
Evaluation of Novel Enhancer Compounds in Gentamicin-Mediated Readthrough of Nonsense Mutations in Rett Syndrome

 , and
, and 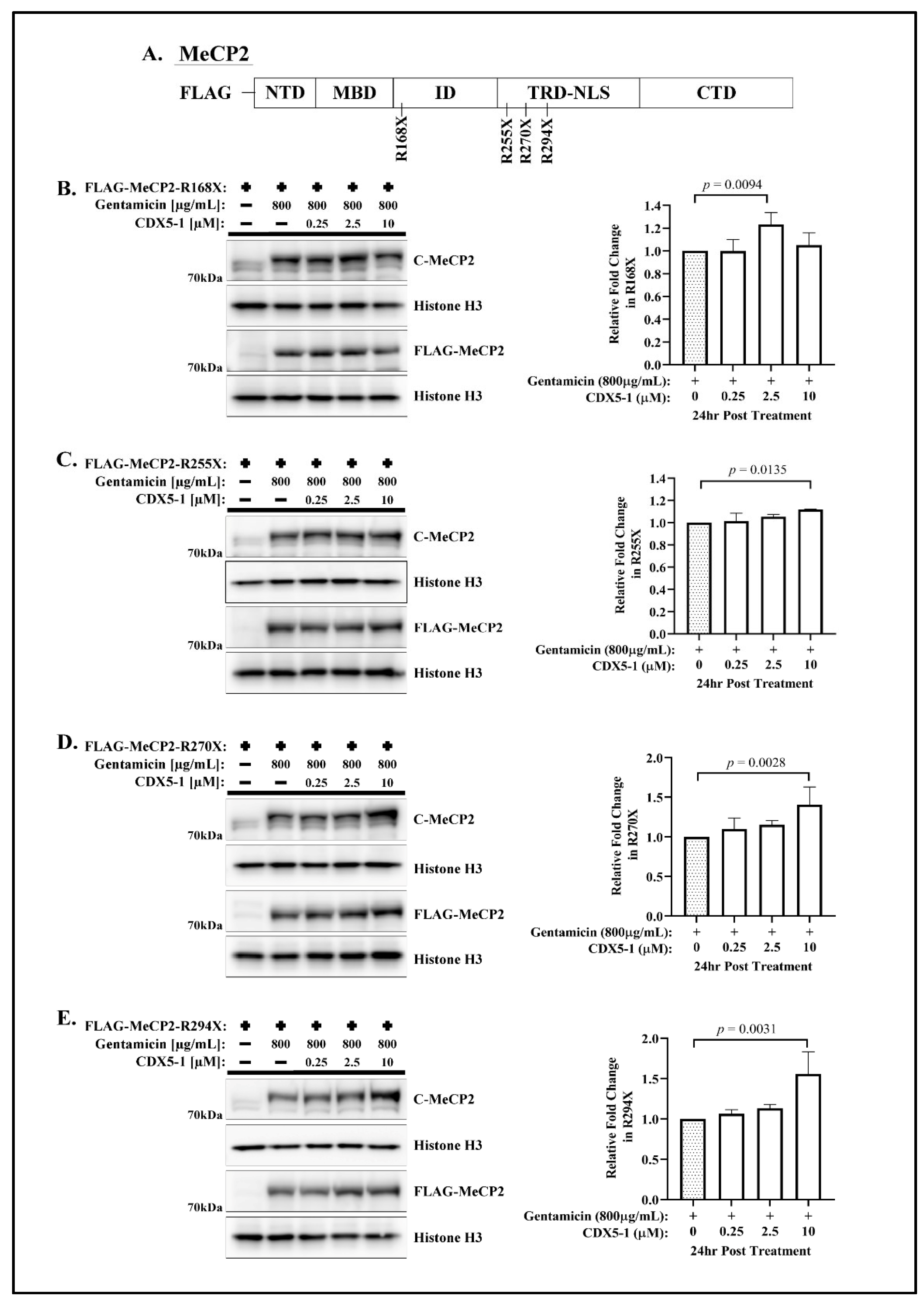{kind=link}
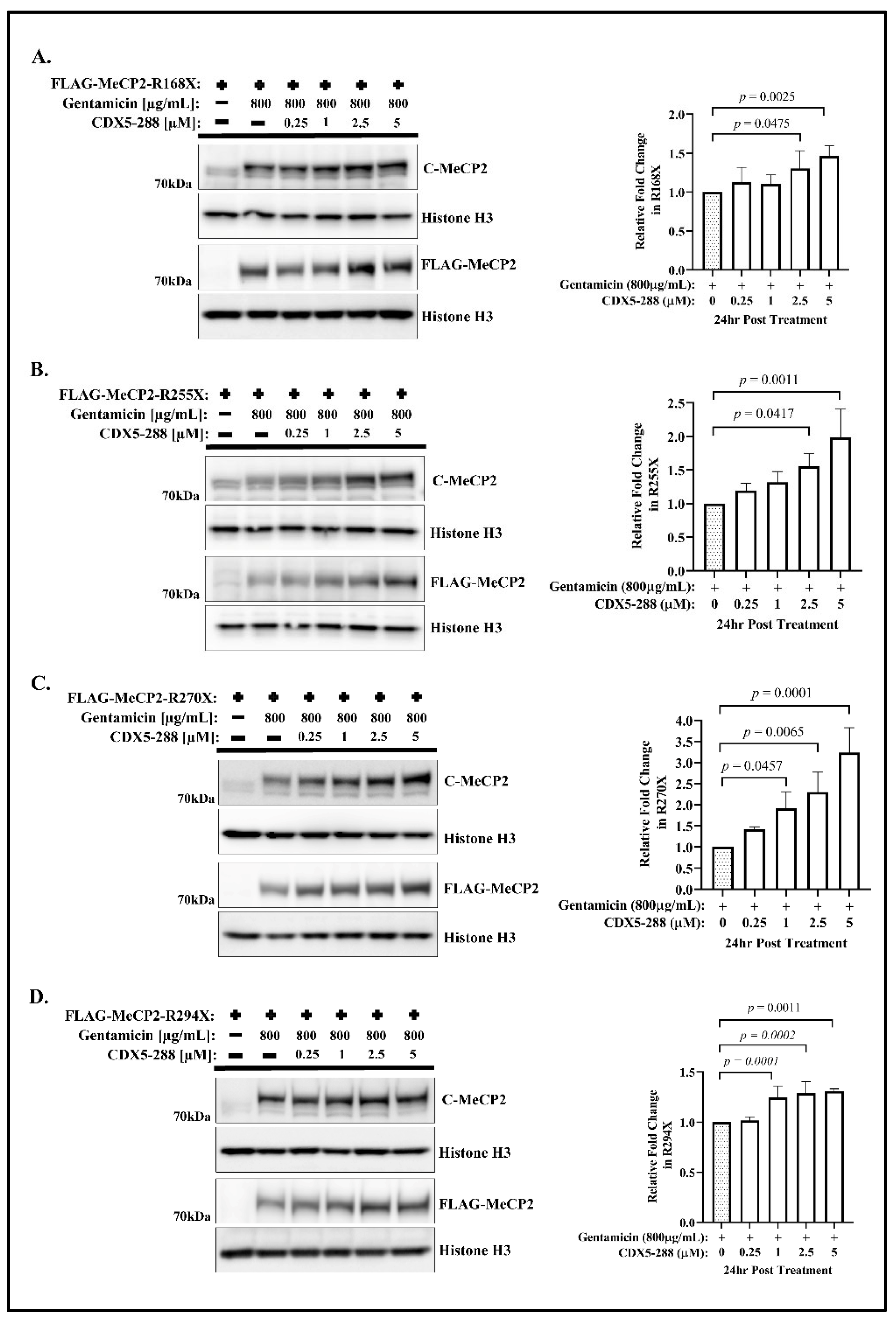{kind=link}
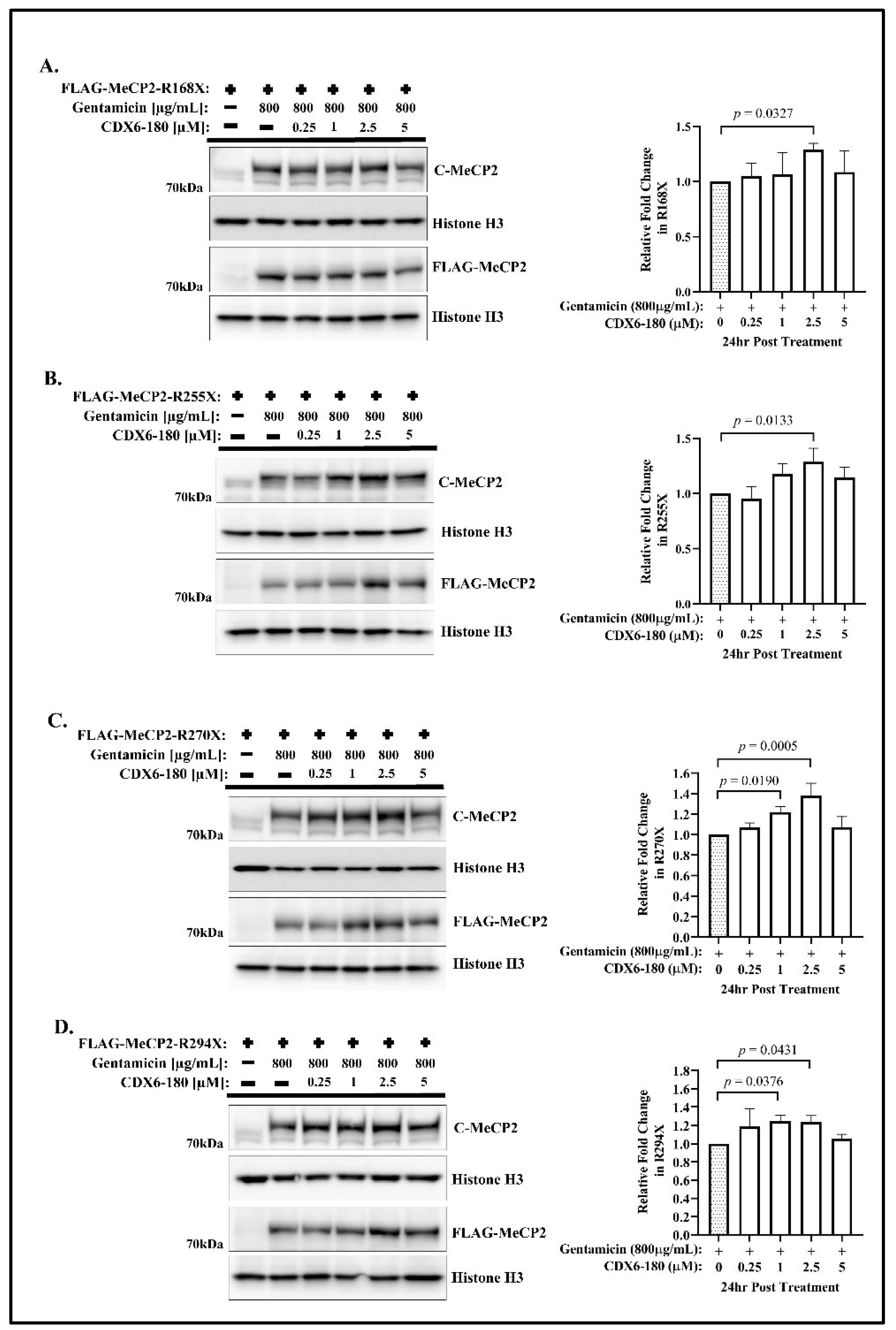{kind=link}
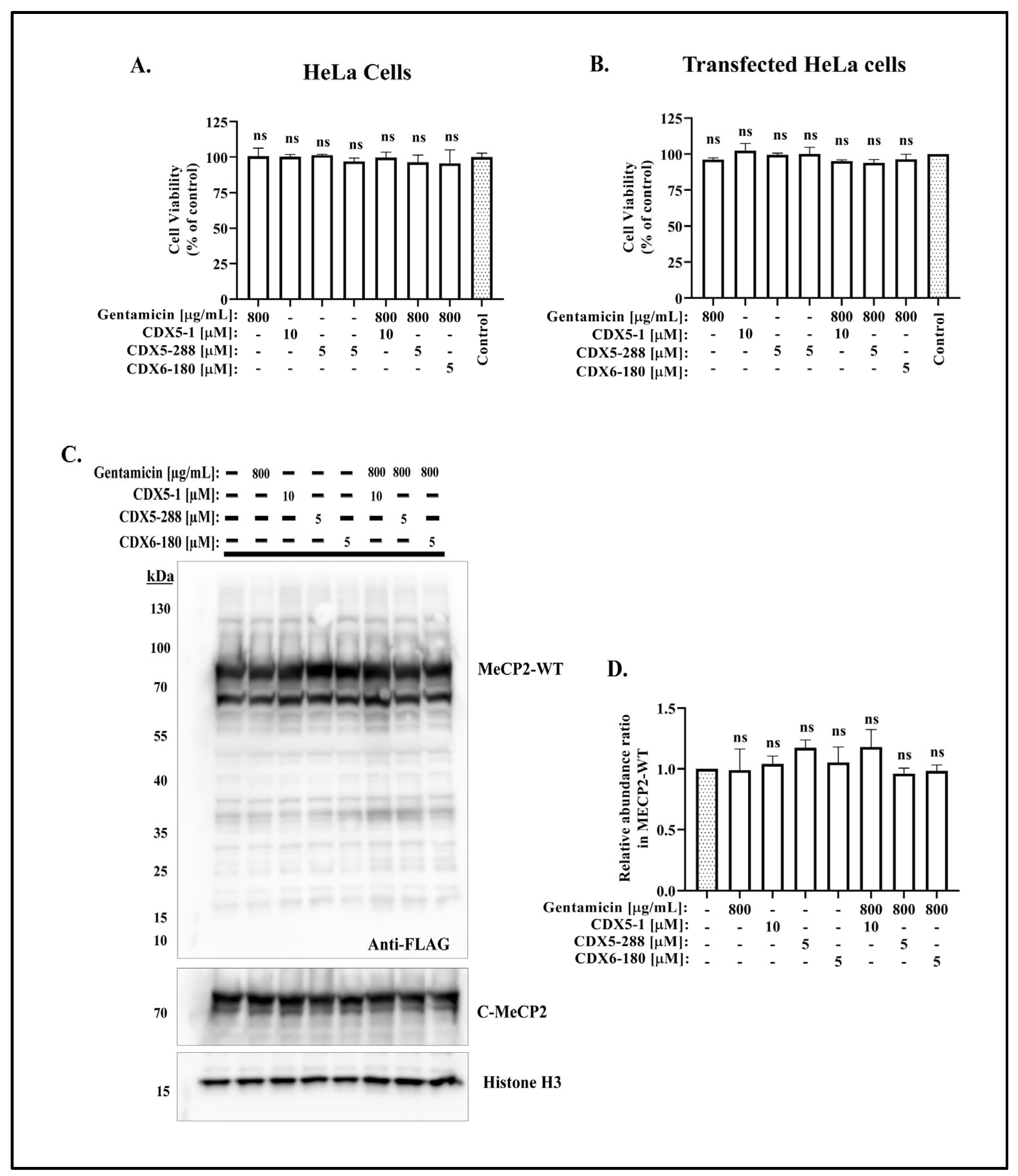{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
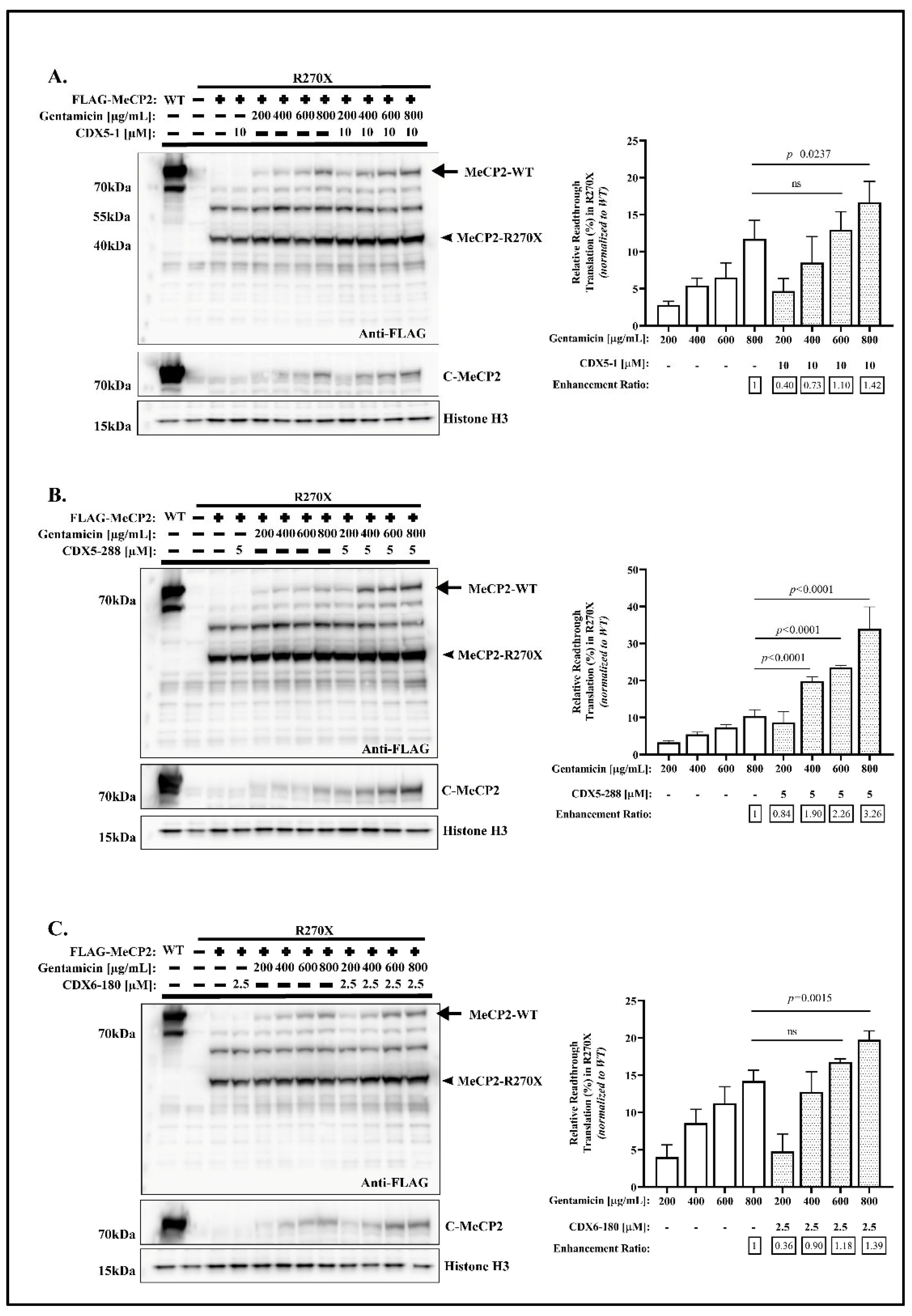
2.1. CDX Compounds Potentiate Gentamicin-Mediated Readthrough in MECP2 Nonsense Mutations
2.2. Co-Treatment of CDX Compounds with Gentamicin Did Not Lead to Cytotoxicity
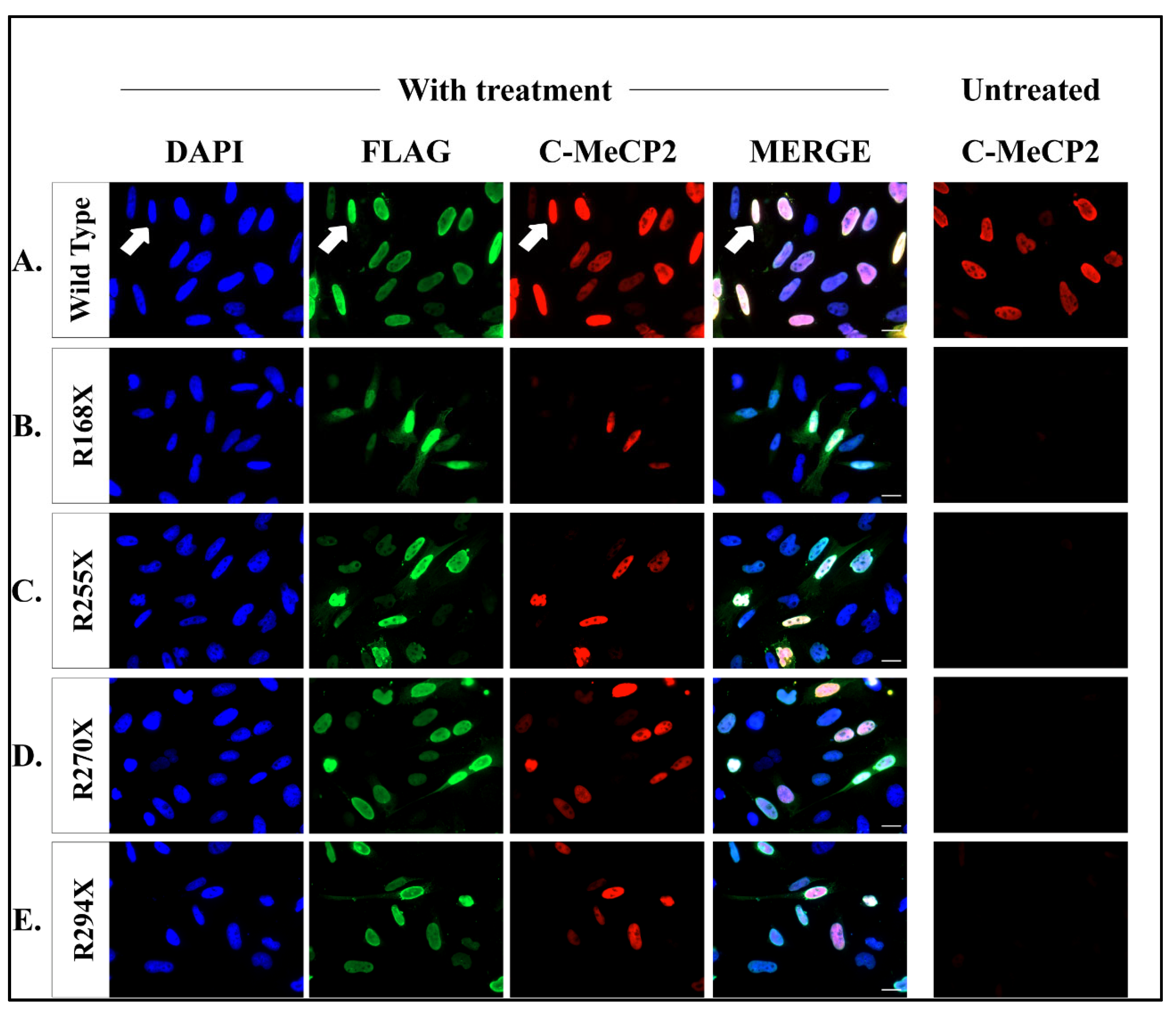
2.3. Readthrough Product Colocalized to Nucleus after Co-Treatment Therapy
2.4. CDX5-288 Allows the Use of Reduced Doses of Gentamicin
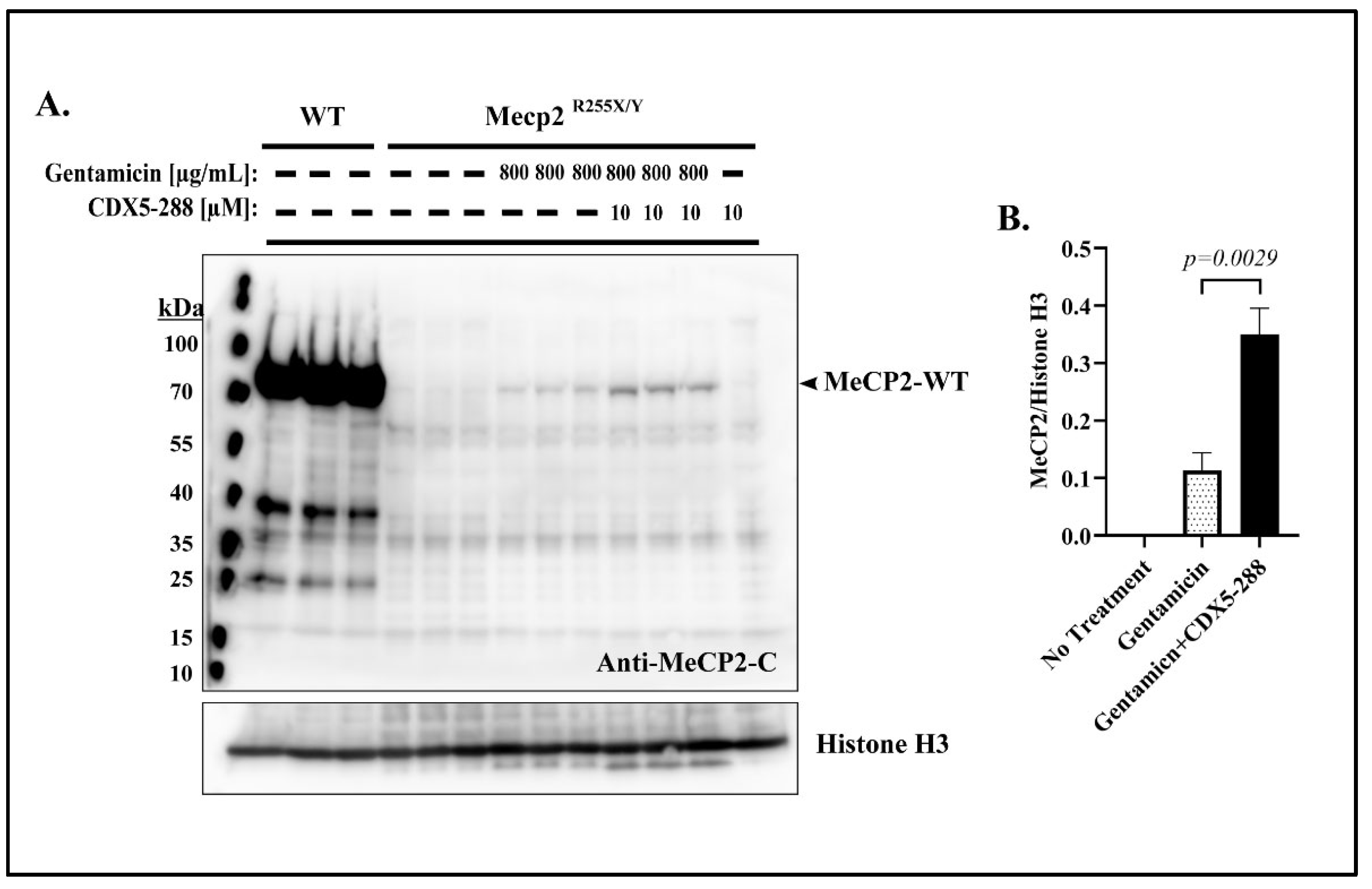
2.5. Co-Treatment Enhances Readthrough in Mecp2R255X/Y Mouse Fibroblasts
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Gentamicin and CDX-Compounds
4.3. Transfection and Drug Treatment
4.4. Western Blotting
4.5. Nuclear Protein Extraction
4.6. Immunofluorescence
4.7. Cell Viability Assay
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rett, A. On a unusual brain atrophy syndrome in hyperammonemia in childhood. Wien. Med. Wochenschr. 1966, 116, 723–726. [Google Scholar] [PubMed]
- Hagberg, B.; Aicardi, J.; Dias, K.; Ramos, O. A progressive syndrome of autism, dementia, ataxia, and loss of purposeful hand use in girls: Rett’s syndrome: Report of 35 cases. Ann. Neurol. 1983, 14, 471–479. [Google Scholar] [CrossRef]
- Huppke, P.; Held, M.; Laccone, F.; Hanefeld, F. The spectrum of phenotypes in females with Rett Syndrome. Brain Dev. 2003, 25, 346–351. [Google Scholar] [CrossRef] [PubMed]
- Neul, J.L.; Kaufmann, W.E.; Glaze, D.G.; Christodoulou, J.; Clarke, A.J.; Bahi-Buisson, N.; Leonard, H.; Bailey, M.E.S.; Schanen, N.C.; Zappella, M.; et al. Rett syndrome: Revised diagnostic criteria and nomenclature. Ann. Neurol. 2010, 68, 944–950. [Google Scholar] [CrossRef] [PubMed]
- Amir, R.E.; Van den Veyver, I.B.; Wan, M.; Tran, C.Q.; Francke, U.; Zoghbi, H.Y. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 1999, 23, 185–188. [Google Scholar] [CrossRef]
- Bienvenu, T.; Carrié, A.; de Roux, N.; Vinet, M.-C.; Jonveaux, P.; Couvert, P.; Villard, L.; Arzimanoglou, A.; Beldjord, C.; Fontes, M.; et al. MECP2 mutations account for most cases of typical forms of Rett syndrome. Hum. Mol. Genet. 2000, 9, 1377–1384. [Google Scholar] [CrossRef]
- Neul, J.L.; Fang, P.; Barrish, J.; Lane, J.; Caeg, E.B.; Smith, E.O.; Zoghbi, H.; Percy, A.; Glaze, D.G. Specific mutations in methyl-CpG-binding protein 2 confer different severity in Rett syndrome. Neurology 2008, 70, 1313–1321. [Google Scholar] [CrossRef]
- Correction to ‘MeCP2 binds to nucleosome free (linker DNA) regions and to H3K9/H3K27 methylated nucleosomes in the brain’. Nucleic Acids Res. 2022, 50, 3599–3600. [CrossRef]
- Lykke-Andersen, S.; Jensen, T.H. Faculty Opinions recommendation of Nonsense-mediated mRNA decay: An intricate machinery that shapes transcriptomes. Nat. Rev. Mol. Cell Biol. 2015, 16, 665–677. [Google Scholar] [CrossRef]
- Howard, M.; Frizzell, R.A.; Bedwell, D.M. Aminoglycoside antibiotics restore CFTR function by overcoming premature stop mutations. Nat. Med. 1996, 2, 467–469. [Google Scholar] [CrossRef]
- Dabrowski, M.; Bukowy-Bieryllo, Z.; Zietkiewicz, E. Advances in therapeutic use of a drug-stimulated translational readthrough of premature termination codons. Mol. Med. 2018, 24, 25. [Google Scholar] [CrossRef] [PubMed]
- Keeling, K.M.; Xue, X.; Gunn, G.; Bedwell, D.M. Therapeutics Based on Stop Codon Readthrough. Annu. Rev. Genom. Hum. Genet. 2014, 15, 371–394. [Google Scholar] [CrossRef] [PubMed]
- Nagel-Wolfrum, K.; Möller, F.; Penner, I.; Baasov, T.; Wolfrum, U. Targeting Nonsense Mutations in Diseases with Translational Read-Through-Inducing Drugs (TRIDs). Biodrugs 2016, 30, 49–74. [Google Scholar] [CrossRef]
- Prokhorova, I.; Altman, R.B.; Djumagulov, M.; Shrestha, J.P.; Urzhumtsev, A.; Ferguson, A.; Chang, C.-W.T.; Yusupov, M.; Blanchard, S.C.; Yusupova, G. Aminoglycoside interactions and impacts on the eukaryotic ribosome. Proc. Natl. Acad. Sci. USA 2017, 114, E10899–E10908. [Google Scholar] [CrossRef] [PubMed]
- Sharma, J.; Keeling, K.M.; Rowe, S.M. Pharmacological approaches for targeting cystic fibrosis nonsense mutations. Eur. J. Med. Chem. 2020, 200, 112436. [Google Scholar] [CrossRef]
- Finkel, R.S. Read-Through Strategies for Suppression of Nonsense Mutations in Duchenne/Becker Muscular Dystrophy: Aminoglycosides and Ataluren (PTC124). J. Child Neurol. 2010, 25, 1158–1164. [Google Scholar] [CrossRef]
- Malik, V.; Rodino-Klapac, L.R.; Viollet, L.; Mendell, J.R. Aminoglycoside-induced mutation suppression (stop codon readthrough) as a therapeutic strategy for Duchenne muscular dystrophy. Ther. Adv. Neurol. Disord. 2010, 3, 379–389. [Google Scholar] [CrossRef]
- Omachi, K.; Kai, H.; Roberge, M.; Miner, J.H. NanoLuc reporters identify COL4A5 nonsense mutations susceptible to drug-induced stop codon readthrough. iScience 2022, 25, 103891. [Google Scholar] [CrossRef]
- Has, C.; Sayar, S.B.; Zheng, S.; Chacón-Solano, E.; Condrat, I.; Yadav, A.; Roberge, M.; Laguzzi, F.L. Read-Through for Nonsense Mutations in Type XVII Collagen-Deficient Junctional Epidermolysis Bullosa. J. Investig. Dermatol. 2021, 142, 1227–1230. [Google Scholar] [CrossRef]
- Floquet, C.; Deforges, J.; Rousset, J.-P.; Bidou, L. Rescue of non-sense mutated p53 tumor suppressor gene by aminoglycosides. Nucleic Acids Res. 2010, 39, 3350–3362. [Google Scholar] [CrossRef]
- Bykov, V.J.N.; Eriksson, S.E.; Bianchi, J.; Wiman, K.G. Targeting mutant p53 for efficient cancer therapy. Nat. Rev. Cancer 2018, 18, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Abreu, R.B.V.; Gomes, T.T.; Nepomuceno, T.C.; Li, X.; Fuchshuber-Moraes, M.; De Gregoriis, G.; Suarez-Kurtz, G.; Monteiro, A.N.A.; Carvalho, M.A. Functional Restoration of BRCA1 Nonsense Mutations by Aminoglycoside-Induced Readthrough. Front. Pharmacol. 2022, 13, 935995. [Google Scholar] [CrossRef] [PubMed]
- Brendel, C.; Klahold, E.; Gärtner, J.; Huppke, P. Suppression of Nonsense Mutations in Rett Syndrome by Aminoglycoside Antibiotics. Pediatr. Res. 2009, 65, 520–523. [Google Scholar] [CrossRef] [PubMed]
- Brendel, C.; Belakhov, V.; Werner, H.; Wegener, E.; Gärtner, J.; Nudelman, I.; Baasov, T.; Huppke, P. Readthrough of nonsense mutations in Rett syndrome: Evaluation of novel aminoglycosides and generation of a new mouse model. J. Mol. Med. 2010, 89, 389–398. [Google Scholar] [CrossRef]
- Popescu, A.C.; Sidorova, E.; Zhang, G.; Eubanks, J.H. Aminoglycoside-mediated partial suppression of MECP2 nonsense mutations responsible for Rett syndrome in vitro. J. Neurosci. Res. 2010, 88, 2316–2324. [Google Scholar] [CrossRef] [PubMed]
- Lacy, M.K.; Nicolau, D.P.; Nightingale, C.H.; Quintiliani, R. The Pharmacodynamics of Aminoglycosides. Clin. Infect. Dis. 1998, 27, 23–27. [Google Scholar] [CrossRef]
- Nudelman, I.; Rebibo-Sabbah, A.; Cherniavsky, M.; Belakhov, V.; Hainrichson, M.; Chen, F.; Schacht, J.; Pilch, D.S.; Ben-Yosef, T.; Baasov, T. Development of Novel Aminoglycoside (NB54) with Reduced Toxicity and Enhanced Suppression of Disease-Causing Premature Stop Mutations. J. Med. Chem. 2009, 52, 2836–2845. [Google Scholar] [CrossRef]
- Nudelman, I.; Glikin, D.; Smolkin, B.; Hainrichson, M.; Belakhov, V.; Baasov, T. Repairing faulty genes by aminoglycosides: Development of new derivatives of geneticin (G418) with enhanced suppression of diseases-causing nonsense mutations. Bioorg. Med. Chem. 2010, 18, 3735–3746. [Google Scholar] [CrossRef]
- Vecsler, M.; Ben Zeev, B.; Nudelman, I.; Anikster, Y.; Simon, A.J.; Amariglio, N.; Rechavi, G.; Baasov, T.; Gak, E. Ex Vivo Treatment with a Novel Synthetic Aminoglycoside NB54 in Primary Fibroblasts from Rett Syndrome Patients Suppresses MECP2 Nonsense Mutations. PLoS ONE 2011, 6, e20733. [Google Scholar] [CrossRef]
- Bidou, L.; Bugaud, O.; Belakhov, V.; Baasov, T.; Namy, O. Characterization of new-generation aminoglycoside promoting premature termination codon readthrough in cancer cells. RNA Biol. 2017, 14, 378–388. [Google Scholar] [CrossRef]
- Baradaran-Heravi, A.; Niesser, J.; Balgi, A.D.; Choi, K.; Zimmerman, C.; South, A.P.; Anderson, H.J.; Strynadka, N.C.; Bally, M.B.; Roberge, M. Gentamicin B1 is a minor gentamicin component with major nonsense mutation suppression activity. Proc. Natl. Acad. Sci. USA 2017, 114, 3479–3484. [Google Scholar] [CrossRef] [PubMed]
- Popadynec, M.; Baradaran-Heravi, A.; Alford, B.; Cameron, S.A.; Clinch, K.; Mason, J.M.; Rendle, P.M.; Zubkova, O.V.; Gan, Z.; Liu, H.; et al. Reducing the Toxicity of Designer Aminoglycosides as Nonsense Mutation Readthrough Agents for Therapeutic Targets. ACS Med. Chem. Lett. 2021, 12, 1486–1492. [Google Scholar] [CrossRef] [PubMed]
- Keeling, K.M.; Wang, D.; Dai, Y.; Murugesan, S.; Chenna, B.; Clark, J.; Belakhov, V.; Kandasamy, J.; Velu, S.E.; Baasov, T.; et al. Attenuation of Nonsense-Mediated mRNA Decay Enhances In Vivo Nonsense Suppression. PLoS ONE 2013, 8, e60478. [Google Scholar] [CrossRef] [PubMed]
- McHugh, D.R.; Cotton, C.U.; Hodges, C.A. Synergy between Readthrough and Nonsense Mediated Decay Inhibition in a Murine Model of Cystic Fibrosis Nonsense Mutations. Int. J. Mol. Sci. 2020, 22, 344. [Google Scholar] [CrossRef]
- Ferguson, M.W.; Gerak, C.; Chow, C.C.T.; Rastelli, E.J.; Elmore, K.E.; Stahl, F.; Hosseini-Farahabadi, S.; Baradaran-Heravi, A.; Coltart, D.M.; Roberge, M. The antimalarial drug mefloquine enhances TP53 premature termination codon readthrough by aminoglycoside G418. PLoS ONE 2019, 14, e0216423. [Google Scholar] [CrossRef]
- Sharma, J.; Du, M.; Wong, E.; Mutyam, V.; Li, Y.; Chen, J.; Wangen, J.; Thrasher, K.; Fu, L.; Peng, N.; et al. A small molecule that induces translational readthrough of CFTR nonsense mutations by eRF1 depletion. Nat. Commun. 2021, 12, 4358. [Google Scholar] [CrossRef]
- Baradaran-Heravi, A.; Balgi, A.D.; Hosseini-Farahabadi, S.; Choi, K.; Has, C.; Roberge, M. Effect of small molecule eRF3 degraders on premature termination codon readthrough. Nucleic Acids Res. 2021, 49, 3692–3708. [Google Scholar] [CrossRef]
- Baradaran-Heravi, A.; Bauer, C.C.; Pickles, I.B.; Hosseini-Farahabadi, S.; Balgi, A.D.; Choi, K.; Linley, D.M.; Beech, D.J.; Roberge, M.; Bon, R.S. Nonselective TRPC channel inhibition and suppression of aminoglycoside-induced premature termination codon readthrough by the small molecule AC1903. J. Biol. Chem. 2022, 298, 101546. [Google Scholar] [CrossRef]
- Baradaran-Heravi, A.; Balgi, A.D.; Zimmerman, C.; Choi, K.; Shidmoossavee, F.S.; Tan, J.S.; Bergeaud, C.; Krause, A.; Flibotte, S.; Shimizu, Y.; et al. Novel small molecules potentiate premature termination codon readthrough by aminoglycosides. Nucleic Acids Res. 2016, 44, 6583–6598. [Google Scholar] [CrossRef]
- Frew, J.; Baradaran-Heravi, A.; Balgi, A.D.; Wu, X.; Yan, T.; Arns, S.; Shidmoossavee, F.S.; Tan, J.; Jaquith, J.B.; Jansen-West, K.R.; et al. Premature termination codon readthrough upregulates progranulin expression and improves lysosomal function in preclinical models of GRN deficiency. Mol. Neurodegener. 2020, 15, 21. [Google Scholar] [CrossRef]
- Bonetti, B.; Fu, L.; Moon, J.; Bedwell, D.M. The Efficiency of Translation Termination is Determined by a Synergistic Interplay Between Upstream and Downstream Sequences inSaccharomyces cerevisiae. J. Mol. Biol. 1995, 251, 334–345. [Google Scholar] [CrossRef] [PubMed]
- Cridge, A.G.; Crowe-McAuliffe, C.; Mathew, S.F.; Tate, W.P. Eukaryotic translational termination efficiency is influenced by the 3′ nucleotides within the ribosomal mRNA channel. Nucleic Acids Res. 2018, 46, 1927–1944. [Google Scholar] [CrossRef] [PubMed]
- Wangen, J.R.; Green, R. Stop codon context influences genome-wide stimulation of termination codon readthrough by aminoglycosides. Elife 2020, 9, e52611. [Google Scholar] [CrossRef] [PubMed]
- Linde, L.; Boelz, S.; Nissim-Rafinia, M.; Oren, Y.S.; Wilschanski, M.; Yaacov, Y.; Virgilis, D.; Neu-Yilik, G.; Kulozik, A.E.; Kerem, E.; et al. Nonsense-mediated mRNA decay affects nonsense transcript levels and governs response of cystic fibrosis patients to gentamicin. J. Clin. Investig. 2007, 117, 683–692. [Google Scholar] [CrossRef]
- Hinzpeter, A.; Aissat, A.; de Becdelièvre, A.; Bieth, E.; Sondo, E.; Martin, N.; Costes, B.; Costa, C.; Goossens, M.; Galietta, L.J.; et al. Alternative Splicing of In-Frame Exon Associated with Premature Termination Codons: Implications for Readthrough Therapies. Hum. Mutat. 2012, 34, 287–291. [Google Scholar] [CrossRef]
- Blanchet, S.; Cornu, D.; Argentini, M.; Namy, O. New insights into the incorporation of natural suppressor tRNAs at stop codons in Saccharomyces cerevisiae. Nucleic Acids Res. 2014, 42, 10061–10072. [Google Scholar] [CrossRef]
- Roy, B.; Leszyk, J.D.; Mangus, D.A.; Jacobson, A. Nonsense suppression by near-cognate tRNAs employs alternative base pairing at codon positions 1 and 3. Proc. Natl. Acad. Sci. USA 2015, 112, 3038–3043. [Google Scholar] [CrossRef]
- Loenarz, C.; Sekirnik, R.; Thalhammer, A.; Ge, W.; Spivakovsky, E.; Mackeen, M.M.; McDonough, M.A.; Cockman, M.E.; Kessler, B.M.; Ratcliffe, P.J.; et al. Hydroxylation of the eukaryotic ribosomal decoding center affects translational accuracy. Proc. Natl. Acad. Sci. USA 2014, 111, 4019–4024. [Google Scholar] [CrossRef]
- Beznosková, P.; Wagner, S.; Jansen, M.E.; von der Haar, T.; Valášek, L.S. Translation initiation factor eIF3 promotes programmed stop codon readthrough. Nucleic Acids Res. 2015, 43, 5099–5111. [Google Scholar] [CrossRef]
- Feng, T.; Yamamoto, A.; Wilkins, S.E.; Sokolova, E.; Yates, L.A.; Münzel, M.; Singh, P.; Hopkinson, R.J.; Fischer, R.; Cockman, M.E.; et al. Optimal Translational Termination Requires C4 Lysyl Hydroxylation of eRF1. Mol. Cell 2014, 53, 645–654. [Google Scholar] [CrossRef]
- Zhang, H.; Lyu, Z.; Fan, Y.; Evans, C.R.; Barber, K.W.; Banerjee, K.; Igoshin, O.A.; Rinehart, J.; Ling, J. Metabolic stress promotes stop-codon readthrough and phenotypic heterogeneity. Proc. Natl. Acad. Sci. USA 2020, 117, 22167–22172. [Google Scholar] [CrossRef] [PubMed]
- McCaughan, K.K.; Brown, C.M.; Dalphin, M.E.; Berry, M.J.; Tate, W.P. Translational termination efficiency in mammals is influenced by the base following the stop codon. Proc. Natl. Acad. Sci. USA 1995, 92, 5431–5435. [Google Scholar] [CrossRef] [PubMed]
- Dabrowski, M.; Bukowy-Bieryllo, Z.; Zietkiewicz, E. Translational readthrough potential of natural termination codons in eucaryotes—The impact of RNA sequence. RNA Biol. 2015, 12, 950–958. [Google Scholar] [CrossRef] [PubMed]
- Floquet, C.; Hatin, I.; Rousset, J.-P.; Bidou, L. Statistical Analysis of Readthrough Levels for Nonsense Mutations in Mammalian Cells Reveals a Major Determinant of Response to Gentamicin. PLoS Genet. 2012, 8, e1002608. [Google Scholar] [CrossRef] [PubMed]
- Manuvakhova, M.; Keeling, K.; Bedwell, D.M. Aminoglycoside antibiotics mediate context-dependent suppression of termination codons in a mammalian translation system. RNA 2000, 6, 1044–1055. [Google Scholar] [CrossRef] [PubMed]
- Howard, M.T.; Shirts, B.H.; Petros, L.M.; Flanigan, K.M.; Gesteland, R.F.; Atkins, J.F. Sequence specificity of aminoglycoside-induced stop condon readthrough: Potential implications for treatment of Duchenne muscular dystrophy. Ann. Neurol. 2000, 48, 164–169. [Google Scholar] [CrossRef]
- Hosseini-Farahabadi, S.; Baradaran-Heravi, A.; Zimmerman, C.; Choi, K.; Flibotte, S.; Roberge, M. Small molecule Y-320 stimulates ribosome biogenesis, protein synthesis, and aminoglycoside-induced premature termination codon readthrough. PLoS Biol. 2021, 19, e3001221. [Google Scholar] [CrossRef]
- Merritt, J.K.; Collins, B.E.; Erickson, K.R.; Dong, H.; Neul, J.L. Pharmacological read-through of R294X Mecp2 in a novel mouse model of Rett syndrome. Hum. Mol. Genet. 2020, 29, 2461–2470. [Google Scholar] [CrossRef]
- Pitcher, M.R.; Herrera, J.A.; Buffington, S.A.; Kochukov, M.Y.; Merritt, J.K.; Fisher, A.R.; Schanen, N.C.; Costa-Mattioli, M.; Neul, J.L. Rett syndrome like phenotypes in the R255X Mecp2 mutant mouse are rescued by MECP2 transgene. Hum. Mol. Genet. 2015, 24, 2662–2672. [Google Scholar] [CrossRef]
- Gunn, G.; Dai, Y.; Du, M.; Belakhov, V.; Kandasamy, J.; Schoeb, T.R.; Baasov, T.; Bedwell, D.M.; Keeling, K.M. Long-term nonsense suppression therapy moderates MPS I-H disease progression. Mol. Genet. Metab. 2014, 111, 374–381. [Google Scholar] [CrossRef]
- Crawford, D.K.; Alroy, I.; Sharpe, N.; Goddeeris, M.M.; Williams, G. ELX-02 Generates Protein via Premature Stop Codon Read-Through without Inducing Native Stop Codon Read-Through Proteins. Experiment 2020, 374, 264–272. [Google Scholar] [CrossRef] [PubMed]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wong, K.M.; Wegener, E.; Baradaran-Heravi, A.; Huppke, B.; Gärtner, J.; Huppke, P. Evaluation of Novel Enhancer Compounds in Gentamicin-Mediated Readthrough of Nonsense Mutations in Rett Syndrome. Int. J. Mol. Sci. 2023, 24, 11665. https://doi.org/10.3390/ijms241411665
Wong KM, Wegener E, Baradaran-Heravi A, Huppke B, Gärtner J, Huppke P. Evaluation of Novel Enhancer Compounds in Gentamicin-Mediated Readthrough of Nonsense Mutations in Rett Syndrome. International Journal of Molecular Sciences. 2023; 24(14):11665. https://doi.org/10.3390/ijms241411665
Chicago/Turabian StyleWong, Keit Men, Eike Wegener, Alireza Baradaran-Heravi, Brenda Huppke, Jutta Gärtner, and Peter Huppke. 2023. "Evaluation of Novel Enhancer Compounds in Gentamicin-Mediated Readthrough of Nonsense Mutations in Rett Syndrome" International Journal of Molecular Sciences 24, no. 14: 11665. https://doi.org/10.3390/ijms241411665
APA StyleWong, K. M., Wegener, E., Baradaran-Heravi, A., Huppke, B., Gärtner, J., & Huppke, P. (2023). Evaluation of Novel Enhancer Compounds in Gentamicin-Mediated Readthrough of Nonsense Mutations in Rett Syndrome. International Journal of Molecular Sciences, 24(14), 11665. https://doi.org/10.3390/ijms241411665