
Calcitonin Gene-Related Peptide mRNA Synthesis in Trigeminal Ganglion Neurons after Cortical Spreading Depolarization


 and
and 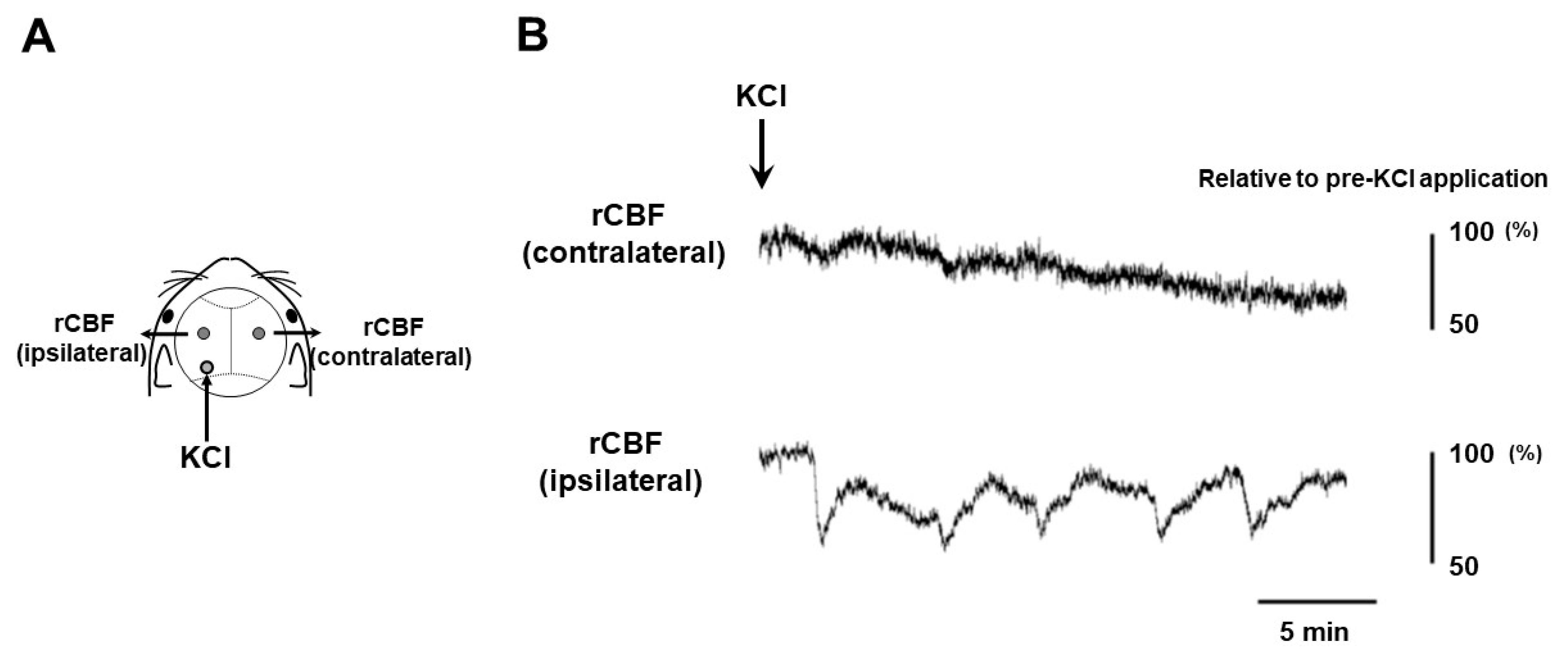{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. CSD Induction
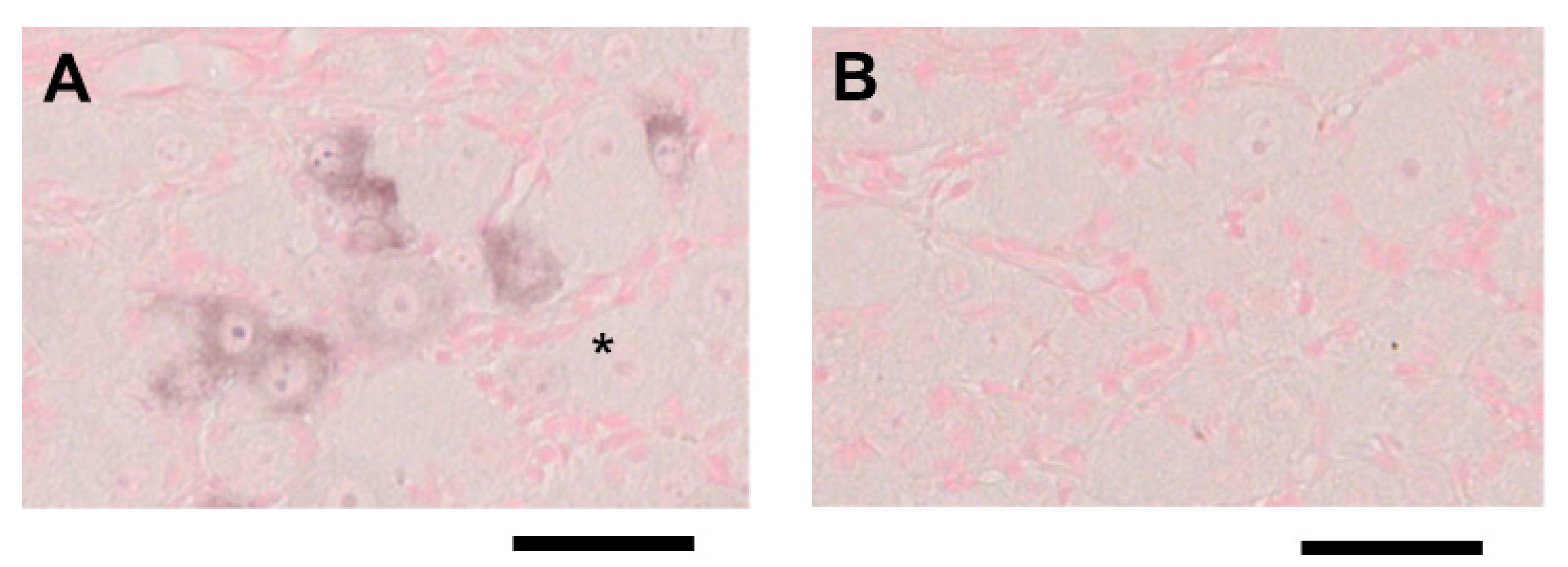
2.2. ISH for Mouse CGRP mRNA in TG Tissue
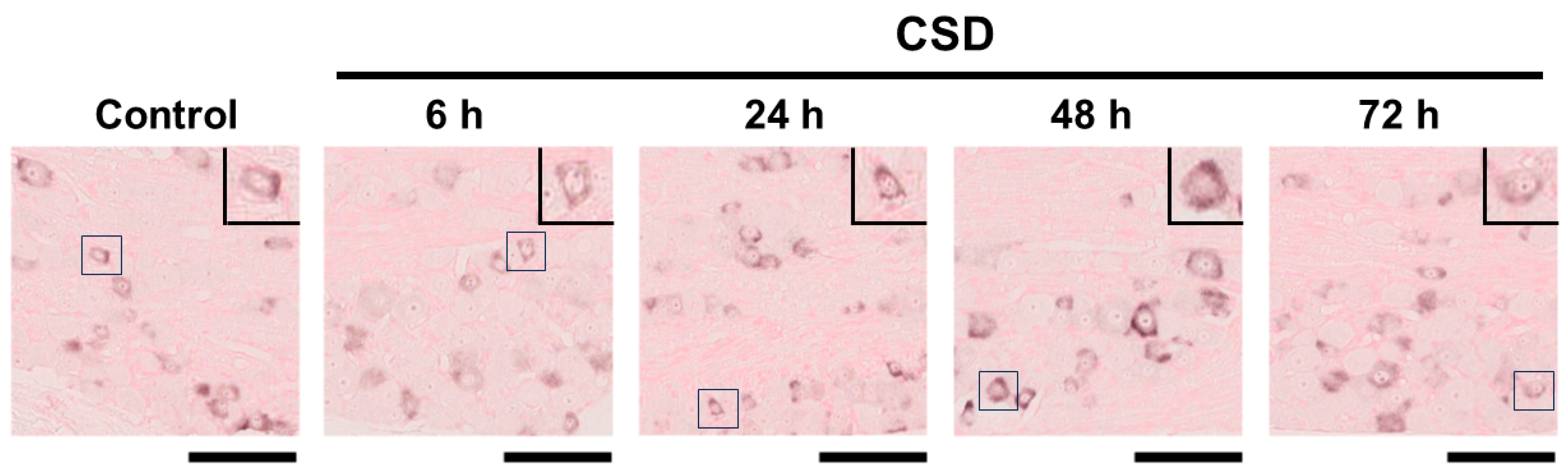
2.3. Density of CGRP mRNA-Synthesizing TG Neurons after CSD
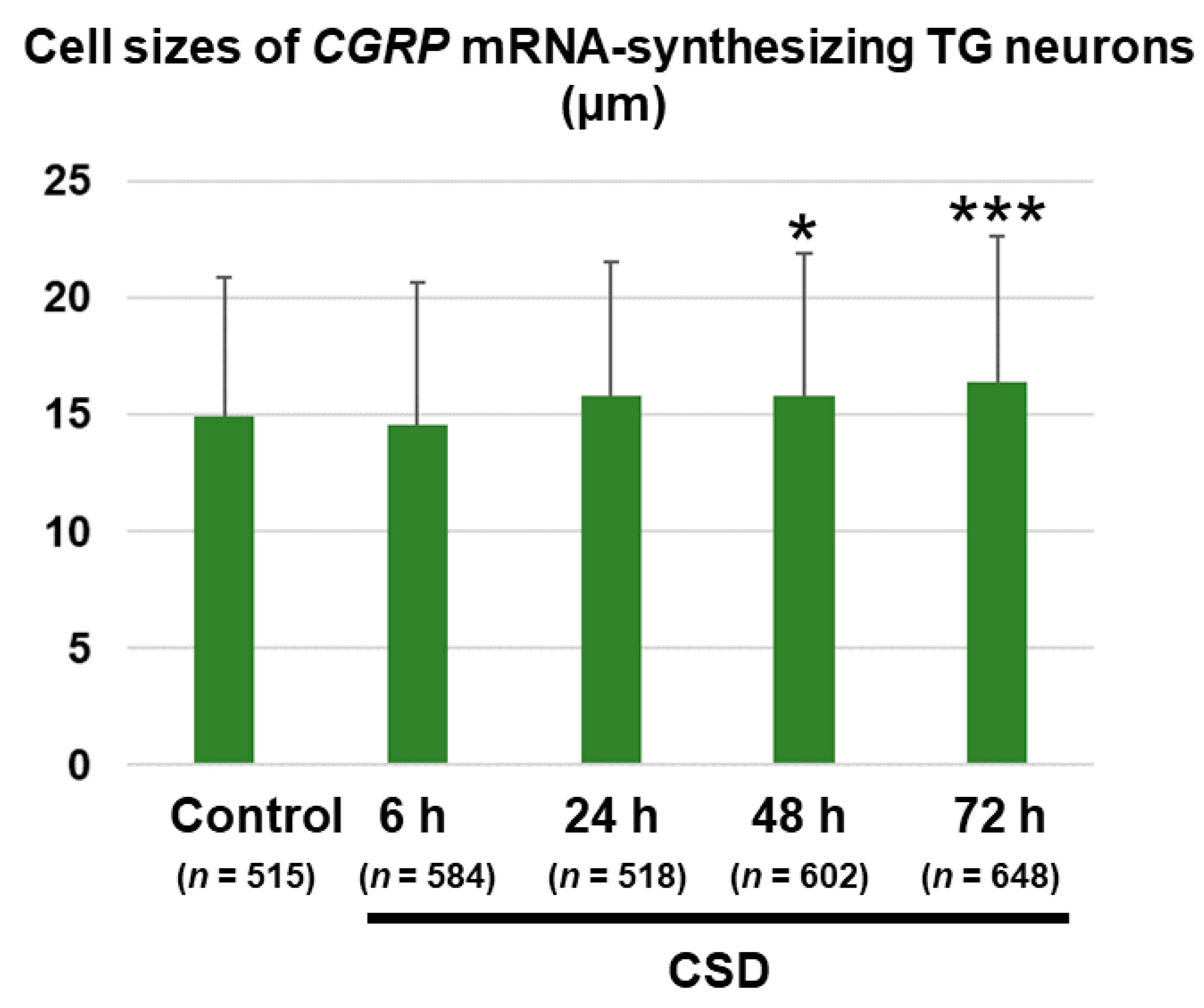
2.4. Cell Size Changes in CGRP mRNA-Synthesizing TG Neurons after CSD
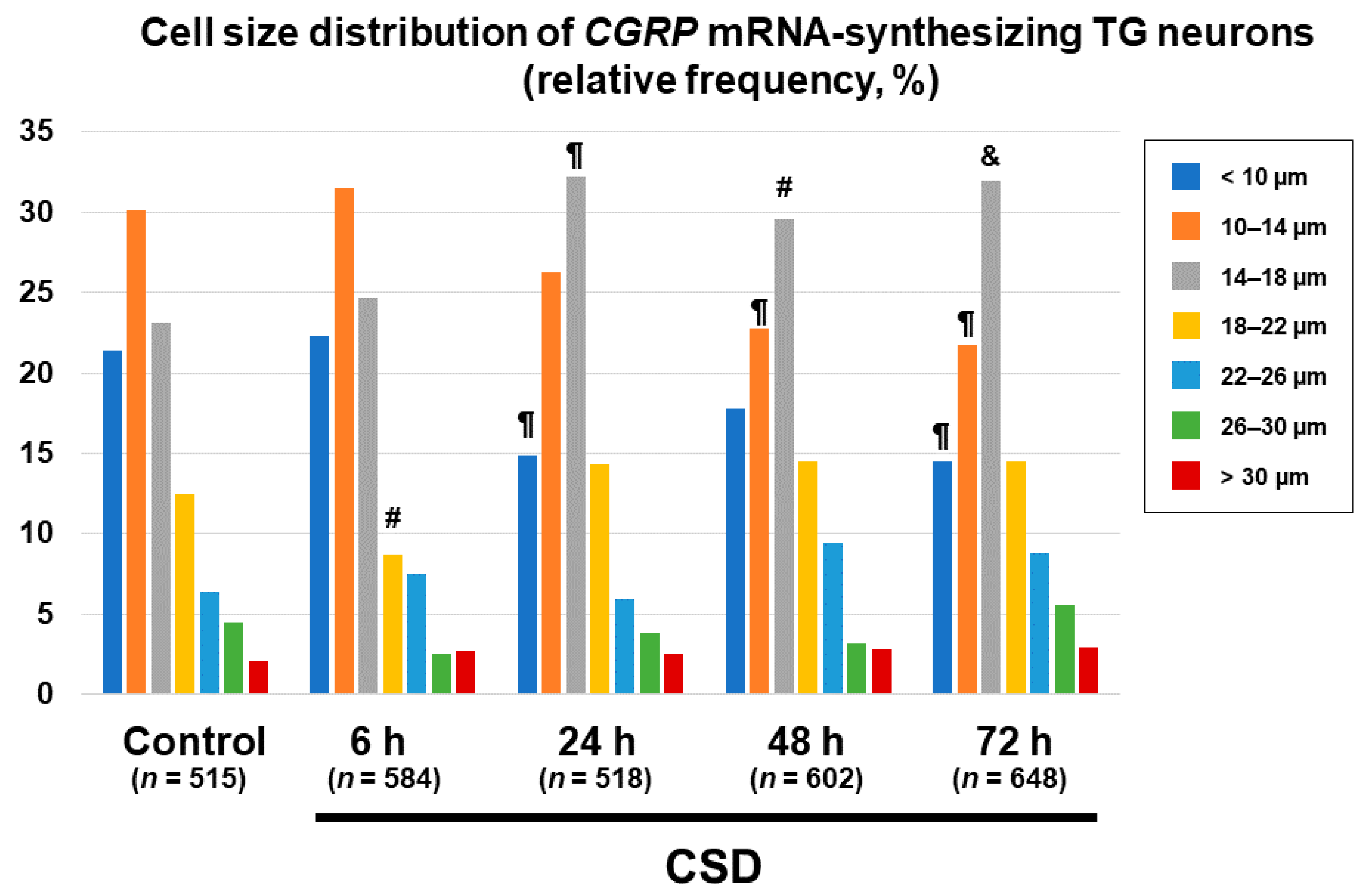
2.5. Changes in Cell Size Distributions of CGRP mRNA-Synthesizing TG Neurons after CSD
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. CSD Induction
4.3. TG Tissue Excision
4.4. Mouse TG ISH
- CGRP: 5′-gaaaggctgatgaaagacacatatatttgcatccttcttagtattgaaaaacccttctccctttgacaggagctaaagctaagtgcagaataagttgcctattgtgcatcgtgttgtatgtgactctgtatccaataaacatgacagcatggttctggcttatctggtagcaaatatggtccccataaaccatcctgttgatgttgatgactctgctaaacctcaaggggatatgaaacactgcctcttgctcttctggggacacatggtaa-3′.
4.5. Statistical Analyses
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Steiner, T.J.; Stovner, L.J.; Jensen, R.; Uluduz, D.; Katsarava, Z. Lifting The Burden: The Global Campaign against Headache. Migraine remains second among the world’s causes of disability, and first among young women: Findings from GBD2019. J. Headache Pain 2020, 21, 137. [Google Scholar] [CrossRef] [PubMed]
- Ashina, M. Migraine. N. Engl. J. Med. 2020, 383, 1866–1876. [Google Scholar] [CrossRef]
- Dreier, J.P.; Fabricius, M.; Ayata, C.; Sakowitz, O.W.; Shuttleworth, C.W.; Dohmen, C.; Graf, R.; Vajkoczy, P.; Helbok, R.; Suzuki, M.; et al. Recording, analysis, and interpretation of spreading depolarizations in neurointensive care: Review and recommendations of the COSBID research group. J. Cereb. Blood Flow Metab. 2017, 37, 1595–1625. [Google Scholar] [CrossRef] [PubMed]
- Lauritzen, M.; Jorgensen, M.B.; Diemer, N.H.; Gjedde, A.; Hansen, A.J. Persistent oligemia of rat cerebral cortex in the wake of spreading depression. Ann. Neurol. 1982, 12, 469–474. [Google Scholar] [CrossRef] [PubMed]
- Olesen, J.; Friberg, L.; Olsen, T.S.; Iversen, H.K.; Lassen, N.A.; Andersen, A.R.; Karle, A. Timing and topography of cerebral blood flow, aura, and headache during migraine attacks. Ann. Neurol. 1990, 28, 791–798. [Google Scholar] [CrossRef]
- Olesen, J.; Larsen, B.; Lauritzen, M. Focal hyperemia followed by spreading oligemia and impaired activation of rCBF in classic migraine. Ann. Neurol. 1981, 9, 344–352. [Google Scholar] [CrossRef]
- Cao, Y.; Welch, K.M.; Aurora, S.; Vikingstad, E.M. Functional MRI-BOLD of visually triggered headache in patients with migraine. Arch. Neurol. 1999, 56, 548–554. [Google Scholar] [CrossRef]
- Andersen, A.R.; Friberg, L.; Olsen, T.S.; Olesen, J. Delayed hyperemia following hypoperfusion in classic migraine. Single photon emission computed tomographic demonstration. Arch. Neurol. 1988, 45, 154–159. [Google Scholar] [CrossRef]
- Bowyer, S.M.; Aurora, K.S.; Moran, J.E.; Tepley, N.; Welch, K.M. Magnetoencephalographic fields from patients with spontaneous and induced migraine aura. Ann. Neurol. 2001, 50, 582–587. [Google Scholar] [CrossRef]
- Hadjikhani, N.; Sanchez Del Rio, M.; Wu, O.; Schwartz, D.; Bakker, D.; Fischl, B.; Kwong, K.K.; Cutrer, F.M.; Rosen, B.R.; Tootell, R.B.; et al. Mechanisms of migraine aura revealed by functional MRI in human visual cortex. Proc. Natl. Acad. Sci. USA 2001, 98, 4687–4692. [Google Scholar] [CrossRef]
- Zhang, X.; Levy, D.; Noseda, R.; Kainz, V.; Jakubowski, M.; Burstein, R. Activation of meningeal nociceptors by cortical spreading depression: Implications for migraine with aura. J. Neurosci. 2010, 30, 8807–8814. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Levy, D.; Kainz, V.; Noseda, R.; Jakubowski, M.; Burstein, R. Activation of central trigeminovascular neurons by cortical spreading depression. Ann. Neurol. 2011, 69, 855–865. [Google Scholar] [CrossRef] [PubMed]
- Karatas, H.; Erdener, S.E.; Gursoy-Ozdemir, Y.; Lule, S.; Eren-Kocak, E.; Sen, Z.D.; Dalkara, T. Spreading depression triggers headache by activating neuronal Panx1 channels. Science 2013, 339, 1092–1095. [Google Scholar] [CrossRef]
- Carneiro-Nascimento, S.; Levy, D. Cortical spreading depression and meningeal nociception. Neurobiol. Pain 2022, 11, 100091. [Google Scholar] [CrossRef] [PubMed]
- Moskowitz, M.A.; Nozaki, K.; Kraig, R.P. Neocortical spreading depression provokes the expression of c-fos protein-like immunoreactivity within trigeminal nucleus caudalis via trigeminovascular mechanisms. J. Neurosci. 1993, 13, 1167–1177. [Google Scholar] [CrossRef] [PubMed]
- Bolay, H.; Reuter, U.; Dunn, A.K.; Huang, Z.; Boas, D.A.; Moskowitz, M.A. Intrinsic brain activity triggers trigeminal meningeal afferents in a migraine model. Nat. Med. 2002, 8, 136–142. [Google Scholar] [CrossRef]
- Ayata, C.; Jin, H.; Kudo, C.; Dalkara, T.; Moskowitz, M.A. Suppression of cortical spreading depression in migraine prophylaxis. Ann. Neurol. 2006, 59, 652–661. [Google Scholar] [CrossRef]
- Ayata, C. Pearls and pitfalls in experimental models of spreading depression. Cephalalgia 2013, 33, 604–613. [Google Scholar] [CrossRef]
- Mayberg, M.; Langer, R.S.; Zervas, N.T.; Moskowitz, M.A. Perivascular meningeal projections from cat trigeminal ganglia: Possible pathway for vascular headaches in man. Science 1981, 213, 228–230. [Google Scholar] [CrossRef]
- Ashina, M.; Hansen, J.M.; Do, T.P.; Melo-Carrillo, A.; Burstein, R.; Moskowitz, M.A. Migraine and the trigeminovascular system-40 years and counting. Lancet Neurol. 2019, 18, 795–804. [Google Scholar] [CrossRef]
- Edvinsson, L.; Haanes, K.A.; Warfvinge, K.; Krause, D.N. CGRP as the target of new migraine therapies—Successful translation from bench to clinic. Nat. Rev. Neurol. 2018, 14, 338–350. [Google Scholar] [CrossRef] [PubMed]
- Buzzi, M.G.; Carter, W.B.; Shimizu, T.; Heath, H., 3rd; Moskowitz, M.A. Dihydroergotamine and sumatriptan attenuate levels of CGRP in plasma in rat superior sagittal sinus during electrical stimulation of the trigeminal ganglion. Neuropharmacology 1991, 30, 1193–1200. [Google Scholar] [CrossRef] [PubMed]
- Messlinger, K.; Hanesch, U.; Kurosawa, M.; Pawlak, M.; Schmidt, R.F. Calcitonin gene related peptide released from dural nerve fibers mediates increase of meningeal blood flow in the rat. Can. J. Physiol. Pharmacol. 1995, 73, 1020–1024. [Google Scholar] [CrossRef] [PubMed]
- Eltorp, C.T.; Jansen-Olesen, I.; Hansen, A.J. Release of calcitonin gene-related peptide (CGRP) from guinea pig dura mater in vitro is inhibited by sumatriptan but unaffected by nitric oxide. Cephalalgia 2000, 20, 838–844. [Google Scholar] [CrossRef]
- Bullock, C.M.; Wookey, P.; Bennett, A.; Mobasheri, A.; Dickerson, I.; Kelly, S. Peripheral calcitonin gene-related peptide receptor activation and mechanical sensitization of the joint in rat models of osteoarthritis pain. Arthritis Rheumatol. 2014, 66, 2188–2200. [Google Scholar] [CrossRef]
- Chatchaisak, D.; Connor, M.; Srikiatkhachorn, A.; Chetsawang, B. The potentiating effect of calcitonin gene-related peptide on transient receptor potential vanilloid-1 activity and the electrophysiological responses of rat trigeminal neurons to nociceptive stimuli. J. Physiol. Sci. 2018, 68, 261–268. [Google Scholar] [CrossRef]
- Cornelison, L.E.; Hawkins, J.L.; Durham, P.L. Elevated levels of calcitonin gene-related peptide in upper spinal cord promotes sensitization of primary trigeminal nociceptive neurons. Neuroscience 2016, 339, 491–501. [Google Scholar] [CrossRef]
- Iyengar, S.; Johnson, K.W.; Ossipov, M.H.; Aurora, S.K. CGRP and the Trigeminal System in Migraine. Headache 2019, 59, 659–681. [Google Scholar] [CrossRef]
- Li, D.; Ren, Y.; Xu, X.; Zou, X.; Fang, L.; Lin, Q. Sensitization of primary afferent nociceptors induced by intradermal capsaicin involves the peripheral release of calcitonin gene-related Peptide driven by dorsal root reflexes. J. Pain 2008, 9, 1155–1168. [Google Scholar] [CrossRef]
- Zhang, L.; Hoff, A.O.; Wimalawansa, S.J.; Cote, G.J.; Gagel, R.F.; Westlund, K.N. Arthritic calcitonin/α calcitonin gene-related peptide knockout mice have reduced nociceptive hypersensitivity. Pain 2001, 89, 265–273. [Google Scholar] [CrossRef]
- Wang, Y.; Tye, A.E.; Zhao, J.; Ma, D.; Raddant, A.C.; Bu, F.; Spector, B.L.; Winslow, N.K.; Wang, M.; Russo, A.F. Induction of calcitonin gene-related peptide expression in rats by cortical spreading depression. Cephalalgia 2019, 39, 333–341. [Google Scholar] [CrossRef]
- Bu, F.; Yuan, M.; Ma, D.; Zhu, Y.; Wang, M. Inhibition of NR2A reduces calcitonin gene-related peptide gene expression induced by cortical spreading depression in rat amygdala. Neuropeptides 2020, 84, 102097. [Google Scholar] [CrossRef] [PubMed]
- Volobueva, M.N.; Suleymanova, E.M.; Smirnova, M.P.; Bolshakov, A.P.; Vinogradova, L.V. A Single Episode of Cortical Spreading Depolarization Increases mRNA Levels of Proinflammatory Cytokines, Calcitonin Gene-Related Peptide and Pannexin-1 Channels in the Cerebral Cortex. Int. J. Mol. Sci. 2022, 24, 85. [Google Scholar] [CrossRef] [PubMed]
- Tozzi, A.; de Iure, A.; Di Filippo, M.; Costa, C.; Caproni, S.; Pisani, A.; Bonsi, P.; Picconi, B.; Cupini, L.M.; Materazzi, S.; et al. Critical role of calcitonin gene-related peptide receptors in cortical spreading depression. Proc. Natl. Acad. Sci. USA 2012, 109, 18985–18990. [Google Scholar] [CrossRef]
- Won, L.; Kraig, R.P. Insulin-like growth factor-1 inhibits spreading depression-induced trigeminal calcitonin gene related peptide, oxidative stress & neuronal activation in rat. Brain Res. 2020, 1732, 146673. [Google Scholar]
- Yisarakun, W.; Chantong, C.; Supornsilpchai, W.; Thongtan, T.; Srikiatkhachorn, A.; Reuangwechvorachai, P.; Maneesri-le Grand, S. Up-regulation of calcitonin gene-related peptide in trigeminal ganglion following chronic exposure to paracetamol in a CSD migraine animal model. Neuropeptides 2015, 51, 9–16. [Google Scholar] [CrossRef]
- Tsai, S.H.; Tew, J.M.; McLean, J.H.; Shipley, M.T. Cerebral arterial innervation by nerve fibers containing calcitonin gene-related peptide (CGRP): I. Distribution and origin of CGRP perivascular innervation in the rat. J. Comp. Neurol. 1988, 271, 435–444. [Google Scholar] [CrossRef]
- Nozaki, K.; Uemura, Y.; Okamoto, S.; Kikuchi, H.; Mizuno, N. Origins and distribution of cerebrovascular nerve fibers showing calcitonin gene-related peptide-like immunoreactivity in the major cerebral artery of the dog. J. Comp. Neurol. 1990, 297, 219–226. [Google Scholar] [CrossRef]
- Eftekhari, S.; Warfvinge, K.; Blixt, F.W.; Edvinsson, L. Differentiation of nerve fibers storing CGRP and CGRP receptors in the peripheral trigeminovascular system. J. Pain 2013, 14, 1289–1303. [Google Scholar] [CrossRef] [PubMed]
- Eftekhari, S.; Salvatore, C.A.; Johansson, S.; Chen, T.B.; Zeng, Z.; Edvinsson, L. Localization of CGRP, CGRP receptor, PACAP and glutamate in trigeminal ganglion. Relation to the blood-brain barrier. Brain Res. 2015, 1600, 93–109. [Google Scholar] [CrossRef]
- Melo-Carrillo, A.; Strassman, A.M.; Nir, R.R.; Schain, A.J.; Noseda, R.; Stratton, J.; Burstein, R. Fremanezumab-A Humanized Monoclonal Anti-CGRP Antibody-Inhibits Thinly Myelinated (Aδ) but Not Unmyelinated (C) Meningeal Nociceptors. J. Neurosci. 2017, 37, 10587–10596. [Google Scholar] [CrossRef] [PubMed]
- Ebine, T.; Toriumi, H.; Shimizu, T.; Unekawa, M.; Takizawa, T.; Kayama, Y.; Shibata, M.; Suzuki, N. Alterations in the threshold of the potassium concentration to evoke cortical spreading depression during the natural estrous cycle in mice. Neurosci. Res. 2016, 112, 57–62. [Google Scholar] [CrossRef] [PubMed]
- van Casteren, D.S.; Kurth, T.; Danser, A.H.J.; Terwindt, G.M.; MaassenVanDenBrink, A. Sex Differences in Response to Triptans: A Systematic Review and Meta-Analysis. Neurology 2021, 96, 162–170. [Google Scholar] [CrossRef]
- White, T.G.; Powell, K.; Shah, K.A.; Woo, H.H.; Narayan, R.K.; Li, C. Trigeminal Nerve Control of Cerebral Blood Flow: A Brief Review. Front. Neurosci. 2021, 15, 649910. [Google Scholar] [CrossRef] [PubMed]
- Takizawa, T.; Qin, T.; Lopes de Morais, A.; Sugimoto, K.; Chung, J.Y.; Morsett, L.; Mulder, I.; Fischer, P.; Suzuki, T.; Anzabi, M.; et al. Non-invasively triggered spreading depolarizations induce a rapid pro-inflammatory response in cerebral cortex. J. Cereb. Blood Flow Metab. 2020, 40, 1117–1131. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shibata, M.; Kitagawa, S.; Unekawa, M.; Takizawa, T.; Nakahara, J. Calcitonin Gene-Related Peptide mRNA Synthesis in Trigeminal Ganglion Neurons after Cortical Spreading Depolarization. Int. J. Mol. Sci. 2023, 24, 11578. https://doi.org/10.3390/ijms241411578
Shibata M, Kitagawa S, Unekawa M, Takizawa T, Nakahara J. Calcitonin Gene-Related Peptide mRNA Synthesis in Trigeminal Ganglion Neurons after Cortical Spreading Depolarization. International Journal of Molecular Sciences. 2023; 24(14):11578. https://doi.org/10.3390/ijms241411578
Chicago/Turabian StyleShibata, Mamoru, Satoshi Kitagawa, Miyuki Unekawa, Tsubasa Takizawa, and Jin Nakahara. 2023. "Calcitonin Gene-Related Peptide mRNA Synthesis in Trigeminal Ganglion Neurons after Cortical Spreading Depolarization" International Journal of Molecular Sciences 24, no. 14: 11578. https://doi.org/10.3390/ijms241411578
APA StyleShibata, M., Kitagawa, S., Unekawa, M., Takizawa, T., & Nakahara, J. (2023). Calcitonin Gene-Related Peptide mRNA Synthesis in Trigeminal Ganglion Neurons after Cortical Spreading Depolarization. International Journal of Molecular Sciences, 24(14), 11578. https://doi.org/10.3390/ijms241411578