
Investigating the Prevalence of RNA-Binding Metabolic Enzymes in E. coli

 , and
, and
Abstract
1. Introduction
2. Results
2.1. Selection of Proteins and Strategy
2.2. iCLIP for Selected Enzyme Candidates Gives No Hints on Specific RNA Binding
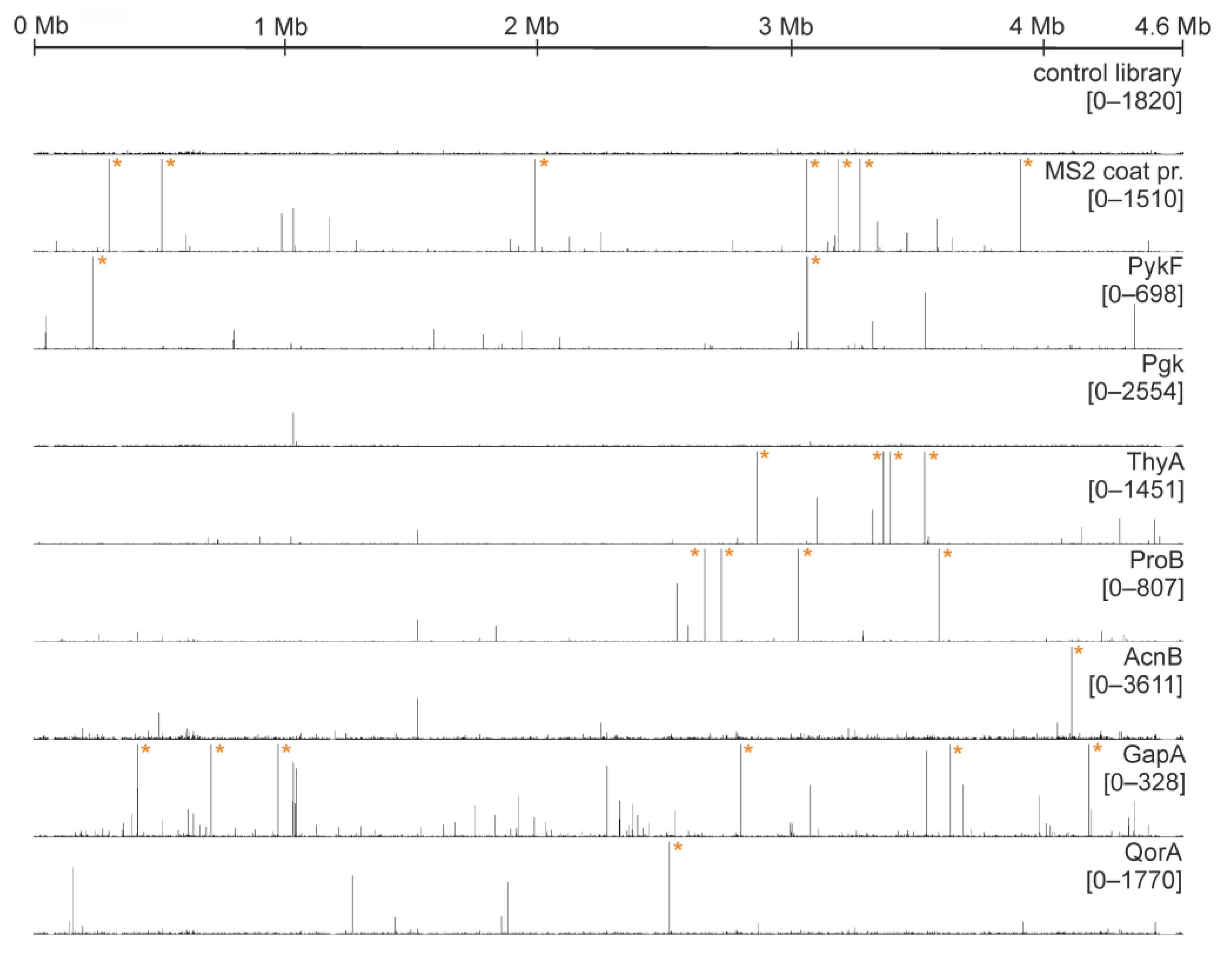
2.3. Multiple Enzymes Show Enrichment of Specific RNAs in Genomic SELEX Experiments
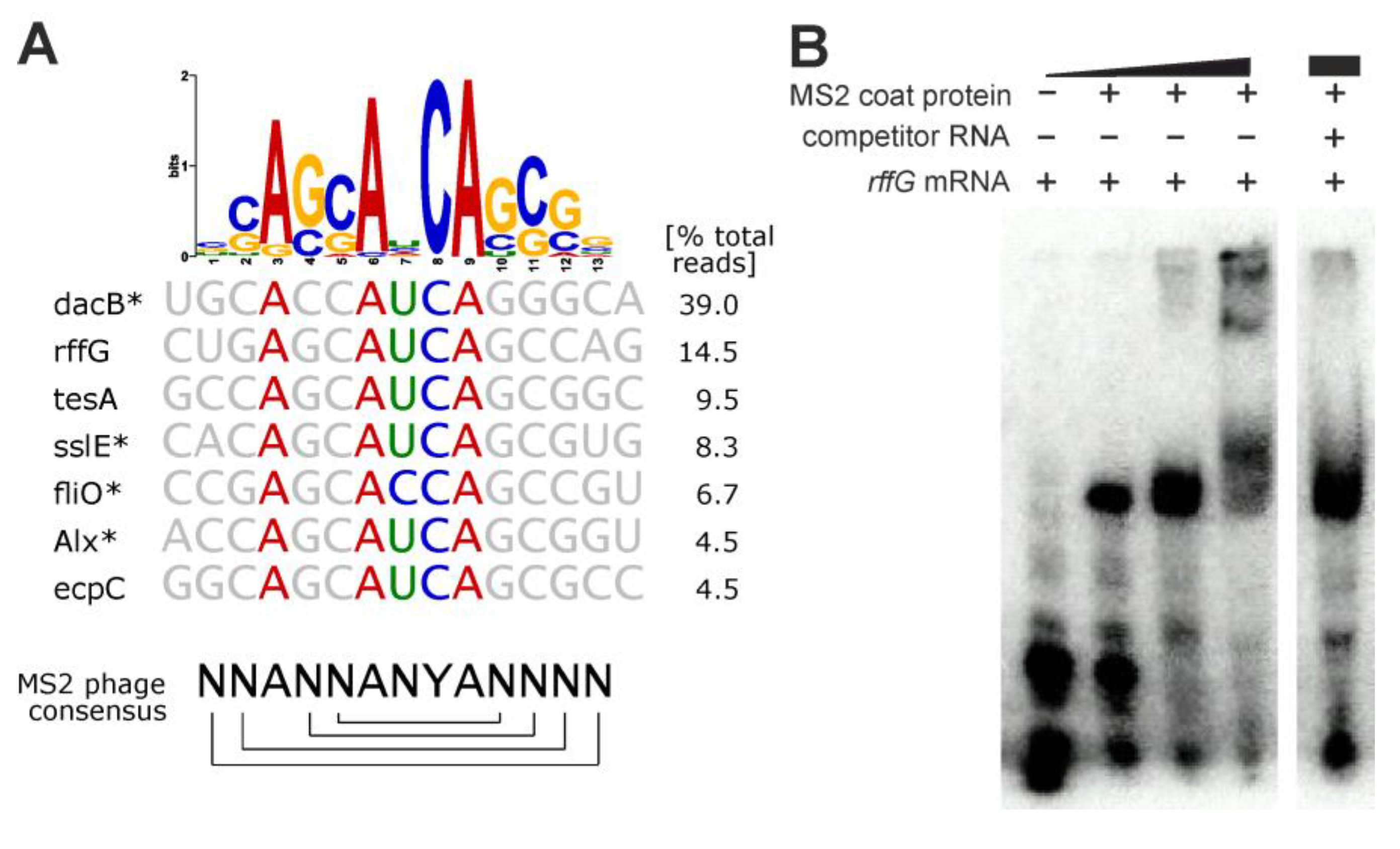
- SELEX results for MS2 phage coat protein
- Pyruvate kinase (PykF)
- Phosphoglycerate kinase (Pgk)
- Glyceralde-3-phosphate dehydrogenase (GapA)
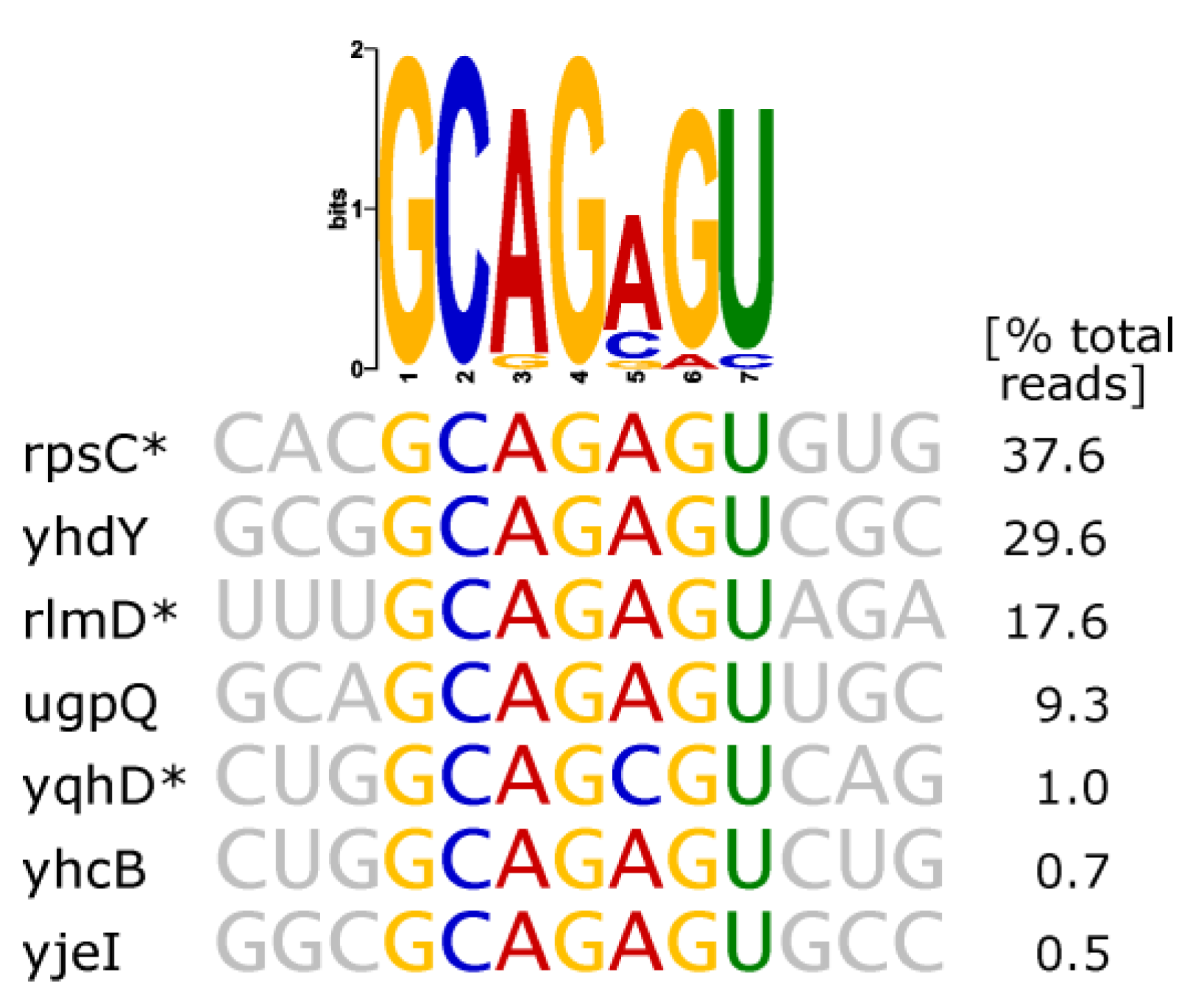
- Thymidylate Synthase (ThyA)
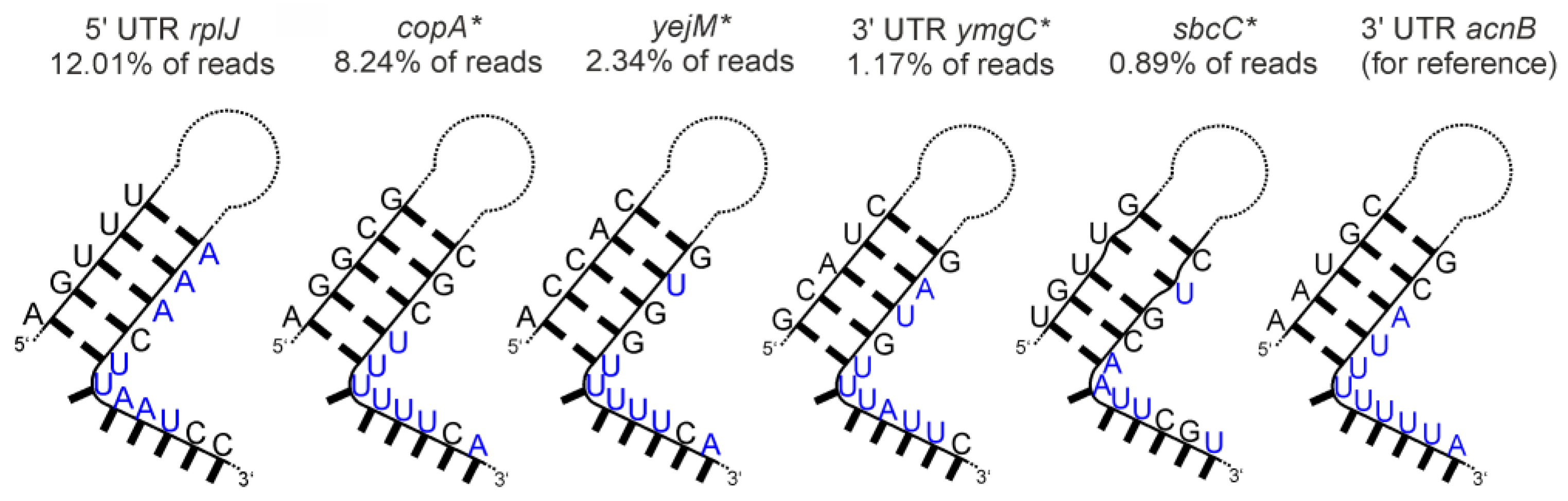
- Aconitase (AcnB)
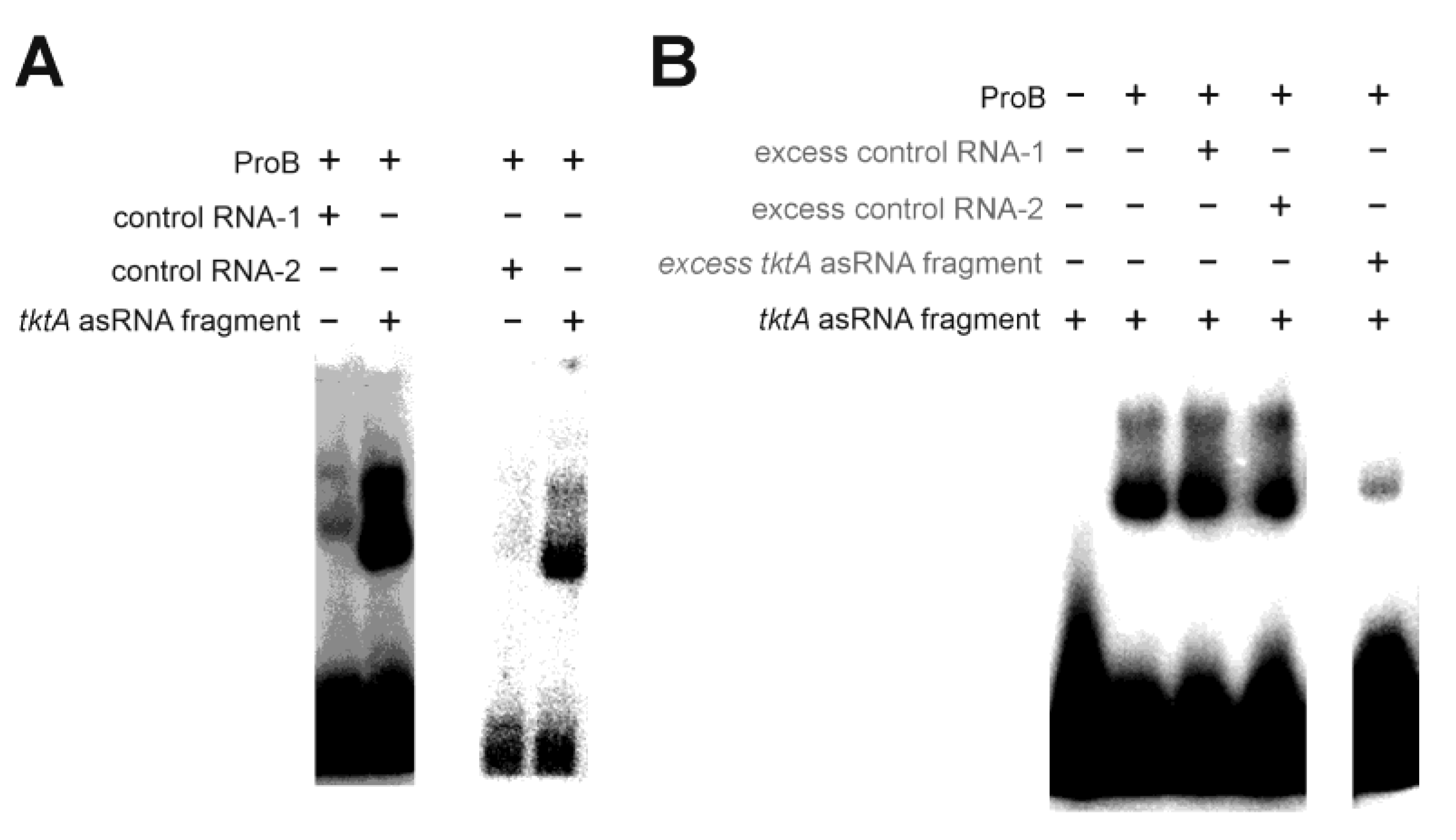
- Glutamate-5-kinase (ProB)
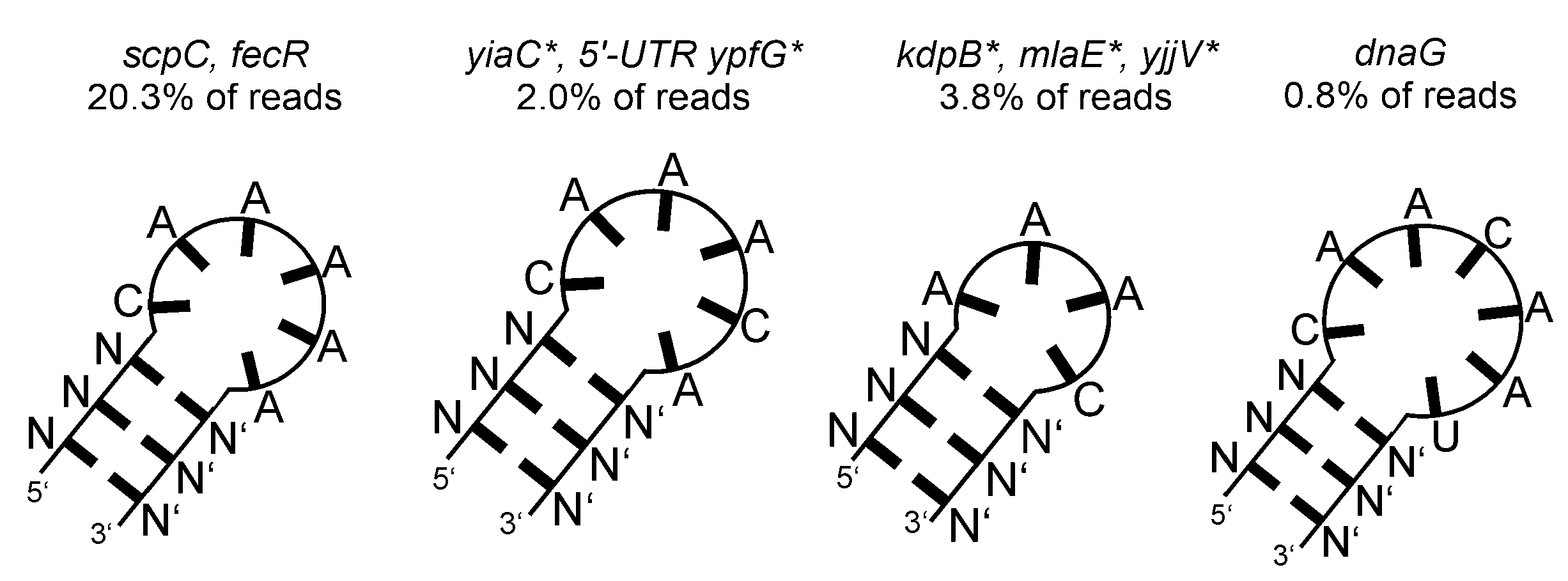
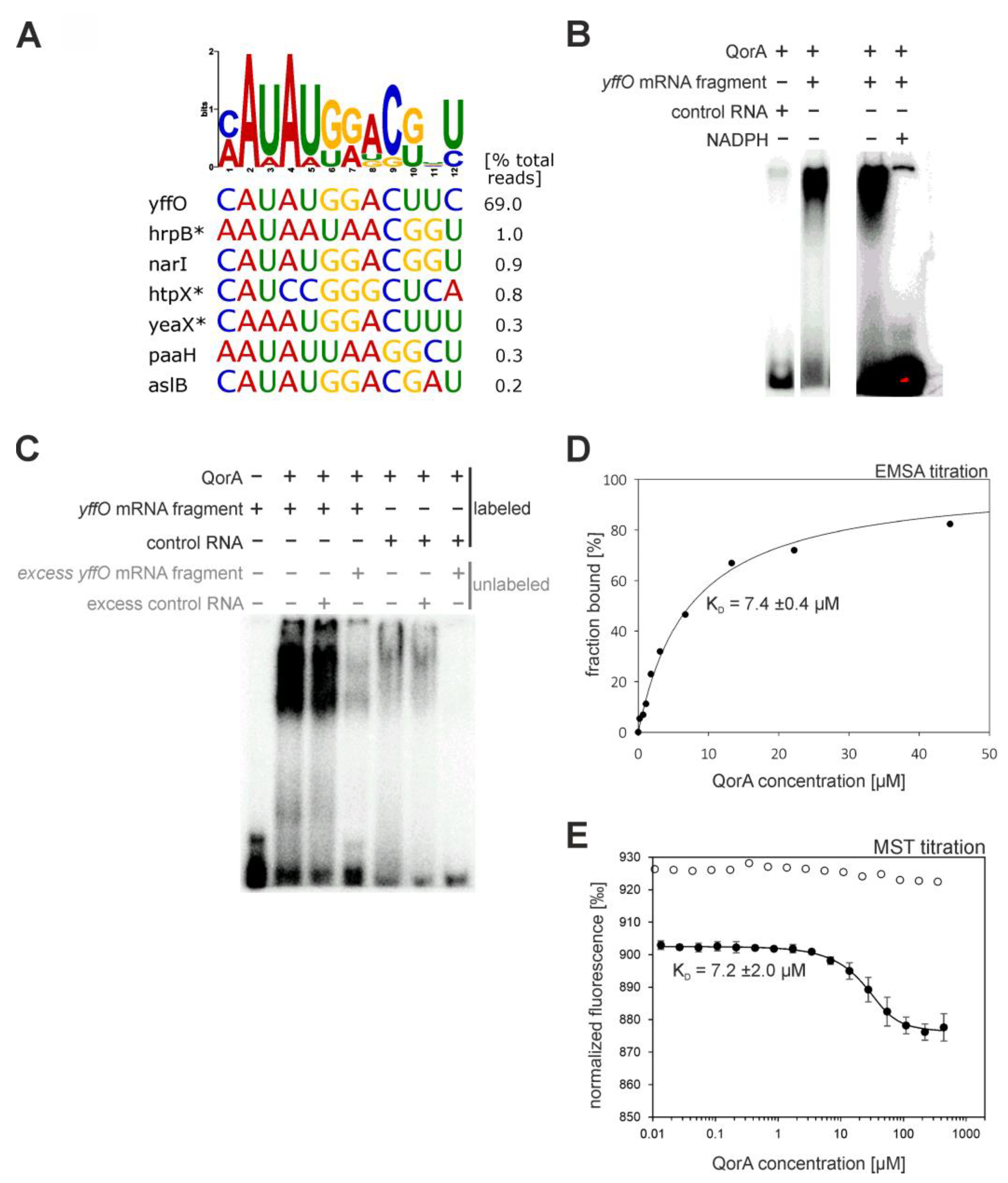
- Quinone Oxidoreductase (QorA)
3. Discussion
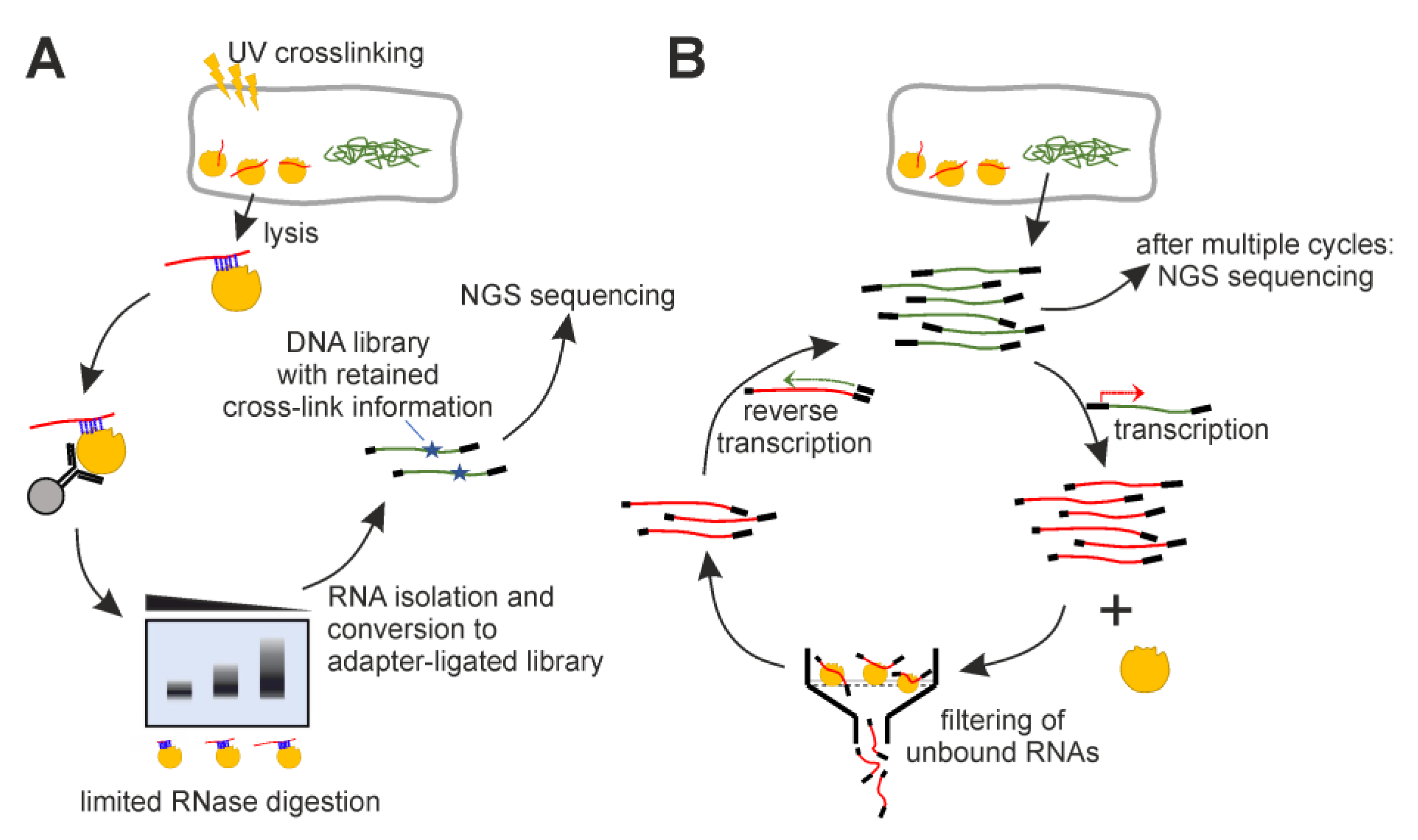
4. Materials and Methods
4.1. Cloning and Protein Purification
4.2. iCLIP
4.3. Genomic SELEX
4.4. Sequencing and Processing of Reads
4.5. Cluster Calling in Mapped Genome Data/Evaluation of Enriched RNAs
4.6. Electrophoretic Mobility Shift Assays
4.7. MST Titration
4.8. Secondary Structure Prediction
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lunde, B.M.; Moore, C.; Varani, G. RNA-binding proteins: Modular design for efficient function. Nat. Rev. Mol. Cell Biol. 2007, 8, 479–490. [Google Scholar] [CrossRef] [PubMed]
- Hacisuleyman, E.; Goff, L.A.; Trapnell, C.; Williams, A.; Henao-Mejia, J.; Sun, L.; McClanahan, P.; Hendrickson, D.G.; Sauvageau, M.; Kelley, D.R.; et al. Topological organization of multichromosomal regions by the long intergenic noncoding RNA Firre. Nat. Struct. Mol. Biol. 2014, 21, 198–206. [Google Scholar] [CrossRef] [PubMed]
- Creamer, K.M.; Lawrence, J.B. XIST RNA: A window into the broader role of RNA in nuclear chromosome architecture. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 2017, 372, 20160360. [Google Scholar] [CrossRef] [PubMed]
- Fuller, G.G.; Han, T.; Freeberg, M.A.; Moresco, J.J.; Ghanbari Niaki, A.; Roach, N.P.; Yates, J.R., 3rd; Myong, S.; Kim, J.K. RNA promotes phase separation of glycolysis enzymes into yeast G bodies in hypoxia. Elife 2020, 9, e48480. [Google Scholar] [CrossRef] [PubMed]
- Ottoz, D.S.M.; Berchowitz, L.E. The role of disorder in RNA binding affinity and specificity. Open Biol. 2020, 10, 200328. [Google Scholar] [CrossRef]
- Mackereth, C.D.; Sattler, M. Dynamics in multi-domain protein recognition of RNA. Curr. Opin. Struct. Biol. 2012, 22, 287–296. [Google Scholar] [CrossRef]
- Lin, Y.; Protter, D.S.W.; Rosen, M.K.; Parker, R. Formation and Maturation of Phase-Separated Liquid Droplets by RNA-Binding Proteins. Mol. Cell 2015, 60, 208–219. [Google Scholar] [CrossRef]
- Hentze, M.W.; Preiss, T. The REM phase of gene regulation. Trends Biochem. Sci. 2010, 35, 423–426. [Google Scholar] [CrossRef]
- Scherrer, T.; Mittal, N.; Janga, S.C.; Gerber, A.P. A screen for RNA-binding proteins in yeast indicates dual functions for many enzymes. PLoS ONE 2010, 5, e15499. [Google Scholar] [CrossRef]
- Castello, A.; Fischer, B.; Eichelbaum, K.; Horos, R.; Beckmann, B.M.; Strein, C.; Davey, N.E.; Humphreys, D.T.; Preiss, T.; Steinmetz, L.M.; et al. Insights into RNA biology from an atlas of mammalian mRNA-binding proteins. Cell 2012, 149, 1393–1406. [Google Scholar] [CrossRef]
- Castello, A.; Horos, R.; Strein, C.; Fischer, B.; Eichelbaum, K.; Steinmetz, L.M.; Krijgsveld, J.; Hentze, M.W. System-wide identification of RNA-binding proteins by interactome capture. Nat. Protoc. 2013, 8, 491–500. [Google Scholar] [CrossRef]
- Tsvetanova, N.G.; Klass, D.M.; Salzman, J.; Brown, P.O. Proteome-wide search reveals unexpected RNA-binding proteins in Saccharomyces cerevisiae. PLoS ONE 2010, 5, e12671. [Google Scholar] [CrossRef] [PubMed]
- Hentze, M.W.; Argos, P. Homology between IRE-BP, a regulatory RNA-binding protein, aconitase, and isopropylmalate isomerase. Nucleic Acids Res. 1991, 19, 1739–1740. [Google Scholar] [CrossRef] [PubMed]
- Constable, A.; Quick, S.; Gray, N.K.; Hentze, M.W. Modulation of the RNA-binding activity of a regulatory protein by iron in vitro: Switching between enzymatic and genetic function? Proc. Natl. Acad. Sci. USA 1992, 89, 4554–4558. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Green, M.R. Sequence-specific binding of transfer RNA by glyceraldehyde-3-phosphate dehydrogenase. Science 1993, 259, 365–368. [Google Scholar] [CrossRef] [PubMed]
- Nagy, E.; Rigby, W.F. Glyceraldehyde-3-phosphate dehydrogenase selectively binds AU-rich RNA in the NAD(+)-binding region (Rossmann fold). J. Biol. Chem. 1995, 270, 2755–2763. [Google Scholar] [CrossRef]
- McGowan, K.; Pekala, P.H. Dehydrogenase binding to the 3′-untranslated region of GLUT1 mRNA. Biochem. Biophys. Res. Commun. 1996, 221, 42–45. [Google Scholar] [CrossRef]
- Schultz, D.E.; Hardin, C.C.; Lemon, S.M. Specific interaction of glyceraldehyde 3-phosphate dehydrogenase with the 5′-nontranslated RNA of hepatitis A virus. J. Biol. Chem. 1996, 271, 14134–14142. [Google Scholar] [CrossRef]
- De, B.P.; Gupta, S.; Zhao, H.; Drazba, J.A.; Banerjee, A.K. Specific interaction in vitro and in vivo of glyceraldehyde-3-phosphate dehydrogenase and LA protein with cis-acting RNAs of human parainfluenza virus type 3. J. Biol. Chem. 1996, 271, 24728–24735. [Google Scholar] [CrossRef]
- Hentze, M.W. Enzymes as RNA-binding proteins: A role for (di)nucleotide-binding domains? Trends Biochem. Sci. 1994, 19, 101–103. [Google Scholar] [CrossRef]
- Carmona, P.; Rodríguez-Casado, A.; Molina, M. Conformational structure and binding mode of glyceraldehyde-3-phosphate dehydrogenase to tRNA studied by Raman and CD spectroscopy. Biochim. Biophys. Acta BBA-Protein Struct. Mol. Enzymol. 1999, 1432, 222–233. [Google Scholar] [CrossRef] [PubMed]
- Kramer, K.; Sachsenberg, T.; Beckmann, B.M.; Qamar, S.; Boon, K.-L.; Hentze, M.W.; Kohlbacher, O.; Urlaub, H. Photo-cross-linking and high-resolution mass spectrometry for assignment of RNA-binding sites in RNA-binding proteins. Nat. Methods 2014, 11, 1064–1070. [Google Scholar] [CrossRef] [PubMed]
- White, M.R.; Garcin, E.D. The sweet side of RNA regulation: Glyceraldehyde-3-phosphate dehydrogenase as a noncanonical RNA-binding protein. Wiley Interdiscip. Rev. RNA 2016, 7, 53–70. [Google Scholar] [CrossRef] [PubMed]
- Chu, E.; Voeller, D.; Koeller, D.M.; Drake, J.C.; Takimoto, C.H.; Maley, G.F.; Maley, F.; Allegra, C.J. Identification of an RNA binding site for human thymidylate synthase. Proc. Natl. Acad. Sci. USA 1993, 90, 517–521. [Google Scholar] [CrossRef]
- Chu, E.; Takimoto, C.H.; Voeller, D.; Grem, J.L.; Allegra, C.J. Specific binding of human dihydrofolate reductase protein to dihydrofolate reductase messenger RNA in vitro. Biochemistry 1993, 32, 4756–4760. [Google Scholar] [CrossRef]
- Nanbu, R.; Kubo, T.; Hashimoto, T.; Natori, S. Purification of an AU-rich RNA binding protein from Sarcophaga peregrina (flesh fly) and its identification as a Thiolase. J. Biochem. 1993, 114, 432–437. [Google Scholar] [CrossRef]
- Preiss, T.; Hall, A.G.; Lightowlers, R.N. Identification of bovine glutamate dehydrogenase as an RNA-binding protein. J. Biol. Chem. 1993, 268, 24523–24526. [Google Scholar] [CrossRef]
- Elzinga, S.D.; Bednarz, A.L.; van Oosterum, K.; Dekker, P.J.; Grivell, L.A. Yeast mitochondrial NAD(+)-dependent isocitrate dehydrogenase is an RNA-binding protein. Nucleic Acids Res. 1993, 21, 5328–5331. [Google Scholar] [CrossRef]
- Pioli, P.A.; Hamilton, B.J.; Connolly, J.E.; Brewer, G.; Rigby, W.F.C. Lactate dehydrogenase is an AU-rich element-binding protein that directly interacts with AUF1. J. Biol. Chem. 2002, 277, 35738–35745. [Google Scholar] [CrossRef]
- Kiri, A.; Goldspink, G. RNA-protein interactions of the 3′ untranslated regions of myosin heavy chain transcripts. J. Muscle Res. Cell Motil. 2002, 23, 119–129. [Google Scholar] [CrossRef]
- Shetty, S.; Muniyappa, H.; Halady, P.K.S.; Idell, S. Regulation of urokinase receptor expression by phosphoglycerate kinase. Am. J. Respir. Cell Mol. Biol. 2004, 31, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Urdaneta, E.C.; Vieira-Vieira, C.H.; Hick, T.; Wessels, H.-H.; Figini, D.; Moschall, R.; Medenbach, J.; Ohler, U.; Granneman, S.; Selbach, M.; et al. Purification of cross-linked RNA-protein complexes by phenol-toluol extraction. Nat. Commun. 2019, 10, 990. [Google Scholar] [CrossRef] [PubMed]
- Queiroz, R.M.L.; Smith, T.; Villanueva, E.; Marti-Solano, M.; Monti, M.; Pizzinga, M.; Mirea, D.-M.; Ramakrishna, M.; Harvey, R.F.; Dezi, V.; et al. Comprehensive identification of RNA-protein interactions in any organism using orthogonal organic phase separation (OOPS). Nat. Biotechnol. 2019, 37, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Shchepachev, V.; Bresson, S.; Spanos, C.; Petfalski, E.; Fischer, L.; Rappsilber, J.; Tollervey, D. Defining the RNA interactome by total RNA-associated protein purification. Mol. Syst. Biol. 2019, 15, e8689. [Google Scholar] [CrossRef] [PubMed]
- Chandran, V.; Luisi, B.F. Recognition of enolase in the Escherichia coli RNA degradosome. J. Mol. Biol. 2006, 358, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Deppert, W.R.; Normann, J.; Wagner, E. Adenylate kinase from plant tissues. J. Chromatogr. A 1992, 625, 13–19. [Google Scholar] [CrossRef]
- Schlattner, U.; Wagner, E.; Greppin, H.; Bonzon, M. Binding of adenylate kinase to RNA. Biochem. Biophys. Res. Commun. 1995, 217, 509–514. [Google Scholar] [CrossRef]
- Voeller, D.M.; Changchien, L.M.; Maley, G.F.; Maley, F.; Takechi, T.; Turner, R.E.; Montfort, W.R.; Allegra, C.J.; Chu, E. Characterization of a specific interaction between Escherichia coli thymidylate synthase and Escherichia coli thymidylate synthase mRNA. Nucleic Acids Res. 1995, 23, 869–875. [Google Scholar] [CrossRef][Green Version]
- Pérez-Arellano, I.; Rubio, V.; Cervera, J. Dissection of Escherichia coli glutamate 5-kinase: Functional impact of the deletion of the PUA domain. FEBS Lett. 2005, 579, 6903–6908. [Google Scholar] [CrossRef]
- Chen, Y.-H.; Liu, S.-J.; Gao, M.-M.; Zeng, T.; Lin, G.-W.; Tan, N.-N.; Tang, H.-L.; Lu, P.; Su, T.; Sun, W.-W.; et al. MDH2 is an RNA binding protein involved in downregulation of sodium channel Scn1a expression under seizure condition. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1492–1499. [Google Scholar] [CrossRef]
- Benjamin, J.-A.M.; Massé, E. The iron-sensing aconitase B binds its own mRNA to prevent sRNA-induced mRNA cleavage. Nucleic Acids Res. 2014, 42, 10023–10036. [Google Scholar] [CrossRef]
- Tang, A.; Curthoys, N.P. Identification of zeta-crystallin/NADPH:quinone reductase as a renal glutaminase mRNA pH response element-binding protein. J. Biol. Chem. 2001, 276, 21375–21380. [Google Scholar] [CrossRef]
- Pickett, G.G.; Peabody, D.S. Encapsidation of heterologous RNAs by bacteriophage MS2 coat protein. Nucleic Acids Res. 1993, 21, 4621–4626. [Google Scholar] [CrossRef] [PubMed]
- Holmqvist, E.; Wright, P.R.; Li, L.; Bischler, T.; Barquist, L.; Reinhardt, R.; Backofen, R.; Vogel, J. Global RNA recognition patterns of post-transcriptional regulators Hfq and CsrA revealed by UV crosslinking in vivo. EMBO J. 2016, 35, 991–1011. [Google Scholar] [CrossRef] [PubMed]
- Potts, A.H.; Vakulskas, C.A.; Pannuri, A.; Yakhnin, H.; Babitzke, P.; Romeo, T. Global role of the bacterial post-transcriptional regulator CsrA revealed by integrated transcriptomics. Nat. Commun. 2017, 8, 1596. [Google Scholar] [CrossRef] [PubMed]
- Holmqvist, E.; Li, L.; Bischler, T.; Barquist, L.; Vogel, J. Global Maps of ProQ Binding In Vivo Reveal Target Recognition via RNA Structure and Stability Control at mRNA 3′ Ends. Mol. Cell 2018, 70, 971–982.e6. [Google Scholar] [CrossRef]
- Bathke, J.; Gauernack, A.S.; Rupp, O.; Weber, L.; Preusser, C.; Lechner, M.; Rossbach, O.; Goesmann, A.; Evguenieva-Hackenberg, E.; Klug, G. iCLIP analysis of RNA substrates of the archaeal exosome. BMC Genom. 2020, 21, 797. [Google Scholar] [CrossRef]
- Buchbender, A.; Mutter, H.; Sutandy, F.X.R.; Körtel, N.; Hänel, H.; Busch, A.; Ebersberger, S.; König, J. Improved library preparation with the new iCLIP2 protocol. Methods 2020, 178, 33–48. [Google Scholar] [CrossRef]
- Datsenko, K.A.; Wanner, B.L. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. USA 2000, 97, 6640–6645. [Google Scholar] [CrossRef]
- Yu, D.; Ellis, H.M.; Lee, E.C.; Jenkins, N.A.; Copeland, N.G.; Court, D.L. An efficient recombination system for chromosome engineering in Escherichia coli. Proc. Natl. Acad. Sci. USA 2000, 97, 5978–5983. [Google Scholar] [CrossRef]
- Roberts, E.; Sethi, A.; Montoya, J.; Woese, C.R.; Luthey-Schulten, Z. Molecular signatures of ribosomal evolution. Proc. Natl. Acad. Sci. USA 2008, 105, 13953–13958. [Google Scholar] [CrossRef] [PubMed]
- Muffler, A.; Fischer, D.; Hengge-Aronis, R. The RNA-binding protein HF-I, known as a host factor for phage Qbeta RNA replication, is essential for rpoS translation in Escherichia coli. Genes Dev. 1996, 10, 1143–1151. [Google Scholar] [CrossRef] [PubMed]
- Van Nostrand, E.L.; Pratt, G.A.; Shishkin, A.A.; Gelboin-Burkhart, C.; Fang, M.Y.; Sundararaman, B.; Blue, S.M.; Nguyen, T.B.; Surka, C.; Elkins, K.; et al. Robust transcriptome-wide discovery of RNA-binding protein binding sites with enhanced CLIP (eCLIP). Nat. Methods 2016, 13, 508–514. [Google Scholar] [CrossRef]
- Shtatland, T.; Gill, S.C.; Javornik, B.E.; Johansson, H.E.; Singer, B.S.; Uhlenbeck, O.C.; Zichi, D.A.; Gold, L. Interactions of Escherichia coli RNA with bacteriophage MS2 coat protein: Genomic SELEX. Nucleic Acids Res. 2000, 28, E93. [Google Scholar] [CrossRef] [PubMed]
- Romaniuk, P.J.; Lowary, P.; Wu, H.N.; Stormo, G.; Uhlenbeck, O.C. RNA binding site of R17 coat protein. Biochemistry 1987, 26, 1563–1568. [Google Scholar] [CrossRef]
- Lowary, P.T.; Uhlenbeck, O.C. An RNA mutation that increases the affinity of an RNA-protein interaction. Nucleic Acids Res. 1987, 15, 10483–10493. [Google Scholar] [CrossRef]
- Young, T.A.; Skordalakes, E.; Marqusee, S. Comparison of proteolytic susceptibility in phosphoglycerate kinases from yeast and E. coli: Modulation of conformational ensembles without altering structure or stability. J. Mol. Biol. 2007, 368, 1438–1447. [Google Scholar] [CrossRef]
- Garcin, E.D. GAPDH as a model non-canonical AU-rich RNA binding protein. Semin. Cell Dev. Biol. 2019, 86, 162–173. [Google Scholar] [CrossRef]
- Bonafé, N.; Gilmore-Hebert, M.; Folk, N.L.; Azodi, M.; Zhou, Y.; Chambers, S.K. Glyceraldehyde-3-phosphate dehydrogenase binds to the AU-Rich 3′ untranslated region of colony-stimulating factor-1 (CSF-1) messenger RNA in human ovarian cancer cells: Possible role in CSF-1 posttranscriptional regulation and tumor phenotype. Cancer Res. 2005, 65, 3762–3771. [Google Scholar] [CrossRef]
- White, M.R.; Khan, M.M.; Deredge, D.; Ross, C.R.; Quintyn, R.; Zucconi, B.E.; Wysocki, V.H.; Wintrode, P.L.; Wilson, G.M.; Garcin, E.D. A dimer interface mutation in glyceraldehyde-3-phosphate dehydrogenase regulates its binding to AU-rich RNA. J. Biol. Chem. 2015, 290, 1770–1785. [Google Scholar] [CrossRef]
- Cho, H.-H.; Cahill, C.M.; Vanderburg, C.R.; Scherzer, C.R.; Wang, B.; Huang, X.; Rogers, J.T. Selective translational control of the Alzheimer amyloid precursor protein transcript by iron regulatory protein-1. J. Biol. Chem. 2010, 285, 31217–31232. [Google Scholar] [CrossRef]
- Zhang, Y.; Yang, S.; Liu, M.; Song, C.; Wu, N.; Ling, P.; Chu, E.; Lin, X. Interaction between thymidylate synthase and its cognate mRNA in zebrafish embryos. PLoS ONE 2010, 5, e10618. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Mizunuma, N.; Chen, T.; Copur, S.M.; Maley, G.F.; Liu, J.; Maley, F.; Chu, E. In vitro selection of an RNA sequence that interacts with high affinity with thymidylate synthase. Nucleic Acids Res. 2000, 28, 4266–4274. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Guest, J.R. Direct evidence for mRNA binding and post-transcriptional regulation by Escherichia coli aconitases. Microbiology 1999, 145 Pt 11, 3069–3079. [Google Scholar] [CrossRef] [PubMed]
- Meydan, S.; Klepacki, D.; Karthikeyan, S.; Margus, T.; Thomas, P.; Jones, J.E.; Khan, Y.; Briggs, J.; Dinman, J.D.; Vázquez-Laslop, N.; et al. Programmed Ribosomal Frameshifting Generates a Copper Transporter and a Copper Chaperone from the Same Gene. Mol. Cell 2017, 65, 207–219. [Google Scholar] [CrossRef]
- Macomber, L.; Imlay, J.A. The iron-sulfur clusters of dehydratases are primary intracellular targets of copper toxicity. Proc. Natl. Acad. Sci. USA 2009, 106, 8344–8349. [Google Scholar] [CrossRef] [PubMed]
- Aravind, L.; Koonin, E.V. Novel predicted RNA-binding domains associated with the translation machinery. J. Mol. Evol. 1999, 48, 291–302. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Arellano, I.; Gallego, J.; Cervera, J. The PUA domain-A structural and functional overview. FEBS J. 2007, 274, 4972–4984. [Google Scholar] [CrossRef]
- Marco-Marín, C.; Gil-Ortiz, F.; Pérez-Arellano, I.; Cervera, J.; Fita, I.; Rubio, V. A novel two-domain architecture within the amino acid kinase enzyme family revealed by the crystal structure of Escherichia coli glutamate 5-kinase. J. Mol. Biol. 2007, 367, 1431–1446. [Google Scholar] [CrossRef]
- Huang, Q.L.; Russell, P.; Stone, S.H.; Zigler, J.S. Zeta-crystallin, a novel lens protein from the guinea pig. Curr. Eye Res. 1987, 6, 725–732. [Google Scholar] [CrossRef]
- Rao, P.V.; Krishna, C.M.; Zigler, J.S., Jr. Identification and characterization of the enzymatic activity of zeta-crystallin from guinea pig lens. A novel NADPH:quinone oxidoreductase. J. Biol. Chem. 1992, 267, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Fernández, M.R.; Porté, S.; Crosas, E.; Barberà, N.; Farrés, J.; Biosca, J.A.; Parés, X. Human and yeast zeta-crystallins bind AU-rich elements in RNA. Cell. Mol. Life Sci. CMLS 2007, 64, 1419–1427. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, H.; Lee, Y.J.; Curthoys, N.P. Renal response to metabolic acidosis: Role of mRNA stabilization. Kidney Int. 2008, 73, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Lapucci, A.; Lulli, M.; Amedei, A.; Papucci, L.; Witort, E.; Di Gesualdo, F.; Bertolini, F.; Brewer, G.; Nicolin, A.; Bevilacqua, A.; et al. zeta-Crystallin is a bcl-2 mRNA binding protein involved in bcl-2 overexpression in T-cell acute lymphocytic leukemia. FASEB J. 2010, 24, 1852–1865. [Google Scholar] [CrossRef] [PubMed]
- Thorn, J.M.; Barton, J.D.; Dixon, N.E.; Ollis, D.L.; Edwards, K.J. Crystal structure of Escherichia coli QOR quinone oxidoreductase complexed with NADPH. J. Mol. Biol. 1995, 249, 785–799. [Google Scholar] [CrossRef] [PubMed]
- Sulzenbacher, G.; Roig-Zamboni, V.; Pagot, F.; Grisel, S.; Salomoni, A.; Valencia, C.; Campanacci, V.; Vincentelli, R.; Tegoni, M.; Eklund, H.; et al. Structure of Escherichia coli YhdH, a putative quinone oxidoreductase. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004, 60 Pt 10, 1855–1862. [Google Scholar] [CrossRef]
- Rudd, K.E. Novel intergenic repeats of Escherichia coli K-12. Res. Microbiol. 1999, 150, 653–664. [Google Scholar] [CrossRef]
- Casjens, S. Prophages and bacterial genomics: What have we learned so far? Mol. Microbiol. 2003, 49, 277–300. [Google Scholar] [CrossRef]
- Wang, X.; Kim, Y.; Ma, Q.; Hong, S.H.; Pokusaeva, K.; Sturino, J.M.; Wood, T.K. Cryptic prophages help bacteria cope with adverse environments. Nat. Commun. 2010, 1, 147. [Google Scholar] [CrossRef]
- Hayashi, K.; Morooka, N.; Yamamoto, Y.; Fujita, K.; Isono, K.; Choi, S.; Ohtsubo, E.; Baba, T.; Wanner, B.L.; Mori, H.; et al. Highly accurate genome sequences of Escherichia coli K-12 strains MG1655 and W3110. Mol. Syst. Biol. 2006, 2, 2006.0007. [Google Scholar] [CrossRef]
- Zhao, H.; Finch, C.J.; Sequeira, R.D.; Johnson, B.A.; Johnson, J.E.; Casjens, S.R.; Tang, L. Crystal structure of the DNA-recognition component of the bacterial virus Sf6 genome-packaging machine. Proc. Natl. Acad. Sci. USA 2010, 107, 1971–1976. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Kamau, Y.N.; Christensen, T.E.; Tang, L. Structural and functional studies of the phage Sf6 terminase small subunit reveal a DNA-spooling device facilitated by structural plasticity. J. Mol. Biol. 2012, 423, 413–426. [Google Scholar] [CrossRef] [PubMed]
- Reeves, R.; Nissen, M.S. The AT-DNA-binding domain of mammalian high mobility group I chromosomal proteins. A novel peptide motif for recognizing DNA structure. J. Biol. Chem. 1990, 265, 8573–8582. [Google Scholar] [CrossRef] [PubMed]
- Stafford, G.P.; Ogi, T.; Hughes, C. Binding and transcriptional activation of non-flagellar genes by the Escherichia coli flagellar master regulator FlhD2C2. Microbiology 2005, 151 Pt 6, 1779–1788. [Google Scholar] [CrossRef]
- Maciag, A.; Peano, C.; Pietrelli, A.; Egli, T.; Bellis, G.d.; Landini, P. In vitro transcription profiling of the σS subunit of bacterial RNA polymerase: Re-definition of the σS regulon and identification of σS-specific promoter sequence elements. Nucleic Acids Res. 2011, 39, 5338–5355. [Google Scholar] [CrossRef]
- Tanaka, K.; Takayanagi, Y.; Fujita, N.; Ishihama, A.; Takahashi, H. Heterogeneity of the principal sigma factor in Escherichia coli: The rpoS gene product, sigma 38, is a second principal sigma factor of RNA polymerase in stationary-phase Escherichia coli. Proc. Natl. Acad. Sci. USA 1993, 90, 3511–3515. [Google Scholar] [CrossRef]
- Dimitrova-Paternoga, L.; Haubrich, K.; Sun, M.; Ephrussi, A.; Hennig, J. Validation and classification of RNA binding proteins identified by mRNA interactome capture. RNA 2021, 27, 1173–1185. [Google Scholar]
- Altuvia, S.; Storz, G.; Papenfort, K. Cross-Regulation between Bacteria and Phages at a Posttranscriptional Level. Microbiol. Spectr. 2018, 6. [Google Scholar] [CrossRef]
- Maruyama, A.; Kumagai, Y.; Morikawa, K.; Taguchi, K.; Hayashi, H.; Ohta, T. Oxidative-stress-inducible qorA encodes an NADPH-dependent quinone oxidoreductase catalysing a one-electron reduction in Staphylococcus aureus. Microbiology 2003, 149 Pt 2, 389–398. [Google Scholar] [CrossRef]
- Kitagawa, M.; Ara, T.; Arifuzzaman, M.; Ioka-Nakamichi, T.; Inamoto, E.; Toyonaga, H.; Mori, H. Complete set of ORF clones of Escherichia coli ASKA library (A Complete Set of E. coli K-12 ORF Archive): Unique Resources for Biological Research. DNA Res. 2005, 12, 291–299. [Google Scholar] [CrossRef]
- Rohweder, B.; Semmelmann, F.; Endres, C.; Sterner, R. Standardized cloning vectors for protein production and generation of large gene libraries in Escherichia coli. Biotechniques 2018, 64, 24–26. [Google Scholar] [CrossRef] [PubMed]
- Gasteiger, E.; Hoogland, C.; Gattiker, A.; Duvaud, S.; Wilkins, M.R.; Appel, R.D.; Bairoch, A. Protein identification and analysis tools on the ExPASy server. In The Proteomics Protocols Handbook; Walker, J.M., Ed.; Humana Press: Totowa, NJ, USA, 2005; pp. 571–607. [Google Scholar]
- Murphy, K.C. Use of bacteriophage lambda recombination functions to promote gene replacement in Escherichia coli. J. Bacteriol. 1998, 180, 2063–2071. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.C.; Ellington, A.D. Automated selection of anti-Protein aptamers. Bioorg. Med. Chem. 2001, 9, 2525–2531. [Google Scholar] [CrossRef] [PubMed]
- Ellington, A.D.; Szostak, J.W. Selection in vitro of single-stranded DNA molecules that fold into specific ligand-binding structures. Nature 1992, 355, 850–852. [Google Scholar] [CrossRef]
- Rio, D.C. Filter-binding assay for analysis of RNA-protein interactions. Cold Spring Harb. Protoc. 2012, 2012, 1078–1081. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. Gigascience 2021, 10, giab008. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Busch, A.; Brüggemann, M.; Ebersberger, S.; Zarnack, K. iCLIP data analysis: A complete pipeline from sequencing reads to RBP binding sites. Methods 2020, 178, 49–62. [Google Scholar] [CrossRef]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef]
- Krakau, S.; Richard, H.; Marsico, A. PureCLIP: Capturing target-specific protein-RNA interaction footprints from single-nucleotide CLIP-seq data. Genome Biol. 2017, 18, 240. [Google Scholar] [CrossRef] [PubMed]
- Bailey, T.L.; Johnson, J.; Grant, C.E.; Noble, W.S. The MEME Suite. Nucleic Acids Res. 2015, 43, W39–W49. [Google Scholar] [CrossRef] [PubMed]
- Gruber, A.R.; Lorenz, R.; Bernhart, S.H.; Neuböck, R.; Hofacker, I.L. The Vienna RNA websuite. Nucleic Acids Res. 2008, 36, W70–W74. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Hamada, M.; Asai, K.; Mituyama, T. CENTROIDFOLD: A web server for RNA secondary structure prediction. Nucleic Acids Res. 2009, 37, W277–W280. [Google Scholar] [CrossRef]
- Sato, K.; Kato, Y.; Hamada, M.; Akutsu, T.; Asai, K. IPknot: Fast and accurate prediction of RNA secondary structures with pseudoknots using integer programming. Bioinformatics 2011, 27, i85–i93. [Google Scholar] [CrossRef]
- Mori, R.; Hamada, M.; Asai, K. Efficient calculation of exact probability distributions of integer features on RNA secondary structures. BMC Genom. 2014, 15 (Suppl. S10), S6. [Google Scholar] [CrossRef]
- Hagio, T.; Sakuraba, S.; Iwakiri, J.; Mori, R.; Asai, K. Capturing alternative secondary structures of RNA by decomposition of base-pairing probabilities. BMC Bioinform. 2018, 19 (Suppl. S1), 38. [Google Scholar] [CrossRef]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022, 50, D439–D444. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Rice, P.; Longden, I.; Bleasby, A. EMBOSS: The European Molecular Biology Open Software Suite. Trends Genet. TIG 2000, 16, 276–277. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metabolic Pathway | Enzyme | Literature References a |
|---|---|---|
| Glycolysis | Pyruvate kinase (PykF) | - |
| Phosphoglycerate kinase (Pgk) | binds coding mRNA regions (euk) [31] | |
| Glyceraldehyde-3-phosphate dehydrogenase (GapA) | binds AU-rich elements (euk) [16,17,18,19] | |
| Enolase (Eno) | part of RNA degradosome (prok) [35] | |
| Phosphoglycerate mutase (GpmA) | - | |
| Pentose phosphate pathway | Transaldolase A (TalA) | - |
| Transaldolase B (TalB) | - | |
| Ribose-5-phosphate isomerase (RpiA) | - | |
| Lipopolysaccharides | 2-dehydro-3-deoxyphosphooctonate aldolase (KdsA) | - |
| Nucleotide metabolism | Uracil phosphoribosyltransferase (Upp) | - |
| Adenylate kinase (Adk) | general RNA co-purification (euk) [36,37] | |
| Thymidylate synthase (ThyA) | binds thyA mRNA (prok) [38] | |
| Amino acid metabolism | L-asparaginase (AnsB) | - |
| Glutamate-5-kinase (ProB) | Features a PUA domain (prok) [39] | |
| Citric acid cycle | Malate dehydrogenase (Mdh) | binds 3′ UTRs of mRNA (euk) [40] |
| Aconitase (AcnB) | binds acnB mRNA (prok) [41] | |
| Oxidoreductases | Superoxide dismutase (SodA) | - |
| Quinone oxidoreductase (QorA) | binds pH-responsive elements (euk) [42] | |
| Pyridoxine/pyridoxamine-5-phosphate oxidase (PdxH) | - | |
| Control | Bacteriophage MS2 coat protein (no metabolic enzyme) | binds phage-specific stem loops (prok) [43] |
| Protein | Description of Enriched RNA Features | Predicted RNA Secondary Structure a |
|---|---|---|
| pyruvate kinase (PykF) | various mRNAs/asRNAs with no apparent communal attribute | - |
| phosphoglycerate kinase (Pgk) | poorly defined hairpins, showing A-rich loops of variable length as common feature |  |
| glyceraldehyde-3-phosphate dehydrogenase (GapA) | poorly defined, AU-rich sequence stretches |  |
| thymidylate synthase (ThyA) | hairpin with strictly conserved sequence GC(AGA)GU (brackets indicating triloop) |  |
| aconitase (AcnB) | poorly defined stem-loops with 3′-adjacent U-rich stretches |  |
| glutamate-5-kinase (ProB) | small number of specific fragments (two mRNAs and one asRNA) | - |
| quinone oxidoreductase (QorA) | few specific mRNAs/asRNAs with a potential motif (see Figure 8A). top-scoring: yffO mRNA fragment. |  |
| positive control MS2 phage coat protein | bacteriophage operator hairpin-like motif NANN(ANCA)N′N′N′ (N and N′ indicate complementary bases, brackets indicate loop) |  |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Klein, T.; Funke, F.; Rossbach, O.; Lehmann, G.; Vockenhuber, M.; Medenbach, J.; Suess, B.; Meister, G.; Babinger, P. Investigating the Prevalence of RNA-Binding Metabolic Enzymes in E. coli. Int. J. Mol. Sci. 2023, 24, 11536. https://doi.org/10.3390/ijms241411536
Klein T, Funke F, Rossbach O, Lehmann G, Vockenhuber M, Medenbach J, Suess B, Meister G, Babinger P. Investigating the Prevalence of RNA-Binding Metabolic Enzymes in E. coli. International Journal of Molecular Sciences. 2023; 24(14):11536. https://doi.org/10.3390/ijms241411536
Chicago/Turabian StyleKlein, Thomas, Franziska Funke, Oliver Rossbach, Gerhard Lehmann, Michael Vockenhuber, Jan Medenbach, Beatrix Suess, Gunter Meister, and Patrick Babinger. 2023. "Investigating the Prevalence of RNA-Binding Metabolic Enzymes in E. coli" International Journal of Molecular Sciences 24, no. 14: 11536. https://doi.org/10.3390/ijms241411536
APA StyleKlein, T., Funke, F., Rossbach, O., Lehmann, G., Vockenhuber, M., Medenbach, J., Suess, B., Meister, G., & Babinger, P. (2023). Investigating the Prevalence of RNA-Binding Metabolic Enzymes in E. coli. International Journal of Molecular Sciences, 24(14), 11536. https://doi.org/10.3390/ijms241411536