
Potential Roles for the GluN2D NMDA Receptor Subunit in Schizophrenia


 , and
, and 
Abstract
1. Schizophrenia—An Overview
2. Glutamatergic Signalling in the Central Nervous System
NMDA Receptor Structure and Function
3. NMDA Receptor Hypothesis of Schizophrenia
4. GluN2D Subunit
4.1. GluN2D Receptor Subunit Expression and Distribution
4.2. GluN2D Receptor Subunit Function
5. Alterations to GluN2D in Schizophrenia
6. Consequences of Loss of GluN2D Function
6.1. Genetic Models
6.2. Pharmacological Manipulations
7. How Might Alterations to the GluN2D Subunit Contribute to Schizophrenia?
7.1. GluN2D Subunit and Parvalbumin-Positive GABAergic Interneurons
7.2. GluN2D Subunit and Dopaminergic Neurons
8. Limitations
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Saha, S.; Chant, D.; Welham, J.; McGrath, J. A systematic review of the prevalence of schizophrenia. PLoS Med. 2005, 2, e141. [Google Scholar] [CrossRef] [PubMed]
- McCutcheon, R.A.; Reis Marques, T.; Howes, O.D. Schizophrenia-An Overview. JAMA Psychiatry 2020, 77, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Kahn, R.; Sommer, I.; Murray, R.; Meyer-Lindenberg, A.; Weinberger, D.; Cannon, T.; O’Donovan, M.; Correl, C.; Kane, J.; van Os, J. Schizophrenia. Nature Reviews Disease Primers. Nov 2015, 12, 15067. [Google Scholar]
- Galderisi, S.; Mucci, A.; Buchanan, R.W.; Arango, C. Negative symptoms of schizophrenia: New developments and unanswered research questions. Lancet Psychiatry 2018, 5, 664–677. [Google Scholar] [CrossRef]
- McCutcheon, R.A.; Keefe, R.S.; McGuire, P.K. Cognitive impairment in schizophrenia: Aetiology, pathophysiology, and treatment. Mol. Psychiatry, 2023; online ahead of print. [Google Scholar] [CrossRef]
- Vigo, D.; Thornicroft, G.; Atun, R. Estimating the true global burden of mental illness. Lancet Psychiatry 2016, 3, 171–178. [Google Scholar] [CrossRef]
- Velligan, D.I.; Brain, C.; Bouérat Duvold, L.; Agid, O. Caregiver burdens associated with treatment-resistant schizophrenia: A quantitative caregiver survey of experiences, attitudes, and perceptions. Front. Psychiatry 2019, 10, 584. [Google Scholar] [CrossRef]
- Cloutier, M.; Aigbogun, M.S.; Guerin, A.; Nitulescu, R.; Ramanakumar, A.V.; Kamat, S.A.; DeLucia, M.; Duffy, R.; Legacy, S.N.; Henderson, C.; et al. The Economic Burden of Schizophrenia in the United States in 2013. J. Clin. Psychiatry 2016, 77, 764–771. [Google Scholar] [CrossRef]
- Neil, A.L.; Carr, V.J.; Mihalopoulos, C.; Mackinnon, A.; Morgan, V.A. Costs of psychosis in 2010: Findings from the second Australian National Survey of Psychosis. Aust. N. Zealand J. Psychiatry 2014, 48, 169–182. [Google Scholar] [CrossRef]
- Ripke, S.; Neale, B.M.; Corvin, A.; Walters, J.T.; Farh, K.-H.; Holmans, P.A.; Lee, P.; Bulik-Sullivan, B.; Collier, D.A.; Huang, H. Biological insights from 108 schizophrenia-associated genetic loci. Nature 2014, 511, 421–427. [Google Scholar]
- Trubetskoy, V.; Pardiñas, A.F.; Qi, T.; Panagiotaropoulou, G.; Awasthi, S.; Bigdeli, T.B.; Bryois, J.; Chen, C.-Y.; Dennison, C.A.; Hall, L.S. Mapping genomic loci implicates genes and synaptic biology in schizophrenia. Nature 2022, 604, 502–508. [Google Scholar] [CrossRef]
- Walsh, T.; McClellan, J.M.; McCarthy, S.E.; Addington, A.M.; Pierce, S.B.; Cooper, G.M.; Nord, A.S.; Kusenda, M.; Malhotra, D.; Bhandari, A.; et al. Rare structural variants disrupt multiple genes in neurodevelopmental pathways in schizophrenia. Science 2008, 320, 539–543. [Google Scholar] [CrossRef]
- Fromer, M.; Roussos, P.; Sieberts, S.K.; Johnson, J.S.; Kavanagh, D.H.; Perumal, T.M.; Ruderfer, D.M.; Oh, E.C.; Topol, A.; Shah, H.R.; et al. Gene expression elucidates functional impact of polygenic risk for schizophrenia. Nat. Neurosci. 2016, 19, 1442–1453. [Google Scholar] [CrossRef]
- Ursini, G.; Punzi, G.; Chen, Q.; Marenco, S.; Robinson, J.F.; Porcelli, A.; Hamilton, E.G.; Mitjans, M.; Maddalena, G.; Begemann, M.; et al. Convergence of placenta biology and genetic risk for schizophrenia. Nat. Med. 2018, 24, 792–801. [Google Scholar] [CrossRef]
- Malaspina, D.; Harlap, S.; Fennig, S.; Heiman, D.; Nahon, D.; Feldman, D.; Susser, E.S. Advancing paternal age and the risk of schizophrenia. Arch. Gen. Psychiatry 2001, 58, 361–367. [Google Scholar] [CrossRef]
- Fountoulakis, K.N.; Gonda, X.; Siamouli, M.; Panagiotidis, P.; Moutou, K.; Nimatoudis, I.; Kasper, S. Paternal and maternal age as risk factors for schizophrenia: A case–control study. Int. J. Psychiatry Clin. Pract. 2018, 22, 170–176. [Google Scholar] [CrossRef] [PubMed]
- Cantor-Graae, E.; Selten, J.-P. Schizophrenia and migration: A meta-analysis and review. Am. J. Psychiatry 2005, 162, 12–24. [Google Scholar] [CrossRef] [PubMed]
- Varese, F.; Smeets, F.; Drukker, M.; Lieverse, R.; Lataster, T.; Viechtbauer, W.; Read, J.; Van Os, J.; Bentall, R.P. Childhood adversities increase the risk of psychosis: A meta-analysis of patient-control, prospective-and cross-sectional cohort studies. Schizophr. Bull. 2012, 38, 661–671. [Google Scholar] [CrossRef] [PubMed]
- Toulopoulou, T.; Picchioni, M.; Mortensen, P.B.; Petersen, L. IQ, the urban environment, and their impact on future schizophrenia risk in men. Schizophr. Bull. 2017, 43, 1056–1063. [Google Scholar] [CrossRef]
- McKenzie, K.; Murray, A.; Booth, T. Do urban environments increase the risk of anxiety, depression and psychosis? An epidemiological study. J. Affect. Disord. 2013, 150, 1019–1024. [Google Scholar] [CrossRef]
- Vaucher, J.; Keating, B.J.; Lasserre, A.M.; Gan, W.; Lyall, D.M.; Ward, J.; Smith, D.J.; Pell, J.P.; Sattar, N.; Paré, G. Cannabis use and risk of schizophrenia: A Mendelian randomization study. Mol. Psychiatry 2018, 23, 1287–1292. [Google Scholar] [CrossRef]
- Leucht, S.; Corves, C.; Arbter, D.; Engel, R.R.; Li, C.; Davis, J.M. Second-generation versus first-generation antipsychotic drugs for schizophrenia: A meta-analysis. Lancet 2009, 373, 31–41. [Google Scholar] [CrossRef]
- Kane, J.M.; Correll, C.U. Pharmacologic treatment of schizophrenia. Dialogues Clin. Neurosci. 2022, 12, 345–357. [Google Scholar] [CrossRef]
- Keefe, R.S.; Bilder, R.M.; Davis, S.M.; Harvey, P.D.; Palmer, B.W.; Gold, J.M.; Meltzer, H.Y.; Green, M.F.; Capuano, G.; Stroup, T.S. Neurocognitive effects of antipsychotic medications in patients with chronic schizophrenia in the CATIE Trial. Arch. Gen. Psychiatry 2007, 64, 633–647. [Google Scholar] [CrossRef] [PubMed]
- Gründer, G.; Cumming, P. The dopamine hypothesis of schizophrenia: Current status. In The Neurobiology of Schizophrenia; Elsevier: Amsterdam, The Netherlands, 2016; pp. 109–124. [Google Scholar]
- Brisch, R.; Saniotis, A.; Wolf, R.; Bielau, H.; Bernstein, H.-G.; Steiner, J.; Bogerts, B.; Braun, K.; Jankowski, Z.; Kumaratilake, J. The role of dopamine in schizophrenia from a neurobiological and evolutionary perspective: Old fashioned, but still in vogue. Front. Psychiatry 2014, 5, 47. [Google Scholar]
- Millard, S.J.; Bearden, C.E.; Karlsgodt, K.H.; Sharpe, M.J. The prediction-error hypothesis of schizophrenia: New data point to circuit-specific changes in dopamine activity. Neuropsychopharmacology 2022, 47, 628–640. [Google Scholar] [CrossRef]
- McCutcheon, R.; Beck, K.; Jauhar, S.; Howes, O.D. Defining the locus of dopaminergic dysfunction in schizophrenia: A meta-analysis and test of the mesolimbic hypothesis. Schizophr. Bull. 2018, 44, 1301–1311. [Google Scholar] [CrossRef] [PubMed]
- Seeman, P.; Lee, T.; Chau-Wong, M.; Wong, K. Antipsychotic drug doses and neuroleptic/dopamine receptors. Nature 1976, 261, 717–719. [Google Scholar] [CrossRef] [PubMed]
- Van Rossum, J. The significance of dopamine-receptor blockade for the mechanism of action of neuroleptic drugs. Arch. Int. Pharm. 1996, 160, 492–494. [Google Scholar]
- Seeman, M.V. History of the dopamine hypothesis of antipsychotic action. World J. Psychiatry 2021, 11, 355. [Google Scholar] [CrossRef]
- Li, P.; L Snyder, G.; E Vanover, K. Dopamine targeting drugs for the treatment of schizophrenia: Past, present and future. Curr. Top. Med. Chem. 2016, 16, 3385–3403. [Google Scholar] [CrossRef]
- Balu, D.T. The NMDA receptor and schizophrenia: From pathophysiology to treatment. In Advances in Pharmacology; Elsevier: Amsterdam, The Netherlands, 2016; Volume 76, pp. 351–382. [Google Scholar]
- Catts, V.S.; Lai, Y.L.; Weickert, C.S.; Weickert, T.W.; Catts, S.V. A quantitative review of the postmortem evidence for decreased cortical N-methyl-D-aspartate receptor expression levels in schizophrenia: How can we link molecular abnormalities to mismatch negativity deficits? Biol. Psychol. 2016, 116, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Nakazawa, K.; Sapkota, K. The origin of NMDA receptor hypofunction in schizophrenia. Pharmacol. Ther. 2020, 205, 107426. [Google Scholar] [CrossRef] [PubMed]
- Snyder, M.A.; Gao, W.-J. NMDA hypofunction as a convergence point for progression and symptoms of schizophrenia. Front. Cell. Neurosci. 2013, 7, 31. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Danbolt, N.C. Glutamate as a neurotransmitter in the healthy brain. J. Neural Transm. 2014, 121, 799–817. [Google Scholar] [CrossRef]
- Willard, S.S.; Koochekpour, S. Glutamate, glutamate receptors, and downstream signaling pathways. Int. J. Biol. Sci. 2013, 9, 948. [Google Scholar] [CrossRef]
- Traynelis, S.F.; Wollmuth, L.P.; McBain, C.J.; Menniti, F.S.; Vance, K.M.; Ogden, K.K.; Hansen, K.B.; Yuan, H.; Myers, S.J.; Dingledine, R. Glutamate receptor ion channels: Structure, regulation, and function. Pharm. Rev. 2010, 62, 405–496. [Google Scholar] [CrossRef]
- Hansen, K.B.; Wollmuth, L.P.; Bowie, D.; Furukawa, H.; Menniti, F.S.; Sobolevsky, A.I.; Swanson, G.T.; Swanger, S.A.; Greger, I.H.; Nakagawa, T. Structure, function, and pharmacology of glutamate receptor ion channels. Pharmacol. Rev. 2021, 73, 1469–1658. [Google Scholar] [CrossRef]
- Reiner, A.; Levitz, J. Glutamatergic signaling in the central nervous system: Ionotropic and metabotropic receptors in concert. Neuron 2018, 98, 1080–1098. [Google Scholar] [CrossRef]
- Yonezawa, K.; Tani, H.; Nakajima, S.; Nagai, N.; Koizumi, T.; Miyazaki, T.; Mimura, M.; Takahashi, T.; Uchida, H. AMPA receptors in schizophrenia: A systematic review of postmortem studies on receptor subunit expression and binding. Schizophr. Res. 2022, 243, 98–109. [Google Scholar] [CrossRef]
- Rubio, M.D.; Drummond, J.B.; Meador-Woodruff, J.H. Glutamate receptor abnormalities in schizophrenia: Implications for innovative treatments. Biomol. Ther. 2012, 20, 1. [Google Scholar] [CrossRef]
- Maksymetz, J.; Moran, S.P.; Conn, P.J. Targeting metabotropic glutamate receptors for novel treatments of schizophrenia. Mol. Brain 2017, 10, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Monyer, H.; Sprengel, R.; Schoepfer, R.; Herb, A.; Higuchi, M.; Lomeli, H.; Burnashev, N.; Sakmann, B.; Seeburg, P.H. Heteromeric NMDA receptors: Molecular and functional distinction of subtypes. Science 1992, 256, 1217–1221. [Google Scholar] [CrossRef] [PubMed]
- Bye, C.M.; McDonald, R.J. A specific role of hippocampal NMDA receptors and arc protein in rapid encoding of novel environmental representations and a more general long-term consolidation function. Front. Behav. Neurosci. 2019, 13, 8. [Google Scholar] [CrossRef]
- Talpalar, A.E.; Kiehn, O. Glutamatergic mechanisms for speed control and network operation in the rodent locomotor CpG. Front. Neural Circuits 2010, 4, 19. [Google Scholar] [CrossRef] [PubMed]
- Lambot, L.; Rodriguez, E.C.; Houtteman, D.; Li, Y.; Schiffmann, S.N.; Gall, D.; de Kerchove d’Exaerde, A. Striatopallidal neuron NMDA receptors control synaptic connectivity, locomotor, and goal-directed behaviors. J. Neurosci. 2016, 36, 4976–4992. [Google Scholar] [CrossRef]
- Li, N.; Lee, B.; Liu, R.-J.; Banasr, M.; Dwyer, J.M.; Iwata, M.; Li, X.-Y.; Aghajanian, G.; Duman, R.S. mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science 2010, 329, 959–964. [Google Scholar] [CrossRef]
- Hou, G.; Zhang, Z.-W. NMDA receptors regulate the development of neuronal intrinsic excitability through cell-autonomous mechanisms. Front. Cell. Neurosci. 2017, 11, 353. [Google Scholar] [CrossRef]
- Frangeul, L.; Kehayas, V.; Sanchez-Mut, J.V.; Fièvre, S.; Krishna, K.K.; Pouchelon, G.; Telley, L.; Bellone, C.; Holtmaat, A.; Gräff, J. Input-dependent regulation of excitability controls dendritic maturation in somatosensory thalamocortical neurons. Nat. Commun. 2017, 8, 2015. [Google Scholar] [CrossRef]
- Karakas, E.; Furukawa, H. Crystal structure of a heterotetrameric NMDA receptor ion channel. Science 2014, 344, 992–997. [Google Scholar] [CrossRef]
- Furukawa, H.; Singh, S.K.; Mancusso, R.; Gouaux, E. Subunit arrangement and function in NMDA receptors. Nature 2005, 438, 185–192. [Google Scholar] [CrossRef]
- Lerma, J.; Zukin, R.S.; Bennett, M. Glycine decreases desensitization of N-methyl-D-aspartate (NMDA) receptors expressed in Xenopus oocytes and is required for NMDA responses. Proc. Natl. Acad. Sci. USA 1990, 87, 2354–2358. [Google Scholar] [CrossRef] [PubMed]
- Greer, P.L.; Greenberg, M.E. From synapse to nucleus: Calcium-dependent gene transcription in the control of synapse development and function. Neuron 2008, 59, 846–860. [Google Scholar] [CrossRef] [PubMed]
- Sugihara, H.; Moriyoshi, K.; Ishii, T.; Masu, M.; Nakanishi, S. Structures and properties of seven isoforms of the NMDA receptor generated by alternative splicing. Biochem. Biophys. Res. Commun. 1992, 185, 826–832. [Google Scholar] [CrossRef] [PubMed]
- Paoletti, P.; Bellone, C.; Zhou, Q. NMDA receptor subunit diversity: Impact on receptor properties, synaptic plasticity and disease. Nat. Rev. Neurosci. 2013, 14, 383–400. [Google Scholar] [CrossRef] [PubMed]
- Schorge, S.; Colquhoun, D. Studies of NMDA receptor function and stoichiometry with truncated and tandem subunits. J. Neurosci. 2003, 23, 1151–1158. [Google Scholar] [CrossRef]
- Henson, M.A.; Roberts, A.C.; Salimi, K.; Vadlamudi, S.; Hamer, R.M.; Gilmore, J.H.; Jarskog, L.F.; Philpot, B.D. Developmental regulation of the NMDA receptor subunits, NR3A and NR1, in human prefrontal cortex. Cereb. Cortex 2008, 18, 2560–2573. [Google Scholar] [CrossRef]
- Tolle, T.; Berthele, A.; Zieglgansberger, W.; Seeburg, P.H.; Wisden, W. The differential expression of 16 NMDA and non-NMDA receptor subunits in the rat spinal cord and in periaqueductal gray. J. Neurosci. 1993, 13, 5009–5028. [Google Scholar] [CrossRef]
- Monyer, H.; Burnashev, N.; Laurie, D.J.; Sakmann, B.; Seeburg, P.H. Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron 1994, 12, 529–540. [Google Scholar] [CrossRef]
- Vicini, S.; Wang, J.F.; Li, J.H.; Zhu, W.J.; Wang, Y.H.; Luo, J.H.; Wolfe, B.B.; Grayson, D.R. Functional and pharmacological differences between recombinant N-methyl-D-aspartate receptors. J. Neurophysiol. 1998, 79, 555–566. [Google Scholar] [CrossRef]
- Krystal, J.H.; Karper, L.P.; Seibyl, J.P.; Freeman, G.K.; Delaney, R.; Bremner, J.D.; Heninger, G.R.; Bowers, M.B., Jr.; Charney, D.S. Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans. Psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch. Gen. Psychiatry 1994, 51, 199–214. [Google Scholar] [CrossRef]
- Javitt, D. Negative schizophrenic symptomatology and the PCP (phencyclidine) model of schizophrenia. Hillside J. Clin. Psychiatry 1987, 9, 12–35. [Google Scholar]
- Luby, E.D.; Cohen, B.D.; Rosenbaum, G.; Gottlieb, J.S.; Kelley, R. Study of a new schizophrenomimetic drug—Sernyl. AMA Arch. Neurol. Psychiatry 1959, 81, 363–369. [Google Scholar] [CrossRef]
- Luby, E.D.; Gottlieb, J.S.; Cohen, B.D.; Rosenbaum, G.; Domino, E.F. Model psychoses and schizophrenia. Am. J. Psychiatry 1962, 119, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Rosenbaum, G.; Cohen, B.D.; Luby, E.D.; Gottlieb, J.S.; Yelen, D. Comparison of sernyl with other drugs: Simulation of schizophrenic performance with sernyl, LSD-25, and amobarbital (amytal) sodium; I. Attention, motor function, and proprioception. AMA Arch. Gen. Psychiatry 1959, 1, 651–656. [Google Scholar] [CrossRef] [PubMed]
- Cohen, B.D.; Luby, E.D.; Rosenbaum, G.; Gottlieb, J.S. Combined sernyl and sensory deprivation. Compr. Psychiatry 1960, 1, 345–348. [Google Scholar] [CrossRef]
- Itil, T.; Keskiner, A.; Kiremitci, N.; Holden, J. Effect of phencyclidine in chronic schizophrenics. Can. Psychiatr. Assoc. J. 1967, 12, 209–212. [Google Scholar] [CrossRef]
- Lahti, A.C.; Koffel, B.; LaPorte, D.; Tamminga, C.A. Subanesthetic doses of ketamine stimulate psychosis in schizophrenia. Neuropsychopharmacology 1995, 13, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Anis, N.A.; Berry, S.C.; Burton, N.R.; Lodge, D. The dissociative anaesthetics, ketamine and phencyclidine, selectively reduce excitation of central mammalian neurones by N-methyl-aspartate. Br. J. Pharm. 1983, 79, 565–575. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kornhuber, H.; Schmid-Burgk, W.; Holzmüller, B. Low cerebrospinal fluid glutamate in schizophrenic patients and a new hypothesis on schizophrenia. Neurosci. Lett. 1980, 20, 379–382. [Google Scholar] [CrossRef]
- Javitt, D.C.; Zukin, S.R. Recent advances in the phencyclidine model of schizophrenia. Am. J. Psychiatry 1991, 148, 1301–1308. [Google Scholar]
- Javitt, D.C. Glutamatergic theories of schizophrenia. Isr. J. Psychiatry Relat. Sci. 2010, 47, 4–16. [Google Scholar]
- Kantrowitz, J.T.; Javitt, D.C. N-methyl-d-aspartate (NMDA) receptor dysfunction or dysregulation: The final common pathway on the road to schizophrenia? Brain Res. Bull. 2010, 83, 108–121. [Google Scholar] [CrossRef] [PubMed]
- Kantrowitz, J.; Javitt, D.C. Glutamatergic transmission in schizophrenia: From basic research to clinical practice. Curr. Opin. Psychiatry 2012, 25, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Mohn, A.R.; Gainetdinov, R.R.; Caron, M.G.; Koller, B.H. Mice with reduced NMDA receptor expression display behaviors related to schizophrenia. Cell 1999, 98, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Halene, T.B.; Ehrlichman, R.S.; Liang, Y.; Christian, E.P.; Jonak, G.J.; Gur, T.L.; Blendy, J.A.; Dow, H.C.; Brodkin, E.S.; Schneider, F. Assessment of NMDA receptor NR1 subunit hypofunction in mice as a model for schizophrenia. Genes Brain Behav. 2009, 8, 661–675. [Google Scholar] [CrossRef] [PubMed]
- Hakami, T.; Jones, N.C.; Tolmacheva, E.A.; Gaudias, J.; Chaumont, J.; Salzberg, M.; O’Brien, T.J.; Pinault, D. NMDA receptor hypofunction leads to generalized and persistent aberrant γ oscillations independent of hyperlocomotion and the state of consciousness. PLoS ONE 2009, 4, e6755. [Google Scholar] [CrossRef] [PubMed]
- Anderson, P.M.; Pinault, D.; O’Brien, T.J.; Jones, N.C. Chronic administration of antipsychotics attenuates ongoing and ketamine-induced increases in cortical γ oscillations. Int. J. Neuropsychopharmacol. 2014, 17, 1895–1904. [Google Scholar] [CrossRef]
- Hudson, M.; Rind, G.; O’brien, T.; Jones, N. Reversal of evoked gamma oscillation deficits is predictive of antipsychotic activity with a unique profile for clozapine. Transl. Psychiatry 2016, 6, e784. [Google Scholar] [CrossRef]
- Belforte, J.E.; Zsiros, V.; Sklar, E.R.; Jiang, Z.; Yu, G.; Li, Y.; Quinlan, E.M.; Nakazawa, K. Postnatal NMDA receptor ablation in corticolimbic interneurons confers schizophrenia-like phenotypes. Nat. Neurosci. 2010, 13, 76–83. [Google Scholar] [CrossRef]
- Cohen, S.M.; Tsien, R.W.; Goff, D.C.; Halassa, M.M. The impact of NMDA receptor hypofunction on GABAergic neurons in the pathophysiology of schizophrenia. Schizophr. Res. 2015, 167, 98–107. [Google Scholar] [CrossRef]
- Nakao, K.; Jeevakumar, V.; Jiang, S.Z.; Fujita, Y.; Diaz, N.B.; Pretell Annan, C.A.; Eskow Jaunarajs, K.L.; Hashimoto, K.; Belforte, J.E.; Nakazawa, K. Schizophrenia-like dopamine release abnormalities in a mouse model of NMDA receptor hypofunction. Schizophr. Bull. 2019, 45, 138–147. [Google Scholar] [CrossRef] [PubMed]
- Akgül, G.; McBain, C.J. Diverse roles for ionotropic glutamate receptors on inhibitory interneurons in developing and adult brain. J. Physiol. 2016, 594, 5471–5490. [Google Scholar] [CrossRef] [PubMed]
- Homayoun, H.; Moghaddam, B. NMDA receptor hypofunction produces opposite effects on prefrontal cortex interneurons and pyramidal neurons. J. Neurosci. 2007, 27, 11496–11500. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Clark, S.; Lewis, D.V.; Wilson, W.A. NMDA receptor antagonists disinhibit rat posterior cingulate and retrosplenial cortices: A potential mechanism of neurotoxicity. J. Neurosci. 2002, 22, 3070–3080. [Google Scholar] [CrossRef]
- Suzuki, Y.; Jodo, E.; Takeuchi, S.; Niwa, S.; Kayama, Y. Acute administration of phencyclidine induces tonic activation of medial prefrontal cortex neurons in freely moving rats. Neuroscience 2002, 114, 769–779. [Google Scholar] [CrossRef]
- Jackson, M.E.; Homayoun, H.; Moghaddam, B. NMDA receptor hypofunction produces concomitant firing rate potentiation and burst activity reduction in the prefrontal cortex. Proc. Natl. Acad. Sci. USA 2004, 101, 8467–8472. [Google Scholar] [CrossRef]
- Moghaddam, B.; Adams, B.; Verma, A.; Daly, D. Activation of glutamatergic neurotransmission by ketamine: A novel step in the pathway from NMDA receptor blockade to dopaminergic and cognitive disruptions associated with the prefrontal cortex. J. Neurosci. 1997, 17, 2921–2927. [Google Scholar] [CrossRef]
- Okada, M.; Fukuyama, K.; Ueda, Y. Lurasidone inhibits NMDA receptor antagonist-induced functional abnormality of thalamocortical glutamatergic transmission via 5-HT7 receptor blockade. Br. J. Pharmacol. 2019, 176, 4002–4018. [Google Scholar] [CrossRef]
- Widman, A.J.; McMahon, L.L. Disinhibition of CA1 pyramidal cells by low-dose ketamine and other antagonists with rapid antidepressant efficacy. Proc. Natl. Acad. Sci. USA 2018, 115, E3007–E3016. [Google Scholar] [CrossRef]
- Schobel, S.A.; Chaudhury, N.H.; Khan, U.A.; Paniagua, B.; Styner, M.A.; Asllani, I.; Inbar, B.P.; Corcoran, C.M.; Lieberman, J.A.; Moore, H. Imaging patients with psychosis and a mouse model establishes a spreading pattern of hippocampal dysfunction and implicates glutamate as a driver. Neuron 2013, 78, 81–93. [Google Scholar] [CrossRef]
- Murray, J.D.; Anticevic, A.; Gancsos, M.; Ichinose, M.; Corlett, P.R.; Krystal, J.H.; Wang, X.-J. Linking microcircuit dysfunction to cognitive impairment: Effects of disinhibition associated with schizophrenia in a cortical working memory model. Cereb. Cortex. 2014, 24, 859–872. [Google Scholar] [CrossRef]
- Shaw, A.D.; Saxena, N.; Jackson, L.E.; Hall, J.E.; Singh, K.D.; Muthukumaraswamy, S.D. Ketamine amplifies induced gamma frequency oscillations in the human cerebral cortex. Eur. Neuropsychopharmacol. 2015, 25, 1136–1146. [Google Scholar] [CrossRef] [PubMed]
- Vollenweider, F.; Leenders, K.; Scharfetter, C.; Antonini, A.; Maguire, P.; Missimer, J.; Angst, J. Metabolic hyperfrontality and psychopathology in the ketamine model of psychosis using positron emission tomography (PET) and [18F] fluorodeoxyglucose (FDG). Eur. Neuropsychopharmacol. 1997, 7, 9–24. [Google Scholar] [CrossRef]
- Breier, A.; Malhotra, A.K.; Pinals, D.A.; Weisenfeld, N.I.; Pickar, D. Association of ketamine-induced psychosis with focal activation of the prefrontal cortex in healthy volunteers. Am. J. Psychiatry 1997, 154, 805–811. [Google Scholar] [PubMed]
- Miyamoto, S.; Leipzig, J.N.; Lieberman, J.A.; Duncan, G.E. Effects of ketamine, MK-801, and amphetamine on regional brain 2-deoxyglucose uptake in freely moving mice. Neuropsychopharmacology 2000, 22, 400–412. [Google Scholar] [CrossRef] [PubMed]
- Duncan, G.E.; Moy, S.S.; Knapp, D.J.; Mueller, R.A.; Breese, G.R. Metabolic mapping of the rat brain after subanesthetic doses of ketamine: Potential relevance to schizophrenia. Brain Res. 1998, 787, 181–190. [Google Scholar] [CrossRef]
- Nair, V.D.; Savelli, J.E.; Mishra, R.K. Modulation of dopamine D 2 receptor expression by an NMDA receptor antagonist in rat brain. J. Mol. Neurosci. 1998, 11, 121–126. [Google Scholar] [CrossRef]
- Micheletti, G.; Lannes, B.; Haby, C.; Borrelli, E.; Kempf, E.; Warter, J.-M.; Zwiller, J. Chronic administration of NMDA antagonists induces D2 receptor synthesis in rat striatum. Mol. Brain Res. 1992, 14, 363–368. [Google Scholar] [CrossRef]
- Kegeles, L.S.; Abi-Dargham, A.; Zea-Ponce, Y.; Rodenhiser-Hill, J.; Mann, J.J.; Van Heertum, R.L.; Cooper, T.B.; Carlsson, A.; Laruelle, M. Modulation of amphetamine-induced striatal dopamine release by ketamine in humans: Implications for schizophrenia. Biol. Psychiatry 2000, 48, 627–640. [Google Scholar] [CrossRef]
- Smith, G.S.; Schloesser, R.; Brodie, J.D.; Dewey, S.L.; Logan, J.; Vitkun, S.A.; Simkowitz, P.; Hurley, A.; Cooper, T.; Volkow, N.D. Glutamate modulation of dopamine measured in vivo with positron emission tomography (PET) and 11C-raclopride in normal human subjects. Neuropsychopharmacology 1998, 18, 18–25. [Google Scholar] [CrossRef]
- Sun, J.; Jia, P.; Fanous, A.H.; Van Den Oord, E.; Chen, X.; Riley, B.P.; Amdur, R.L.; Kendler, K.S.; Zhao, Z. Schizophrenia gene networks and pathways and their applications for novel candidate gene selection. PLoS ONE 2010, 5, e11351. [Google Scholar] [CrossRef] [PubMed]
- Greenwood, T.A.; Light, G.A.; Swerdlow, N.R.; Radant, A.D.; Braff, D.L. Association analysis of 94 candidate genes and schizophrenia-related endophenotypes. PLoS ONE 2012, 7, e29630. [Google Scholar] [CrossRef] [PubMed]
- Balu, D.T.; Coyle, J.T. Neuroplasticity signaling pathways linked to the pathophysiology of schizophrenia. Neurosci. Biobehav. Rev. 2011, 35, 848–870. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, K.; Fukushima, T.; Shimizu, E.; Komatsu, N.; Watanabe, H.; Shinoda, N.; Nakazato, M.; Kumakiri, C.; Okada, S.; Hasegawa, H.; et al. Decreased serum levels of D-serine in patients with schizophrenia: Evidence in support of the N-methyl-D-aspartate receptor hypofunction hypothesis of schizophrenia. Arch. Gen. Psychiatry 2003, 60, 572–576. [Google Scholar] [CrossRef]
- Weickert, C.S.; Fung, S.J.; Catts, V.S.; Schofield, P.R.; Allen, K.M.; Moore, L.T.; Newell, K.A.; Pellen, D.; Huang, X.F.; Catts, S.V.; et al. Molecular evidence of N-methyl-D-aspartate receptor hypofunction in schizophrenia. Mol. Psychiatry 2013, 18, 1185–1192. [Google Scholar] [CrossRef]
- Labrie, V.; Lipina, T.; Roder, J.C. Mice with reduced NMDA receptor glycine affinity model some of the negative and cognitive symptoms of schizophrenia. Psychopharmacology 2008, 200, 217–230. [Google Scholar] [CrossRef]
- Zhao, X.; Li, H.; Shi, Y.; Tang, R.; Chen, W.; Liu, J.; Feng, G.; Shi, J.; Yan, L.; Liu, H. Significant association between the genetic variations in the 5′ end of the N-methyl-D-aspartate receptor subunit gene GRIN1 and schizophrenia. Biol. Psychiatry 2006, 59, 747–753. [Google Scholar] [CrossRef]
- Poltavskaya, E.G.; Fedorenko, O.Y.; Kornetova, E.G.; Loonen, A.J.; Kornetov, A.N.; Bokhan, N.A.; Ivanova, S.A. Study of early onset schizophrenia: Associations of GRIN2A and GRIN2B polymorphisms. Life 2021, 11, 997. [Google Scholar] [CrossRef]
- Catts, V.S.; Derminio, D.S.; Hahn, C.-G.; Weickert, C.S. Postsynaptic density levels of the NMDA receptor NR1 subunit and PSD-95 protein in prefrontal cortex from people with schizophrenia. Npj Schizophrenia 2015, 1, 1–8. [Google Scholar] [CrossRef]
- Ishii, T.; Moriyoshi, K.; Sugihara, H.; Sakurada, K.; Kadotani, H.; Yokoi, M.; Akazawa, C.; Shigemoto, R.; Mizuno, N.; Masu, M. Molecular characterization of the family of the N-methyl-D-aspartate receptor subunits. J. Biol. Chem. 1993, 268, 2836–2843. [Google Scholar] [CrossRef] [PubMed]
- Akazawa, C.; Shigemoto, R.; Bessho, Y.; Nakanishi, S.; Mizuno, N. Differential expression of five N-methyl-D-aspartate receptor subunit mRNAs in the cerebellum of developing and adult rats. J. Comp. Neurol. 1994, 347, 150–160. [Google Scholar] [CrossRef]
- Wenzel, A.; Villa, M.; Mohler, H.; Benke, D. Developmental and regional expression of NMDA receptor subtypes containing the NR2D subunit in rat brain. J. Neurochem. 1996, 66, 1240–1248. [Google Scholar] [CrossRef] [PubMed]
- Dunah, A.W.; Yasuda, R.P.; Wang, Y.h.; Luo, J.; Dávila-García, M.I.; Gbadegesin, M.; Vicini, S.; Wolfe, B.B. Regional and ontogenic expression of the NMDA receptor subunit NR2D protein in rat brain using a subunit-specific antibody. J. Neurochem. 1996, 67, 2335–2345. [Google Scholar] [CrossRef] [PubMed]
- Standaert, D.G.; Landwehrmeyer, G.B.; Kerner, J.A.; Penney Jr, J.B.; Young, A.B. Expression of NMDAR2D glutamate receptor subunit mRNA in neurochemically identified interneurons in the rat neostriatum, neocortex and hippocampus. Mol. Brain Res. 1996, 42, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Cannon, M.; Murray, R.M. Neonatal origins of schizophrenia. Arch. Dis. Child. 1998, 78, 1–3. [Google Scholar] [CrossRef]
- Birnbaum, R.; Weinberger, D.R. Genetic insights into the neurodevelopmental origins of schizophrenia. Nat. Rev. Neurosci. 2017, 18, 727–740. [Google Scholar] [CrossRef] [PubMed]
- Engelhardt, J.v.; Bocklisch, C.; Tönges, L.; Herb, A.; Mishina, M.; Monyer, H. GluN2D-containing NMDA receptors-mediate synaptic currents in hippocampal interneurons and pyramidal cells in juvenile mice. Front. Cell Neurosci. 2015, 9, 95. [Google Scholar] [CrossRef]
- Garst-Orozco, J.; Malik, R.; Lanz, T.A.; Weber, M.L.; Xi, H.; Arion, D.; Enwright III, J.F.; Lewis, D.A.; O’Donnell, P.; Sohal, V.S. GluN2D-mediated excitatory drive onto medial prefrontal cortical PV+ fast-spiking inhibitory interneurons. PLoS ONE 2020, 15, e0233895. [Google Scholar] [CrossRef]
- Perszyk, R.E.; DiRaddo, J.O.; Strong, K.L.; Low, C.M.; Ogden, K.K.; Khatri, A.; Vargish, G.A.; Pelkey, K.A.; Tricoire, L.; Liotta, D.C.; et al. GluN2D-Containing N-methyl-d-Aspartate Receptors Mediate Synaptic Transmission in Hippocampal Interneurons and Regulate Interneuron Activity. Mol. Pharm. 2016, 90, 689–702. [Google Scholar] [CrossRef]
- Alsaad, H.A.; DeKorver, N.W.; Mao, Z.; Dravid, S.M.; Arikkath, J.; Monaghan, D.T. In the telencephalon, GluN2C NMDA receptor subunit mRNA is predominately expressed in glial cells and GluN2D mRNA in interneurons. Neurochem. Res. 2019, 44, 61–77. [Google Scholar] [CrossRef]
- Camp, C.R.; Yuan, H. GRIN2D/GluN2D NMDA receptor: Unique features and its contribution to pediatric developmental and epileptic encephalopathy. Eur. J. Paediatr. Neurol. 2020, 24, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, M.; Okada, R.; Takasaki, C.; Toki, S.; Fukaya, M.; Natsume, R.; Sakimura, K.; Mishina, M.; Shirakawa, T.; Watanabe, M. Opposing role of NMDA receptor GluN2B and GluN2D in somatosensory development and maturation. J. Neurosci. 2014, 34, 11534–11548. [Google Scholar] [CrossRef] [PubMed]
- Ritter, L.M.; Unis, A.S.; Meador-Woodruff, J.H. Ontogeny of ionotropic glutamate receptor expression in human fetal brain. Dev. Brain Res. 2001, 127, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Scherzer, C.R.; Landwehrmeyer, G.B.; Kerner, J.A.; Counihan, T.J.; Kosinski, C.M.; Standaert, D.G.; Daggett, L.P.; Velicelebi, G.; Penney, J.B.; Young, A.B. Expression of N-methyl-D-aspartate receptor subunit mRNAs in the human brain: Hippocampus and cortex. J. Comp. Neurol. 1998, 390, 75–90. [Google Scholar] [CrossRef]
- Akbarian, S.; Sucher, N.J.; Bradley, D.; Tafazzoli, A.; Trinh, D.; Hetrick, W.P.; Potkin, S.G.; Sandman, C.A.; Bunney, W.E., Jr.; Jones, E.G. Selective alterations in gene expression for NMDA receptor subunits in prefrontal cortex of schizophrenics. J. Neurosci. 1996, 16, 19–30. [Google Scholar] [CrossRef]
- Dunah, A.W.; Luo, J.; Wang, Y.-H.; Yasuda, R.P.; Wolfe, B.B. Subunit Composition ofN-Methyl-D-aspartate Receptors in the Central Nervous System that Contain the NR2D Subunit. Mol. Pharmacol. 1998, 53, 429–437. [Google Scholar] [CrossRef]
- Brickley, S.G.; Misra, C.; Mok, M.S.; Mishina, M.; Cull-Candy, S.G. NR2B and NR2D subunits coassemble in cerebellar Golgi cells to form a distinct NMDA receptor subtype restricted to extrasynaptic sites. J. Neurosci. 2003, 23, 4958–4966. [Google Scholar] [CrossRef]
- Jones, S.; Gibb, A.J. Functional NR2B-and NR2D-containing NMDA receptor channels in rat substantia nigra dopaminergic neurones. J. Physiol. 2005, 569, 209–221. [Google Scholar] [CrossRef]
- Erreger, K.; Geballe, M.T.; Kristensen, A.; Chen, P.E.; Hansen, K.B.; Lee, C.J.; Yuan, H.; Le, P.; Lyuboslavsky, P.N.; Micale, N. Subunit-specific agonist activity at NR2A-, NR2B-, NR2C-, and NR2D-containing N-methyl-D-aspartate glutamate receptors. Mol. Pharmacol. 2007, 72, 907–920. [Google Scholar] [CrossRef]
- Chen, P.E.; Geballe, M.T.; Katz, E.; Erreger, K.; Livesey, M.R.; O’toole, K.K.; Le, P.; Lee, C.J.; Snyder, J.P.; Traynelis, S.F. Modulation of glycine potency in rat recombinant NMDA receptors containing chimeric NR2A/2D subunits expressed in Xenopus laevis oocytes. J. Physiol. 2008, 586, 227–245. [Google Scholar] [CrossRef] [PubMed]
- Kuner, T.; Schoepfer, R. Multiple structural elements determine subunit specificity of Mg2+ block in NMDA receptor channels. J. Neurosci. 1996, 16, 3549–3558. [Google Scholar] [CrossRef] [PubMed]
- Clarke, R.J.; Johnson, J.W. NMDA receptor NR2 subunit dependence of the slow component of magnesium unblock. J Neurosci 2006, 26, 5825–5834. [Google Scholar] [CrossRef]
- Kotermanski, S.E.; Johnson, J.W. Mg2+ imparts NMDA receptor subtype selectivity to the Alzheimer’s drug memantine. J. Neurosci. 2009, 29, 2774–2779. [Google Scholar] [CrossRef]
- Sapkota, K.; Mao, Z.; Synowicki, P.; Lieber, D.; Liu, M.; Ikezu, T.; Gautam, V.; Monaghan, D.T. GluN2D N-Methyl-d-Aspartate Receptor Subunit Contribution to the Stimulation of Brain Activity and Gamma Oscillations by Ketamine: Implications for Schizophrenia. J. Pharm. Exp. 2016, 356, 702–711. [Google Scholar] [CrossRef] [PubMed]
- Wyllie, D.J.; Behe, P.; Colquhoun, D. Single-channel activations and concentration jumps: Comparison of recombinant NR1a/NR2A and NR1a/NR2D NMDA receptors. J. Physiol. 1998, 510 Pt 1, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Krupp, J.J.; Vissel, B.; Heinemann, S.F.; Westbrook, G.L. Calcium-dependent inactivation of recombinant N-methyl-D-aspartate receptors is NR2 subunit specific. Mol. Pharmacol. 1996, 50, 1680–1688. [Google Scholar]
- Cull-Candy, S.; Brickley, S.; Farrant, M. NMDA receptor subunits: Diversity, development and disease. Curr. Opin. Neurobiol. 2001, 11, 327–335. [Google Scholar] [CrossRef]
- Hanson, E.; Armbruster, M.; Lau, L.A.; Sommer, M.E.; Klaft, Z.-J.; Swanger, S.A.; Traynelis, S.F.; Moss, S.J.; Noubary, F.; Chadchankar, J. Tonic activation of GluN2C/GluN2D-containing NMDA receptors by ambient glutamate facilitates cortical interneuron maturation. J. Neurosci. 2019, 39, 3611–3626. [Google Scholar] [CrossRef]
- Swanger, S.A.; Vance, K.M.; Pare, J.-F.; Sotty, F.; Fog, K.; Smith, Y.; Traynelis, S.F. NMDA receptors containing the GluN2D subunit control neuronal function in the subthalamic nucleus. J. Neurosci. 2015, 35, 15971–15983. [Google Scholar] [CrossRef]
- Pearlstein, E.; Gouty-Colomer, L.-A.; Michel, F.J.; Cloarec, R.; Hammond, C. Glutamatergic synaptic currents of nigral dopaminergic neurons follow a postnatal developmental sequence. Front. Cell Neurosci. 2015, 9, 210. [Google Scholar] [CrossRef]
- Andrade-Talavera, Y.; Duque-Feria, P.; Paulsen, O.; Rodríguez-Moreno, A. Presynaptic spike timing-dependent long-term depression in the mouse hippocampus. Cereb. Cortex. 2016, 26, 3637–3654. [Google Scholar] [CrossRef] [PubMed]
- Feldman, D.E. The spike-timing dependence of plasticity. Neuron 2012, 75, 556–571. [Google Scholar] [CrossRef] [PubMed]
- Dubois, C.J.; Lachamp, P.M.; Sun, L.; Mishina, M.; Liu, S.J. Presynaptic GluN2D receptors detect glutamate spillover and regulate cerebellar GABA release. J. Neurophysiol. 2016, 115, 271–285. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.; Inoue, S.; Hiroi, H.; Orimo, A.; Muramatsu, M. NMDA receptor type 2D gene as target for estrogen receptor in the brain. Mol. Brain Res. 1999, 63, 375–379. [Google Scholar] [CrossRef]
- Riecher-Rössler, A.; Häfner, H.; Stumbaum, M.; Maurer, K.; Schmidt, R. Can estradiol modulate schizophrenic symptomatology? Schizophr. Bull. 1994, 20, 203–214. [Google Scholar] [CrossRef]
- McCarthny, C.R.; Du, X.; Wu, Y.C.; Hill, R.A. Investigating the interactive effects of sex steroid hormones and brain-derived neurotrophic factor during adolescence on hippocampal NMDA receptor expression. Int. J. Endocrinol. 2018, 2018, 7231915. [Google Scholar] [CrossRef]
- Makino, C.; Shibata, H.; Ninomiya, H.; Tashiro, N.; Fukumaki, Y. Identification of single-nucleotide polymorphisms in the human N-methyl-D-aspartate receptor subunit NR2D gene, GRIN2D, and association study with schizophrenia. Psychiatr. Genet. 2005, 15, 215–221. [Google Scholar] [CrossRef]
- Yu, Y.; Lin, Y.; Takasaki, Y.; Wang, C.; Kimura, H.; Xing, J.; Ishizuka, K.; Toyama, M.; Kushima, I.; Mori, D. Rare loss of function mutations in N-methyl-D-aspartate glutamate receptors and their contributions to schizophrenia susceptibility. Transl. Psychiatry 2018, 8, 1–9. [Google Scholar] [CrossRef]
- Beneyto, M.; Meador-Woodruff, J.H. Lamina-specific abnormalities of NMDA receptor-associated postsynaptic protein transcripts in the prefrontal cortex in schizophrenia and bipolar disorder. Neuropsychopharmacology 2008, 33, 2175–2186. [Google Scholar] [CrossRef]
- Kristiansen, L.; Beneyto, M.; Haroutunian, V.; Meador-Woodruff, J. Changes in NMDA receptor subunits and interacting PSD proteins in dorsolateral prefrontal and anterior cingulate cortex indicate abnormal regional expression in schizophrenia. Mol. Psychiatry 2006, 11, 737–747. [Google Scholar] [CrossRef]
- Sodhi, M.S.; Simmons, M.; McCullumsmith, R.; Haroutunian, V.; Meador-Woodruff, J.H. Glutamatergic gene expression is specifically reduced in thalamocortical projecting relay neurons in schizophrenia. Biol. Psychiatry 2011, 70, 646–654. [Google Scholar] [CrossRef] [PubMed]
- Andreasen, N.C. A unitary model of schizophrenia: Bleuler’s fragmented phrene as schizencephaly. Arch. Gen. Psychiatry 1999, 56, 781–787. [Google Scholar] [CrossRef]
- Anticevic, A.; Cole, M.W.; Repovs, G.; Murray, J.D.; Brumbaugh, M.S.; Winkler, A.M.; Savic, A.; Krystal, J.H.; Pearlson, G.D.; Glahn, D.C. Characterizing thalamo-cortical disturbances in schizophrenia and bipolar illness. Cereb. Cortex. 2014, 24, 3116–3130. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Ye, E.; Jin, X.; Zhu, Y.; Wang, L. Association between thalamocortical functional connectivity abnormalities and cognitive deficits in schizophrenia. Sci. Rep. 2019, 9, 2952. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, A.; Koschel, J.; Zink, M.; Bauer, M.; Sommer, C.; Frank, J.; Treutlein, J.; Schulze, T.; Schneider-Axmann, T.; Parlapani, E. Gene expression of NMDA receptor subunits in the cerebellum of elderly patients with schizophrenia. Eur. Arch. Psychiatry Clin. Neurosci. 2010, 260, 101–111. [Google Scholar] [CrossRef]
- Ikeda, K.; Araki, K.; Takayama, C.; Inoue, Y.; Yagi, T.; Aizawa, S.; Mishina, M. Reduced spontaneous activity of mice defective in the ε4 subunit of the NMDA receptor channel. Mol. Brain Res. 1995, 33, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, Y.; Yamada, K.; Noda, Y.; Mori, H.; Mishina, M.; Nabeshima, T. Lower sensitivity to stress and altered monoaminergic neuronal function in mice lacking the NMDA receptor ε4 subunit. J. Neurosci. 2002, 22, 2335–2342. [Google Scholar] [CrossRef]
- Obiang, P.; Macrez, R.; Jullienne, A.; Bertrand, T.; Lesept, F.; Ali, C.; Maubert, E.; Vivien, D.; Agin, V. GluN2D subunit-containing NMDA receptors control tissue plasminogen activator-mediated spatial memory. J. Neurosci. 2012, 32, 12726–12734. [Google Scholar] [CrossRef]
- Ide, S.; Ikekubo, Y.; Mishina, M.; Hashimoto, K.; Ikeda, K. Cognitive impairment that is induced by (R)-ketamine is abolished in NMDA GluN2D receptor subunit knockout mice. Int. J. Neuropsychopharmacol. 2019, 22, 449–452. [Google Scholar] [CrossRef]
- Hagino, Y.; Kasai, S.; Han, W.; Yamamoto, H.; Nabeshima, T.; Mishina, M.; Ikeda, K. Essential role of NMDA receptor channel ε4 subunit (GluN2D) in the effects of phencyclidine, but not methamphetamine. PLoS ONE 2010, 5, e13722. [Google Scholar] [CrossRef]
- Takeuchi, T.; Kiyama, Y.; Nakamura, K.; Tsujita, M.; Matsuda, I.; Mori, H.; Munemoto, Y.; Kuriyama, H.; Natsume, R.; Sakimura, K. Roles of the glutamate receptor ε2 and δ2 subunits in the potentiation and prepulse inhibition of the acoustic startle reflex. Eur. J. Neurosci. 2001, 14, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, H.; Kamegaya, E.; Sawada, W.; Hasegawa, R.; Yamamoto, T.; Hagino, Y.; Takamatsu, Y.; Imai, K.; Koga, H.; Mishina, M. Involvement of the N-methyl-D-aspartate receptor GluN2D subunit in phencyclidine-induced motor impairment, gene expression, and increased Fos immunoreactivity. Mol. Brain 2013, 6, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Nakayama, T.; Yamaguchi, J.; Matsuzawa, M.; Mishina, M.; Ikeda, K.; Yamamoto, H. Role of the NMDA receptor GluN2D subunit in the expression of ketamine-induced behavioral sensitization and region-specific activation of neuronal nitric oxide synthase. Neurosci. Lett. 2016, 610, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Duncan, G.E.; Miyamoto, S.; Leipzig, J.N.; Lieberman, J.A. Comparison of brain metabolic activity patterns induced by ketamine, MK-801 and amphetamine in rats: Support for NMDA receptor involvement in responses to subanesthetic dose of ketamine. Brain Res. 1999, 843, 171–183. [Google Scholar] [CrossRef] [PubMed]
- Mao, Z.; He, S.; Mesnard, C.; Synowicki, P.; Zhang, Y.; Chung, L.; Wiesman, A.I.; Wilson, T.W.; Monaghan, D.T. NMDA receptors containing GluN2C and GluN2D subunits have opposing roles in modulating neuronal oscillations; potential mechanism for bidirectional feedback. Brain Res. 2020, 1727, 146571. [Google Scholar] [CrossRef]
- Hrabetova, S.; Serrano, P.; Blace, N.; Heong, W.T.; Skifter, D.A.; Jane, D.E.; Monaghan, D.T.; Sacktor, T.C. Distinct NMDA receptor subpopulations contribute to long-term potentiation and long-term depression induction. J. Neurosci. 2000, 20, RC81. [Google Scholar] [CrossRef]
- Lozovaya, N.A.; Grebenyuk, S.E.; Tsintsadze, T.S.; Feng, B.; Monaghan, D.T.; Krishtal, O.A. Extrasynaptic NR2B and NR2D subunits of NMDA receptors shape ‘superslow’afterburst EPSC in rat hippocampus. J. Physiol. 2004, 558, 451–463. [Google Scholar] [CrossRef]
- Eapen, A.V.; Fernández-Fernández, D.; Georgiou, J.; Bortolotto, Z.A.; Lightman, S.; Jane, D.E.; Volianskis, A.; Collingridge, G.L. Multiple roles of GluN2D-containing NMDA receptors in short-term potentiation and long-term potentiation in mouse hippocampal slices. Neuropharmacology 2021, 201, 108833. [Google Scholar] [CrossRef]
- Ingram, R.; Kang, H.; Lightman, S.; Jane, D.E.; Bortolotto, Z.A.; Collingridge, G.L.; Lodge, D.; Volianskis, A. Some distorted thoughts about ketamine as a psychedelic and a novel hypothesis based on NMDA receptor-mediated synaptic plasticity. Neuropharmacology 2018, 142, 30–40. [Google Scholar] [CrossRef]
- Bygrave, A.M.; Kilonzo, K.; Kullmann, D.M.; Bannerman, D.M.; Kätzel, D. Can N-methyl-D-aspartate receptor hypofunction in schizophrenia be localized to an individual cell type? Front. Psychiatry 2019, 10, 835. [Google Scholar] [CrossRef]
- Bygrave, A.M.; Masiulis, S.; Nicholson, E.; Berkemann, M.; Barkus, C.; Sprengel, R.; Harrison, P.J.; Kullmann, D.M.; Bannerman, D.M.; Kätzel, D. Knockout of NMDA-receptors from parvalbumin interneurons sensitizes to schizophrenia-related deficits induced by MK-801. Transl. Psychiatry 2016, 6, e778. [Google Scholar] [CrossRef]
- Korotkova, T.; Fuchs, E.C.; Ponomarenko, A.; von Engelhardt, J.; Monyer, H. NMDA receptor ablation on parvalbumin-positive interneurons impairs hippocampal synchrony, spatial representations, and working memory. Neuron 2010, 68, 557–569. [Google Scholar] [CrossRef] [PubMed]
- DeFelipe, J.; López-Cruz, P.L.; Benavides-Piccione, R.; Bielza, C.; Larrañaga, P.; Anderson, S.; Burkhalter, A.; Cauli, B.; Fairén, A.; Feldmeyer, D. New insights into the classification and nomenclature of cortical GABAergic interneurons. Nat. Rev. Neurosci. 2013, 14, 202–216. [Google Scholar] [CrossRef]
- Kaar, S.J.; Angelescu, I.; Marques, T.R.; Howes, O.D. Pre-frontal parvalbumin interneurons in schizophrenia: A meta-analysis of post-mortem studies. J. Neural Transm. 2019, 126, 1637–1651. [Google Scholar] [CrossRef] [PubMed]
- Chung, D.W.; Fish, K.N.; Lewis, D.A. Pathological basis for deficient excitatory drive to cortical parvalbumin interneurons in schizophrenia. Am. J. Psychiatry 2016, 173, 1131–1139. [Google Scholar] [CrossRef] [PubMed]
- Nakazawa, K.; Zsiros, V.; Jiang, Z.; Nakao, K.; Kolata, S.; Zhang, S.; Belforte, J.E. GABAergic interneuron origin of schizophrenia pathophysiology. Neuropharmacology 2012, 62, 1574–1583. [Google Scholar] [CrossRef] [PubMed]
- Uhlhaas, P.J.; Singer, W. Abnormal neural oscillations and synchrony in schizophrenia. Nat. Rev. Neurosci. 2010, 11, 100–113. [Google Scholar] [CrossRef]
- Gonzalez-Burgos, G.; Cho, R.Y.; Lewis, D.A. Alterations in cortical network oscillations and parvalbumin neurons in schizophrenia. Biol. Psychiatry 2015, 77, 1031–1040. [Google Scholar] [CrossRef]
- Antonoudiou, P.; Tan, Y.L.; Kontou, G.; Upton, A.L.; Mann, E.O. Parvalbumin and somatostatin interneurons contribute to the generation of hippocampal gamma oscillations. J. Neurosci. 2020, 40, 7668–7687. [Google Scholar] [CrossRef]
- Kriener, B.; Hu, H.; Vervaeke, K. Parvalbumin interneuron dendrites enhance gamma oscillations. Cell Rep. 2022, 39, 110948. [Google Scholar] [CrossRef]
- Buzsaki, G.; Wang, X.J. Mechanisms of gamma oscillations. Annu. Rev. Neurosci. 2012, 35, 203–225. [Google Scholar] [CrossRef] [PubMed]
- Alekseichuk, I.; Turi, Z.; de Lara, G.A.; Antal, A.; Paulus, W. Spatial working memory in humans depends on theta and high gamma synchronization in the prefrontal cortex. Curr. Biol. 2016, 26, 1513–1521. [Google Scholar] [CrossRef] [PubMed]
- Uhlhaas, P.J.; Singer, W. Oscillations and neuronal dynamics in schizophrenia: The search for basic symptoms and translational opportunities. Biol. Psychiatry 2015, 77, 1001–1009. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-M.A.; Stanford, A.D.; Mao, X.; Abi-Dargham, A.; Shungu, D.C.; Lisanby, S.H.; Schroeder, C.E.; Kegeles, L.S. GABA level, gamma oscillation, and working memory performance in schizophrenia. NeuroImage Clin. 2014, 4, 531–539. [Google Scholar] [CrossRef] [PubMed]
- Spencer, K.M.; Niznikiewicz, M.A.; Nestor, P.G.; Shenton, M.E.; McCarley, R.W. Left auditory cortex gamma synchronization and auditory hallucination symptoms in schizophrenia. BMC Neurosci. 2009, 10, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Spencer, K.M. Baseline gamma power during auditory steady-state stimulation in schizophrenia. Front. Hum. Neurosci. 2012, 5, 190. [Google Scholar] [CrossRef] [PubMed]
- Hunt, M.J.; Kasicki, S. A systematic review of the effects of NMDA receptor antagonists on oscillatory activity recorded in vivo. J. Psychopharmacol. 2013, 27, 972–986. [Google Scholar] [CrossRef] [PubMed]
- Kocsis, B.; Brown, R.E.; McCarley, R.W.; Hajos, M. Impact of ketamine on neuronal network dynamics: Translational modeling of schizophrenia-relevant deficits. CNS Neurosci. Ther. 2013, 19, 437–447. [Google Scholar] [CrossRef]
- Hamm, J.P.; Gilmore, C.S.; Clementz, B.A. Augmented gamma band auditory steady-state responses: Support for NMDA hypofunction in schizophrenia. Schizophr. Res. 2012, 138, 1–7. [Google Scholar] [CrossRef]
- Sokolenko, E.; Hudson, M.R.; Nithianantharajah, J.; Jones, N.C. The mGluR2/3 agonist LY379268 reverses NMDA receptor antagonist effects on cortical gamma oscillations and phase coherence, but not working memory impairments, in mice. J. Psychopharmacol. 2019, 33, 1588–1599. [Google Scholar] [CrossRef]
- Lemercier, C.E.; Holman, C.; Gerevich, Z. Aberrant alpha and gamma oscillations ex vivo after single application of the NMDA receptor antagonist MK-801. Schizophr. Res. 2017, 188, 118–124. [Google Scholar] [CrossRef]
- Schuelert, N.; Dorner-Ciossek, C.; Brendel, M.; Rosenbrock, H. A comprehensive analysis of auditory event-related potentials and network oscillations in an NMDA receptor antagonist mouse model using a novel wireless recording technology. Physiol. Rep. 2018, 6, e13782. [Google Scholar] [CrossRef] [PubMed]
- Gawande, D.Y.; Narasimhan, K.K.S.; Shelkar, G.P.; Pavuluri, R.; Stessman, H.A.; Dravid, S.M. GluN2D Subunit in Parvalbumin Interneurons Regulates Prefrontal Cortex Feedforward Inhibitory Circuit and Molecular Networks Relevant to Schizophrenia. Biol. Psychiatry, 2023; In Press. [Google Scholar]
- Suryavanshi, P.S.; Ugale, R.; Yilmazer-Hanke, D.; Stairs, D.J.; Dravid, S. GluN2C/GluN2D subunit-selective NMDA receptor potentiator CIQ reverses MK-801-induced impairment in prepulse inhibition and working memory in Y-maze test in mice. Br. J. Pharmacol. 2014, 171, 799–809. [Google Scholar] [CrossRef] [PubMed]
- de la Fuente-Sandoval, C.; León-Ortiz, P.; Azcárraga, M.; Stephano, S.; Favila, R.; Díaz-Galvis, L.; Alvarado-Alanis, P.; Ramírez-Bermúdez, J.; Graff-Guerrero, A. Glutamate levels in the associative striatum before and after 4 weeks of antipsychotic treatment in first-episode psychosis: A longitudinal proton magnetic resonance spectroscopy study. JAMA Psychiatry 2013, 70, 1057–1066. [Google Scholar] [CrossRef] [PubMed]
- Levitt, J.J.; Nestor, P.G.; Levin, L.; Pelavin, P.; Lin, P.; Kubicki, M.; McCarley, R.W.; Shenton, M.E.; Rathi, Y. Reduced structural connectivity in frontostriatal white matter tracts in the associative loop in schizophrenia. Am. J. Psychiatry 2017, 174, 1102–1111. [Google Scholar] [CrossRef]
- Egerton, A.; Chaddock, C.A.; Winton-Brown, T.T.; Bloomfield, M.A.; Bhattacharyya, S.; Allen, P.; McGuire, P.K.; Howes, O.D. Presynaptic striatal dopamine dysfunction in people at ultra-high risk for psychosis: Findings in a second cohort. Biol. Psychiatry 2013, 74, 106–112. [Google Scholar] [CrossRef]
- Kegeles, L.S.; Abi-Dargham, A.; Frankle, W.G.; Gil, R.; Cooper, T.B.; Slifstein, M.; Hwang, D.-R.; Huang, Y.; Haber, S.N.; Laruelle, M. Increased synaptic dopamine function in associative regions of the striatum in schizophrenia. Arch. Gen. Psychiatry 2010, 67, 231–239. [Google Scholar] [CrossRef]
- McCutcheon, R.A.; Jauhar, S.; Pepper, F.; Nour, M.M.; Rogdaki, M.; Veronese, M.; Turkheimer, F.E.; Egerton, A.; McGuire, P.; Mehta, M.M. The topography of striatal dopamine and symptoms in psychosis: An integrative positron emission tomography and magnetic resonance imaging study. Biol. Psychiatry Cogn. Neurosci. Neuroimaging 2020, 5, 1040–1051. [Google Scholar] [CrossRef]
- Morris, P.G.; Mishina, M.; Jones, S. Altered synaptic and extrasynaptic NMDA receptor properties in substantia nigra dopaminergic neurons from mice lacking the GluN2D subunit. Front. Cell Neurosci. 2018, 12, 354. [Google Scholar] [CrossRef]
- Huang, Z.; Gibb, A.J. Mg2+ block properties of triheteromeric GluN1–GluN2B–GluN2D NMDA receptors on neonatal rat substantia nigra pars compacta dopaminergic neurones. J. Physiol. 2014, 592, 2059–2078. [Google Scholar] [CrossRef]
- Sitzia, G.; Mantas, I.; Zhang, X.; Svenningsson, P.; Chergui, K. NMDA receptors are altered in the substantia nigra pars reticulata and their blockade ameliorates motor deficits in experimental parkinsonism. Neuropharmacology 2020, 174, 108136. [Google Scholar] [CrossRef]
- Byrial, P.; Nyboe, L.; Thomsen, P.H.; Clausen, L. Motor impairments in early onset schizophrenia. Early Interv. Psychiatry 2022, 16, 481–491. [Google Scholar] [CrossRef]
- Nadesalingam, N.; Chapellier, V.; Lefebvre, S.; Pavlidou, A.; Stegmayer, K.; Alexaki, D.; Gama, D.B.; Maderthaner, L.; von Känel, S.; Wüthrich, F. Motor abnormalities are associated with poor social and functional outcomes in schizophrenia. Compr. Psychiatry 2022, 115, 152307. [Google Scholar] [CrossRef] [PubMed]
- Peralta, V.; Campos, M.S.; De Jalón, E.G.; Cuesta, M.J. Motor behavior abnormalities in drug-naïve patients with schizophrenia spectrum disorders. Mov. Disord. 2010, 25, 1068–1076. [Google Scholar] [CrossRef]
- van Hooijdonk, C.F.; van der Pluijm, M.; Bosch, I.; van Amelsvoort, T.A.; Booij, J.; de Haan, L.; Selten, J.-P.; van de Giessen, E. The substantia nigra in the pathology of schizophrenia: A review on post-mortem and molecular imaging findings. Eur. Neuropsychopharmacol. 2023, 68, 57–77. [Google Scholar] [CrossRef]
- Abram, S.V.; Roach, B.J.; Holroyd, C.B.; Paulus, M.P.; Ford, J.M.; Mathalon, D.H.; Fryer, S.L. Reward processing electrophysiology in schizophrenia: Effects of age and illness phase. NeuroImage Clin. 2020, 28, 102492. [Google Scholar] [CrossRef]
- Strauss, G.P.; Waltz, J.A.; Gold, J.M. A review of reward processing and motivational impairment in schizophrenia. Schizophr. Bull. 2014, 40 (Suppl. 2), S107–S116. [Google Scholar] [CrossRef] [PubMed]
- White, D.M.; Kraguljac, N.V.; Reid, M.A.; Lahti, A.C. Contribution of substantia nigra glutamate to prediction error signals in schizophrenia: A combined magnetic resonance spectroscopy/functional imaging study. Npj Schizophrenia 2015, 1, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Salimando, G.J.; Hyun, M.; Boyt, K.M.; Winder, D.G. BNST GluN2D-containing NMDA receptors influence anxiety-and depressive-like behaviors and modulatecell-specific excitatory/inhibitory synaptic balance. J. Neurosci. 2020, 40, 3949–3968. [Google Scholar] [CrossRef]


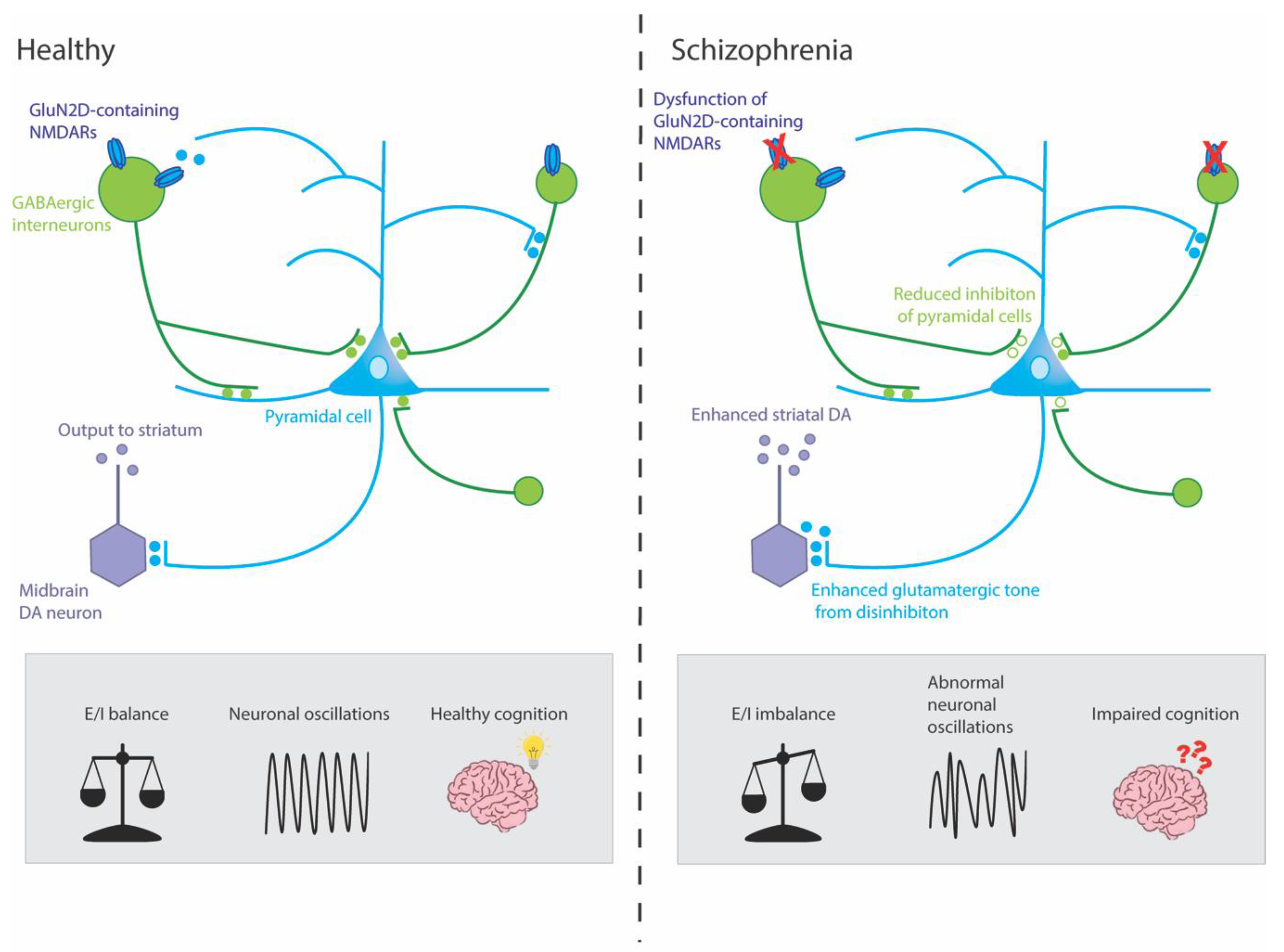{kind=link}
| Reference | Outcome Measure | Brain Region | Direction of Change |
|---|---|---|---|
| [128] | mRNA | Prefrontal cortex and cerebellum | ↑ in PFC ↔ in cerebellum |
| [152] | Receptor binding and mRNA | Dorsolateral prefrontal cortex | ↔ |
| [153] | Protein | Dorsolateral prefrontal cortex and anterior cingulate cortex | ↔ |
| [154] | mRNA | Medial dorsal thalamus | ↓ in glutamatergic relay neurons ↔ in mixed glial-interneuronal cells |
| [158] | Receptor binding and mRNA | Cerebellum | ↑ mRNA in the right cerebellum ↔ mRNA in left cerebellum and receptor binding |
| Reference | Sex | Pharmacological Manipulation and Dose | Behavioural Domain Examined | Behavioural Tests Used | Main Findings |
|---|---|---|---|---|---|
| [159] | Not specified | N/A | Locomotion, anxiety, novelty preference | Open field test, novelty preference test, light–dark compartment test, elevated plus-maze | ↓ spontaneous locomotion, ↓ novelty preference, ↔ change in anxiety |
| [160] | Not specified | N/A | Locomotion, anxiety | Open field test, light–dark compartment test, elevated plus-maze, forced swim test | ↓ spontaneous locomotion, ↓ anxiety |
| [163] | Male | Acute and chronic PCP (3 mg/kg) | Locomotion | Open field test | ↓ PCP-induced hyperlocomotion |
| [161] | Male | N/A | Locomotion, contextual fear memory, spatial memory | Open field test, fear conditioning test, Y-maze | ↓ spontaneous locomotion, ↓ contextual fear memory, ↓ spatial memory |
| [165] | Male and female | Acute PCP (3 mg/kg) | Motor function | Rotarod | ↓ PCP-induced motor impairment |
| [137] | Male | Acute ketamine (30 mg/kg) | Locomotion, spatial memory | Open field test, Morris water maze | ↓ ketamine-induced hyperlocomotion, ↓ spatial memory acquisition |
| [166] | Male | Subchronic ketamine (25 mg/kg) | Locomotion | Open field test | ↓ ketamine-induced hyperlocomotion |
| [162] | Not specified | Acute (RS)-ketamine (10 or 20 mg/kg), (R)-ketamine (10 or 20 mg/kg), (S)-ketamine (10 or 20 mg/kg) | Novel object recognition task | Novel object recognition task | ↓ (R)-ketamine-induced novel object recognition deficits |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vinnakota, C.; Hudson, M.R.; Jones, N.C.; Sundram, S.; Hill, R.A. Potential Roles for the GluN2D NMDA Receptor Subunit in Schizophrenia. Int. J. Mol. Sci. 2023, 24, 11835. https://doi.org/10.3390/ijms241411835
Vinnakota C, Hudson MR, Jones NC, Sundram S, Hill RA. Potential Roles for the GluN2D NMDA Receptor Subunit in Schizophrenia. International Journal of Molecular Sciences. 2023; 24(14):11835. https://doi.org/10.3390/ijms241411835
Chicago/Turabian StyleVinnakota, Chitra, Matthew R. Hudson, Nigel C. Jones, Suresh Sundram, and Rachel A. Hill. 2023. "Potential Roles for the GluN2D NMDA Receptor Subunit in Schizophrenia" International Journal of Molecular Sciences 24, no. 14: 11835. https://doi.org/10.3390/ijms241411835
APA StyleVinnakota, C., Hudson, M. R., Jones, N. C., Sundram, S., & Hill, R. A. (2023). Potential Roles for the GluN2D NMDA Receptor Subunit in Schizophrenia. International Journal of Molecular Sciences, 24(14), 11835. https://doi.org/10.3390/ijms241411835