos, M.T.; Schleif, R.; Bairoch, A.; Hofmann, K.; Ramos, J.L. Arac/XylS Family of Transcriptional Regulators. Microbiol. Mol. Biol. Rev. 1997, 61, 393–410. [Google Scholar] [CrossRef] [PubMed]
- Tian, Z.; Cheng, S.; Xia, B.; Jin, Y.; Bai, F.; Cheng, Z.; Jin, S.; Liu, X.; Wu, W. Pseudomonas aeruginosa ExsA Regulates a Metalloprotease, ImpA, That Inhibits Phagocytosis of Macrophages. Infect. Immun. 2019, 87, e69519. [Google Scholar] [CrossRef] [PubMed]
- Urbanowski, M.L.; Brutinel, E.D.; Yahr, T.L. Translocation of ExsE into Chinese Hamster Ovary Cells Is Required for Transcriptional Induction of the Pseudomonas aeruginosa Type III Secretion System. Infect. Immun. 2007, 75, 4432–4439. [Google Scholar] [CrossRef] [PubMed]
- McCaw, M.L.; Lykken, G.L.; Singh, P.K.; Yahr, T.L. ExsD Is a Negative Regulator of the Pseudomonas aeruginosa Type III Secretion Regulon. Mol. Microbiol. 2002, 46, 1123–1133. [Google Scholar] [CrossRef] [PubMed]
- Karna, S.L.R.; Nguyen, J.Q.; Evani, S.J.; Qian, L.W.; Chen, P.; Abercrombie, J.J.; Sebastian, E.A.; Fourcaudot, A.B.; Leung, K.P. T3SS and Alginate Biosynthesis of Pseudomonas aeruginosa Impair Healing of Infected Rabbit Wounds. Microb. Pathog. 2020, 147, 104254. [Google Scholar] [CrossRef]
- Allsopp, L.P.; Collins, A.C.Z.; Hawkins, E.; Wood, T.E.; Filloux, A. RpoN/Sfa2-Dependent Activation of the Pseudomonas aeruginosa H2-T6SS and Its Cognate Arsenal of Antibacterial Toxins. Nucleic Acids Res. 2022, 50, 227–243. [Google Scholar] [CrossRef]
- Sana, T.G.; Soscia, C.; Tonglet, C.M.; Garvis, S.; Bleves, S. Divergent Control of Two Type VI Secretion Systems by RpoN in Pseudomonas aeruginosa. PLoS ONE 2013, 8, e76030. [Google Scholar] [CrossRef]
- Germán Sánchez, D.; Primo, E.D.; Damiani, M.T.; Lisa, A.T. Pseudomonas aeruginosa GbdR Gene Is Transcribed from a Σ54-Dependent Promoter under the Control of NtrC/CbrB, IHF and BetI. Microbiology 2017, 163, 1343–1354. [Google Scholar] [CrossRef]
- Pan, X.; Fan, Z.; Chen, L.; Liu, C.; Bai, F.; Wei, Y.; Tian, Z.; Dong, Y.; Shi, J.; Chen, H.; et al. PvrA Is a Novel Regulator That Contributes to Pseudomonas aeruginosa Pathogenesis by Controlling Bacterial Utilization of Long Chain Fatty Acids. Nucleic Acids Res. 2021, 48, 5967–5985. [Google Scholar] [CrossRef] [PubMed]
- LaBauve, A.E.; Wargo, M.J. Detection of Host-Derived Sphingosine by Pseudomonas aeruginosa Is Important for Survival in the Murine Lung. PLoS Pathog. 2014, 10, e1003889. [Google Scholar] [CrossRef] [PubMed]
- Maddocks, S.E.; Oyston, P.C.F. Structure and Function of the LysR-Type Transcriptional Regulator (LTTR) Family Proteins. Microbiology 2008, 154, 3609–3623. [Google Scholar] [CrossRef] [PubMed]
- Daddaoua, A.; Krell, T.; Ramos, J.L. Transcriptional Control by Two Interacting Regulatory Proteins: Identification of the PtxS Binding Site at PtxR. Nucleic Acids Res. 2013, 41, 10150–10156. [Google Scholar] [CrossRef]
- Hobman, J.L. MerR Family Transcription Activators: Similar Designs, Different Specificities. Mol. Microbiol. 2007, 63, 1275–1278. [Google Scholar] [CrossRef]
- Dietrich, L.E.P.; Price-Whelan, A.; Petersen, A.; Whiteley, M.; Newman, D.K. The Phenazine Pyocyanin Is a Terminal Signalling Factor in the Quorum Sensing Network of Pseudomonas aeruginosa. Mol. Microbiol. 2006, 61, 1308–1321. [Google Scholar] [CrossRef]
- Fujikawa, M.; Kobayashi, K.; Tsutsui, Y.; Tanaka, T.; Kozawa, T. Rational Tuning of Superoxide Sensitivity in SoxR, the [2Fe-2S] Transcription Factor: Implications of Species-Specific Lysine Residues. Biochemistry 2017, 56, 403–410. [Google Scholar] [CrossRef]
- Palma, M.; Zurita, J.; Ferreras, J.A.; Worgall, S.; Larone, D.H.; Shi, L.; Campagne, F.; Quadri, L.E.N. Pseudomonas aeruginosa SoxR Does Not Conform to the Archetypal Paradigm for SoxR-Dependent Regulation of the Bacterial Oxidative Stress Adaptive Response. Infect. Immun. 2005, 73, 2958–2966. [Google Scholar] [CrossRef]
- Aendekerk, S.; Ghysels, B.; Cornelis, P.; Baysse, C. Characterization of a New Efflux Pump, MexGHI-OpmD, from Pseudomonas aeruginosa That Confers Resistance to Vanadium. Microbiology 2002, 148, 2371–2381. [Google Scholar] [CrossRef]
- Sakhtah, H.; Koyama, L.; Zhang, Y.; Morales, D.K.; Fields, B.L.; Price-Whelan, A.; Hogan, D.A.; Shepard, K.; Dietrich, L.E.P. The Pseudomonas aeruginosa Efflux Pump MexGHI-OpmD Transports a Natural Phenazine That Controls Gene Expression and Biofilm Development. Proc. Natl. Acad. Sci. USA 2016, 113, E3538–E3547. [Google Scholar] [CrossRef]
- Ha, U.; Jin, S. Expression of the SoxR Gene of Pseudomonas aeruginosa Is Inducible during Infection of Burn Wounds in Mice and Is Required To Cause Efficient Bacteremia. Infect. Immun. 1999, 67, 5324. [Google Scholar] [CrossRef]
- Gifford, D.R.; Furió, V.; Papkou, A.; Vogwill, T.; Oliver, A.; MacLean, R.C. Identifying and Exploiting Genes That Potentiate the Evolution of Antibiotic Resistance. Nat. Ecol. Evol. 2018, 2, 1033–1039. [Google Scholar] [CrossRef]
- Dik, D.A.; Domínguez-Gil, T.; Lee, M.; Hesek, D.; Byun, B.; Fishovitz, J.; Boggess, B.; Hellman, L.M.; Fisher, J.F.; Hermoso, J.A.; et al. Muropeptide Binding and the X-ray Structure of the Effector Domain of the Transcriptional Regulator AmpR of Pseudomonas aeruginosa. J. Am. Chem. Soc. 2017, 139, 1448. [Google Scholar] [CrossRef] [PubMed]
- Balasubramanian, D.; Schneper, L.; Merighi, M.; Smith, R.; Narasimhan, G.; Lory, S.; Mathee, K. The Regulatory Repertoire of Pseudomonas aeruginosa AmpC SS-Lactamase Regulator AmpR Includes Virulence Genes. PLoS ONE 2012, 7, e34067. [Google Scholar] [CrossRef]
- Stock, A.M.; Robinson, V.L.; Goudreau, P.N. Two-Component Signal Transduction. Annu. Rev. Biochem. 2000, 69, 183–215. [Google Scholar] [CrossRef] [PubMed]
- Groisman, E.A. Feedback Control of Two-Component Regulatory Systems. Annu. Rev. Microbiol. 2016, 70, 103–124. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Huang, J.; Liu, Z.; Lin, Q.; Xu, Z. The Two-Component System FleS/FleR Represses H1-T6SS Via. Appl. Environ. Microbiol. 2022, 88, e01655-21. [Google Scholar] [CrossRef]
- Zhou, T.; Huang, J.; Liu, Z.; Xu, Z.; Zhang, L.H. Molecular Mechanisms Underlying the Regulation of Biofilm Formation and Swimming Motility by FleS/FleR in Pseudomonas aeruginosa. Front. Microbiol. 2021, 12, 707711. [Google Scholar] [CrossRef] [PubMed]
- Colley, B.; Dederer, V.; Carnell, M.; Kjelleberg, S.; Rice, S.A.; Klebensberger, J. SiaA/D Interconnects c-Di-GMP and RsmA Signaling to Coordinate Cellular Aggregation of Pseudomonas aeruginosa in Response to Environmental Conditions. Front. Microbiol. 2016, 7, 179. [Google Scholar] [CrossRef]
- Rao, F.; Yang, Y.; Qi, Y.; Liang, Z.X. Catalytic Mechanism of Cyclic Di-GMP-Specific Phosphodiesterase: A Study of the EAL Domain-Containing RocR from Pseudomonas aeruginosa. J. Bacteriol. 2008, 190, 3622–3631. [Google Scholar] [CrossRef]
- He, J.; Baldini, R.L.; Déziel, E.; Saucier, M.; Zhang, Q.; Liberati, N.T.; Lee, D.; Urbach, J.; Goodman, H.M.; Rahme, L.G. The Broad Host Range Pathogen Pseudomonas aeruginosa Strain PA14 Carries Two Pathogenicity Islands Harboring Plant and Animal Virulence Genes. Proc. Natl. Acad. Sci. USA 2004, 101, 2530–2535. [Google Scholar] [CrossRef]
- Okkotsu, Y.; Tieku, P.; Fitzsimmons, L.F.; Churchill, M.E.; Schurr, M.J. Pseudomonas aeruginosa AlgR Phosphorylation Modulates Rhamnolipid Production and Motility. J. Bacteriol. 2013, 195, 5499–5515. [Google Scholar] [CrossRef]
- Matilla, M.A.; Martín-Mora, D.; Gavira, J.A.; Krell, T. Pseudomonas aeruginosa as a Model To Study Chemosensory Pathway Signaling. Microbiol. Mol. Biol. Rev. 2021, 85, e00151-20. [Google Scholar] [CrossRef]
- Marko, V.A.; Kilmury, S.L.N.; MacNeil, L.T.; Burrows, L.L. Pseudomonas aeruginosa Type IV Minor Pilins and PilY1 Regulate Virulence by Modulating FimS-AlgR Activity. PLoS Pathog. 2018, 14, e1007074. [Google Scholar] [CrossRef]
- Chand, N.S.; Lee, J.S.W.; Clatworthy, A.E.; Golas, A.J.; Smith, R.S.; Hung, D.T. The Sensor Kinase KinB Regulates Virulence in Acute Pseudomonas aeruginosa Infection. J. Bacteriol. 2011, 193, 2989–2999. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chand, N.S.; Clatworthy, A.E.; Hung, D.T. The Two-Component Sensor KinB Acts as a Phosphatase to Regulate Pseudomonas aeruginosa Virulence. J. Bacteriol. 2012, 194, 6537–6547. [Google Scholar] [CrossRef] [PubMed]
- Damron, F.H.; Owings, J.P.; Okkotsu, Y.; Varga, J.J.; Schurr, J.R.; Goldberg, J.B.; Schurr, M.J.; Yu, H.D. Analysis of the Pseudomonas aeruginosa Regulon Controlled by the Sensor Kinase KinB and Sigma Factor RpoN. J. Bacteriol. 2012, 194, 1317–1330. [Google Scholar] [CrossRef] [PubMed]
- Mikkelsen, H.; McMullan, R.; Filloux, A. The Pseudomonas aeruginosa Reference Strain PA14 Displays Increased Virulence Due to a Mutation in LadS. PLoS ONE 2011, 6, e29113. [Google Scholar] [CrossRef]
- Monteagudo-Cascales, E.; Santero, E.; Canosa, I. The Regulatory Hierarchy Following Signal Integration by the CbrAB Two-Component System: Diversity of Responses and Functions. Genes 2022, 13, 375. [Google Scholar] [CrossRef]
- Ryan Kaler, K.M.; Nix, J.C.; Schubot, F.D. RetS Inhibits Pseudomonas aeruginosa Biofilm Formation by Disrupting the Canonical Histidine Kinase Dimerization Interface of GacS. J. Biol. Chem. 2021, 297, 101193. [Google Scholar] [CrossRef]
- Wang, B.X.; Wheeler, K.M.; Cady, K.C.; Lehoux, S.; Cummings, R.D.; Laub, M.T.; Ribbeck, K. Mucin Glycans Signal through the Sensor Kinase RetS to Inhibit Virulence-Associated Traits in Pseudomonas aeruginosa. Curr. Biol. 2021, 31, 90–102.e7. [Google Scholar] [CrossRef]
- Fadel, F.; Bassim, V.; Francis, V.I.; Porter, S.L.; Botzanowski, T.; Legrand, P.; Perez, M.M.; Bourne, Y.; Cianférani, S.; Vincent, F. Insights into the Atypical Autokinase Activity of the Pseudomonas aeruginosa GacS Histidine Kinase and Its Interaction with RetS. Structure 2022, 30, 1285–1297.e5. [Google Scholar] [CrossRef] [PubMed]
- Petrova, O.E.; Sauer, K. SagS Contributes to the Motile-Sessile Switch and Acts in Concert with BfiSR to Enable Pseudomonas aeruginosa Biofilm Formation. J. Bacteriol. 2011, 193, 6614–6628. [Google Scholar] [CrossRef]
- Cao, Q.; Wang, Y.; Chen, F.; Xia, Y.; Lou, J.; Zhang, X.; Yang, N.; Sun, X.; Zhang, Q.; Zhuo, C.; et al. A Novel Signal Transduction Pathway That Modulates Rhl Quorum Sensing and Bacterial Virulence in Pseudomonas aeruginosa. PLoS Pathog. 2014, 10, e1004340. [Google Scholar] [CrossRef] [PubMed]
- Fan, K.; Cao, Q.; Lan, L. Genome-Wide Mapping Reveals Complex Regulatory Activities of Bfmr in Pseudomonas aeruginosa. Microorganisms 2021, 9, 485. [Google Scholar] [CrossRef]
- Sarwar, Z.; Wang, M.X.; Lundgren, B.R.; Nomura, C.T. MifS, a DctB Family Histidine Kinase, Is a Specific Regulator of α-Ketoglutarate Response in Pseudomonas aeruginosa PAO1. Microbiology 2020, 166, 867. [Google Scholar] [CrossRef]
- Xiong, W.; Perna, A.; Jacob, I.B.; Lundgren, B.R.; Wang, G. The Enhancer-Binding Protein MifR, an Essential Regulator of a-Ketoglutarate Transport, Is Required for Full Virulence of Pseudomonas aeruginosa PAO1 in a Mouse Model of Pneumonia. Infect. Immun. 2022, 90, e00136-22. [Google Scholar] [CrossRef]
- Xu, C.; Cao, Q.; Lan, L. Glucose-Binding of Periplasmic Protein Gltb Activates Gtrs-Gltr Two-Component System in Pseudomonas aeruginosa. Microorganisms 2021, 9, 447. [Google Scholar] [CrossRef] [PubMed]
- Quesada, J.M.; Otero-Asman, J.R.; Bastiaansen, K.C.; Civantos, C.; Llamas, M.A. The Activity of the Pseudomonas aeruginosa Virulence Regulator ΣVreI Is Modulated by the Anti-σ Factor VreR and the Transcription Factor PhoB. Front. Microbiol. 2016, 7, 1159. [Google Scholar] [CrossRef]
- Lamarche, M.G.; Wanner, B.L.; Crépin, S.; Harel, J. The Phosphate Regulon and Bacterial Virulence: A Regulatory Network Connecting Phosphate Homeostasis and Pathogenesis. FEMS Microbiol. Rev. 2008, 32, 461–473. [Google Scholar] [CrossRef]
- Ball, G.; Durand, É.; Lazdunski, A.; Filloux, A. A Novel Type II Secretion System in Pseudomonas aeruginosa. Mol. Microbiol. 2002, 43, 475–485. [Google Scholar] [CrossRef]
- Otero-Asman, J.R.; Quesada, J.M.; Jim, K.K.; Ocampo-Sosa, A.; Civantos, C.; Bitter, W.; Llamas, M.A. The Extracytoplasmic Function Sigma Factor ΣVreI Is Active during Infection and Contributes to Phosphate Starvation-Induced Virulence of Pseudomonas aeruginosa. Sci. Rep. 2020, 10, 3139. [Google Scholar] [CrossRef]
- Chekabab, S.M.; Harel, J.; Dozois, C.M. Interplay between Genetic Regulation of Phosphate Homeostasis and Bacterial Virulence. Virulence 2014, 5, 786–793. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Xu, C.; Qu, J.; Jin, Y.; Bai, F.; Cheng, Z.; Wu, W.; Pan, X. PitA Controls the H2- and H3-T6SSs through PhoB in Pseudomonas aeruginosa. Appl. Environ. Microbiol. 2023, 89, e2094-22. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Zhang, X.-F.; An, S.-W.; Xu, J.-L.; Zhang, L.-H. A Novel Two-Component System BqsS-BqsR Modulates Quorum Sensing-Dependent Biofilm Decay in Pseudomonas aeruginosa. Commun. Integr. Biol. 2008, 1, 88–96. [Google Scholar] [CrossRef]
- Schmidberger, A.; Henkel, M.; Hausmann, R.; Schwartz, T. Influence of Ferric Iron on Gene Expression and Rhamnolipid Synthesis during Batch Cultivation of Pseudomonas aeruginosa PAO1. Appl. Microbiol. Biotechnol. 2014, 98, 6725–6737. [Google Scholar] [CrossRef] [PubMed]
- Kreamer, N.N.; Phillips, R.; Newman, D.K.; Boedicker, J.Q. Predicting the Impact of Promoter Variability on Regulatory Outputs. Sci. Rep. 2015, 5, 18238. [Google Scholar] [CrossRef] [PubMed]
- Otero-Asman, J.R.; Wettstadt, S.; Bernal, P.; Llamas, M.A. Diversity of Extracytoplasmic Function Sigma (ΣECF) Factor-Dependent Signaling in Pseudomonas. Mol. Microbiol. 2019, 112, 356–373. [Google Scholar] [CrossRef]
- Bouillet, S.; Ba, M.; Houot, L.; Iobbi-Nivol, C.; Bordi, C. Connected Partner-Switches Control the Life Style of Pseudomonas aeruginosa through RpoS Regulation. Sci. Rep. 2019, 9, 6496. [Google Scholar] [CrossRef]
- Lo, Y.L.; Shen, L.; Chang, C.H.; Bhuwan, M.; Chiu, C.H.; Chang, H.Y. Regulation of Motility and Phenazine Pigment Production by FliA Is Cyclic-Di-Gmp Dependent in Pseudomonas aeruginosa PAO1. PLoS ONE 2016, 11, e155397. [Google Scholar] [CrossRef]
- Ranjani, J.; Sivakumar, R.; Gunasekaran, P.; Velmurugan, G.; Ramasamy, S.; Rajendhran, J. Genome-Wide Identification of Genetic Requirements of Pseudomonas aeruginosa PAO1 for Rat Cardiomyocyte (H9C2) Infection by Insertion Sequencing. Infect. Genet. Evol. 2022, 98, 105231. [Google Scholar] [CrossRef] [PubMed]
- Blanka, A.; Schulz, S.; Eckweiler, D.; Franke, R.; Bielecka, A.; Nicolai, T.; Casilag, F.; Düvel, J.; Abraham, W.R.; Kaever, V.; et al. Identification of the Alternative Sigma Factor SigX Regulon and Its Implications for Pseudomonas aeruginosa Pathogenicity. J. Bacteriol. 2014, 196, 345. [Google Scholar] [CrossRef] [PubMed]
- Chevalier, S.; Bouffartigues, E.; Tortuel, D.; David, A.; Tahrioui, A.; Labbé, C.; Barreau, M.; Tareau, A.S.; Louis, M.; Lesouhaitier, O.; et al. Cell Envelope Stress Response in Pseudomonas aeruginosa. In Pseudomonas aeruginosa: Biology, Pathogenesis and Control Strategies; Springer: Berlin/Heidelberg, Germany, 2022; Volume 1386, ISBN 9783031084911. [Google Scholar]
- Wilderman, P.J.; Sowa, N.A.; FitzGerald, D.J.; FitzGerald, P.C.; Gottesman, S.; Ochsner, U.A.; Vasil, M.L. Identification of Tandem Duplicate Regulatory Small RNAs in Pseudomonas aeruginosa Involved in Iron Homeostasis. Proc. Natl. Acad. Sci. USA 2004, 101, 9792–9797. [Google Scholar] [CrossRef] [PubMed]
- Duchesne, R.; Bouffartigues, E.; Oxaran, V.; Maillot, O.; Bénard, M.; Feuilloley, M.G.J.; Orange, N.; Chevalier, S. A Proteomic Approach of SigX Function in Pseudomonas aeruginosa Outer Membrane Composition. J. Proteomics 2013, 94, 451–459. [Google Scholar] [CrossRef]
- Chevalier, S.; Bouffartigues, E.; Bazire, A.; Tahrioui, A.; Duchesne, R.; Tortuel, D.; Maillot, O.; Clamens, T.; Orange, N.; Feuilloley, M.G.J.; et al. Extracytoplasmic Function Sigma Factors in Pseudomonas aeruginosa. Biochim. Biophys. Acta—Gene Regul. Mech. 2019, 1862, 706–721. [Google Scholar] [CrossRef]
- Anthony, M.; Rose, B.; Pegler, M.B.; Elkins, M.; Service, H.; Thamotharampillai, K.; Watson, J.; Robinson, M.; Bye, P.; Merlino, J.; et al. Genetic Analysis of Pseudomonas aeruginosa Isolates from the Sputa of Australian Adult Cystic Fibrosis Patients. J. Clin. Microbiol. 2002, 40, 2772–2778. [Google Scholar] [CrossRef]
- Qaisar, U.; Kruczek, C.J.; Azeem, M.; Javaid, N.; Colmer-Hamood, J.A.; Hamood, A.N. The Pseudomonas aeruginosa Extracellular Secondary Metabolite, Paerucumarin, Chelates Iron and Is Not Localized to Extracellular Membrane Vesicles. J. Microbiol. 2016, 54, 573–581. [Google Scholar] [CrossRef]
- Wood, L.F.; Ohman, D.E. Use of Cell Wall Stress to Characterize Σ22 (AlgT/U) Activation by Regulated Proteolysis and Its Regulon in Pseudomonas aeruginosa. Mol. Microbiol. 2009, 72, 183–201. [Google Scholar] [CrossRef]
- Jones, A.K.; Fulcher, N.B.; Balzer, G.J.; Urbanowski, M.L.; Pritchett, C.L.; Schurr, M.J.; Yahr, T.L.; Wolfgang, M.C. Activation of the Pseudomonas aeruginosa AlgU Regulon through MucA Mutation Inhibits Cyclic AMP/Vfr Signaling. J. Bacteriol. 2010, 192, 5709–5717. [Google Scholar] [CrossRef]
- James, G.A.; Ge Zhao, A.; Usui, M.; Underwood, R.A.; Nguyen, H.; Beyenal, H.; Delancey Pulcini, E.; Agostinho Hunt, A.; Bernstein, H.C.; Fleckman, P.; et al. Microsensor and Transcriptomic Signatures of Oxygen Depletion in Biofilms Associated with Chronic Wounds. Wound Repair Regen. 2016, 24, 373–383. [Google Scholar] [CrossRef]
- Schuster, M.; Hawkins, A.C.; Harwood, C.S.; Greenberg, E.P. The Pseudomonas aeruginosa RpoS Regulon and Its Relationship to Quorum Sensing. Mol. Microbiol. 2004, 51, 973–985. [Google Scholar] [CrossRef] [PubMed]
- Duan, X.; Pan, Y.; Cai, Z.; Liu, Y.; Zhang, Y.; Liu, M.; Liu, Y.; Wang, K.; Zhang, L.; Yang, L. RpoS-Mutation Variants are Selected in Pseudomonas aeruginosa Biofilms under Imipenem Pressure. Cell Biosci. 2021, 11, 138. [Google Scholar] [CrossRef] [PubMed]
- Dong, T.; Schellhorn, H.E. Role of RpoS in Virulence of Pathogens. Infect. Immun. 2010, 78, 887–897. [Google Scholar] [CrossRef] [PubMed]
- Wood, L.F.; Leech, A.J.; Ohman, D.E. Cell Wall-Inhibitory Antibiotics Activate the Alginate Biosynthesis Operon in Pseudomonas aeruginosa: Roles of σ22 (AlgT) and the AlgW and Prc Proteases. Mol. Microbiol. 2006, 62, 412–426. [Google Scholar] [CrossRef] [PubMed]
- Llamas, M.A.; Van Der Sar, A.; Chu, B.C.H.; Sparrius, M.; Vogel, H.J.; Bitter, W. A Novel Extracytoplasmic Function (ECF) Sigma Factor Regulates Virulence in Pseudomonas aeruginosa. PLoS Pathog. 2009, 5, e1000572. [Google Scholar] [CrossRef]
- Ball, G.; Viarre, V.; Garvis, S.; Voulhoux, R.; Filloux, A. Type II-Dependent Secretion of a Pseudomonas aeruginosa DING Protein. Res. Microbiol. 2012, 163, 457–469. [Google Scholar] [CrossRef]
- Kang, D.; Kirienko, N.V. High-Throughput Genetic Screen Reveals That Early Attachment and Biofilm Formation are Necessary for Full Pyoverdine Production by Pseudomonas aeruginosa. Front. Microbiol. 2017, 8, e01707. [Google Scholar] [CrossRef]
- Palma, M.; DeLuca, D.; Worgall, S.; Quadri, L.E.N. Transcriptome Analysis of the Response of Pseudomonas aeruginosa to Hydrogen Peroxide. J. Bacteriol. 2004, 186, 248–252. [Google Scholar] [CrossRef]
- Lamont, I.L.; Beare, P.A.; Ochsner, U.; Vasil, A.I.; Vasil, M.L. Siderophore-Mediated Signaling Regulates Virulence Factor Production in Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA 2002, 99, 7072–7077. [Google Scholar] [CrossRef]
- Imperi, F.; Massai, F.; Facchini, M.; Frangipani, E.; Visaggio, D.; Leoni, L.; Bragonzi, A.; Visca, P. Repurposing the Antimycotic Drug Flucytosine for Suppression of Pseudomonas aeruginosa Pathogenicity. Proc. Natl. Acad. Sci. USA 2013, 110, 7458–7463. [Google Scholar] [CrossRef]
- Llamas, M.A.; Imperi, F.; Visca, P.; Lamont, I.L. Cell-Surface Signaling in Pseudomonas: Stress Responses, Iron Transport, and Pathogenicity. FEMS Microbiol. Rev. 2014, 38, 569–597. [Google Scholar] [CrossRef] [PubMed]
- Damron, F.H.; Oglesby-Sherrouse, A.G.; Wilks, A.; Barbier, M. Dual-Seq Transcriptomics Reveals the Battle for Iron during Pseudomonas aeruginosa Acute Murine Pneumonia. Sci. Rep. 2016, 6, 39172. [Google Scholar] [CrossRef] [PubMed]
- Turner, K.H.; Everett, J.; Trivedi, U.; Rumbaugh, K.P.; Whiteley, M. Requirements for Pseudomonas aeruginosa Acute Burn and Chronic Surgical Wound Infection. PLoS Genet. 2014, 10, e1004518. [Google Scholar] [CrossRef] [PubMed]
- Reig, S.; Le Gouellec, A.; Bleves, S. What Is New in the Anti–Pseudomonas aeruginosa Clinical Development Pipeline Since the 2017 WHO Alert? Front. Cell. Infect. Microbiol. 2022, 12, 862. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sánchez-Jiménez, A.; Llamas, M.A.; Marcos-Torres, F.J. Transcriptional Regulators Controlling Virulence in Pseudomonas aeruginosa. Int. J. Mol. Sci. 2023, 24, 11895. https://doi.org/10.3390/ijms241511895
Sánchez-Jiménez A, Llamas MA, Marcos-Torres FJ. Transcriptional Regulators Controlling Virulence in Pseudomonas aeruginosa. International Journal of Molecular Sciences. 2023; 24(15):11895. https://doi.org/10.3390/ijms241511895
Chicago/Turabian StyleSánchez-Jiménez, Ana, María A. Llamas, and Francisco Javier Marcos-Torres. 2023. "Transcriptional Regulators Controlling Virulence in Pseudomonas aeruginosa" International Journal of Molecular Sciences 24, no. 15: 11895. https://doi.org/10.3390/ijms241511895
APA StyleSánchez-Jiménez, A., Llamas, M. A., & Marcos-Torres, F. J. (2023). Transcriptional Regulators Controlling Virulence in Pseudomonas aeruginosa. International Journal of Molecular Sciences, 24(15), 11895. https://doi.org/10.3390/ijms241511895