
Development of a Simple Direct and Hot-Start PCR Using Escherichia coli-Expressing Taq DNA Polymerase

 ,
,
Abstract
1. Introduction
2. Results
2.1. Activity of EcoliTaq in Commercial PCR Buffer
2.2. Activity of EcoliTaq Compared with That of Commercial Taq DNA Polymerase
2.3. Activity of EcoliTaq at Different Storage Temperatures
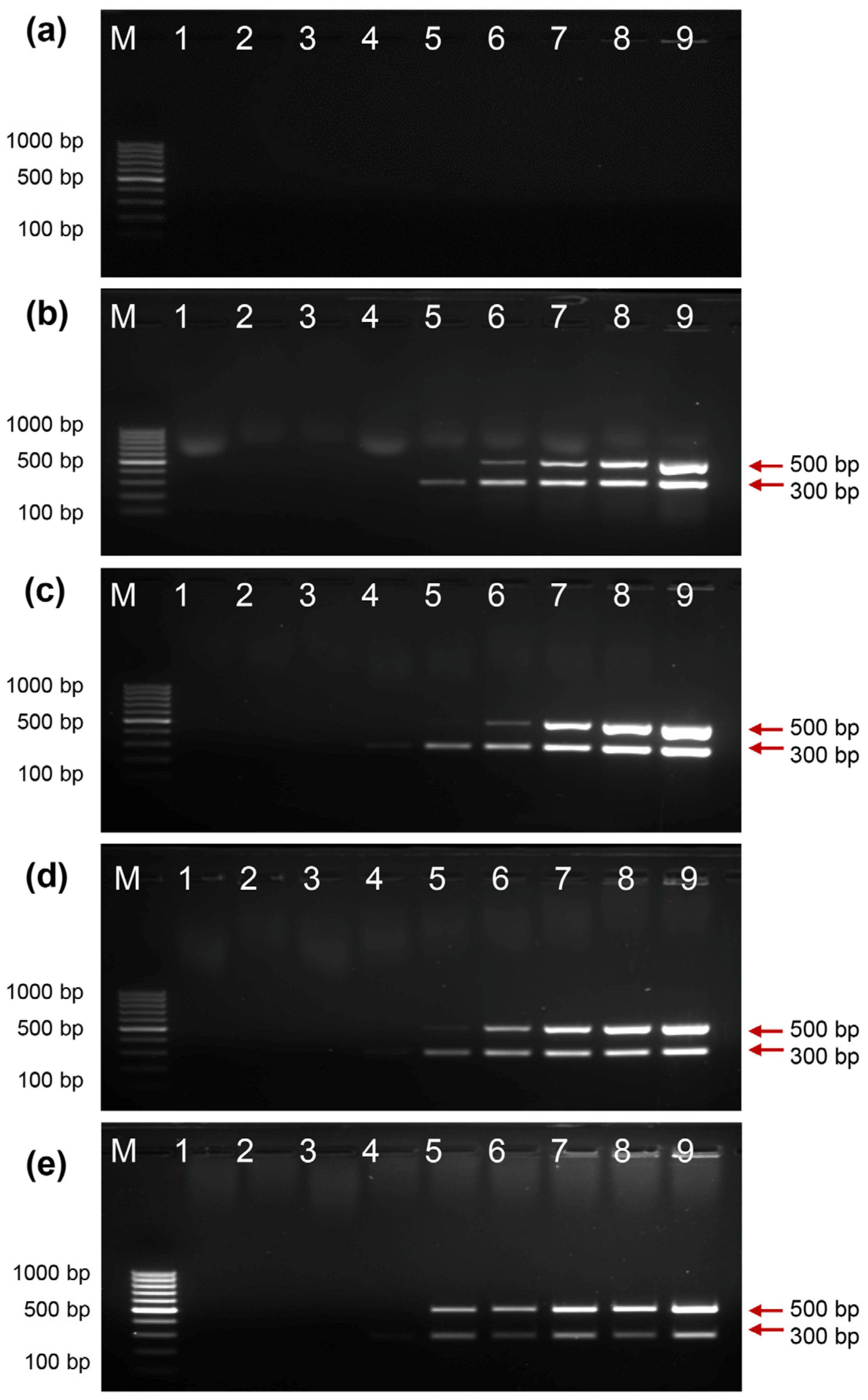
2.4. Activity of EcoliTaq in the Presence of Whole Blood
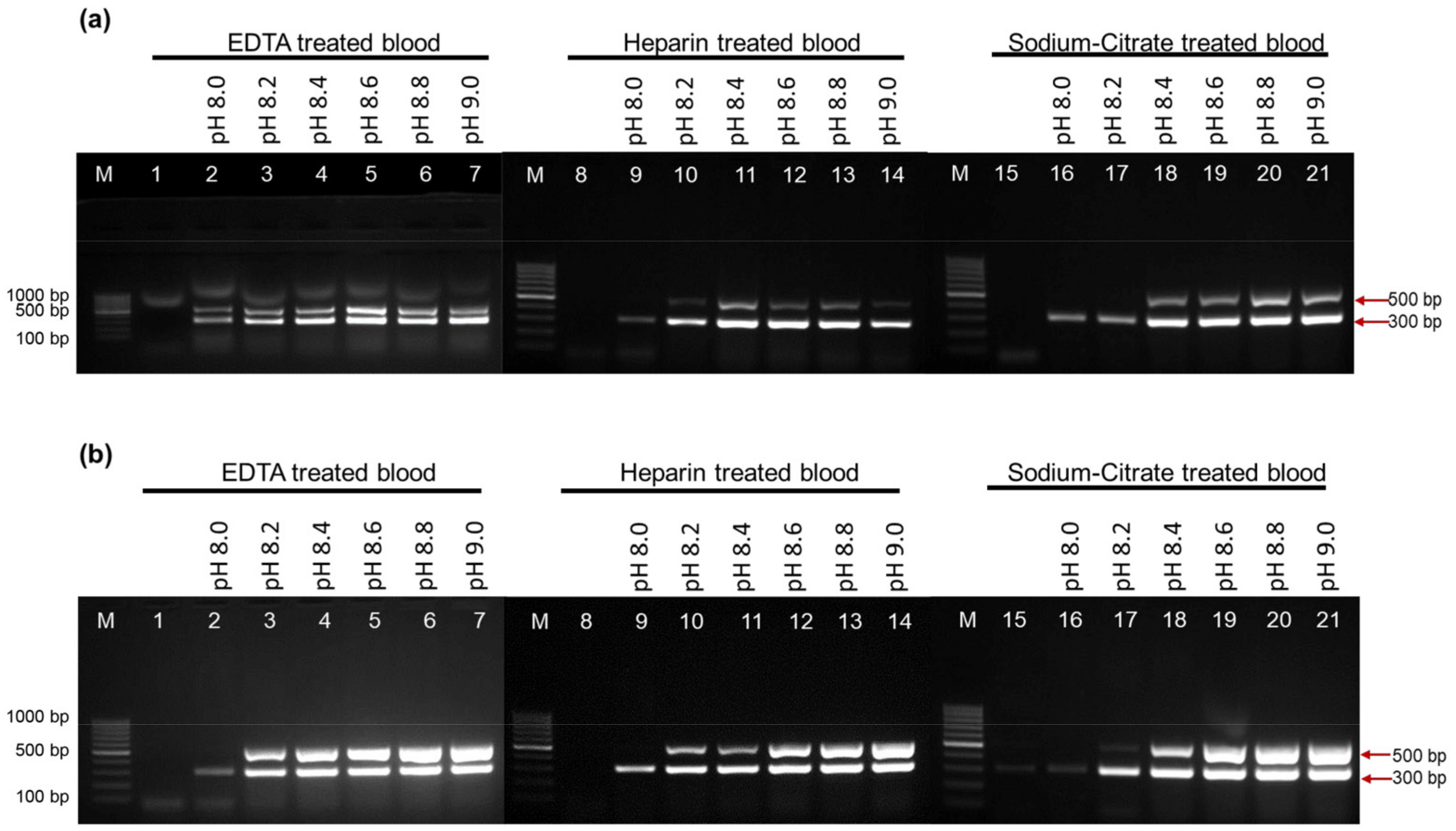
2.5. Activity of EcoliTaq in the Presence of Whole Blood Containing Three Anticoagulants at Different pH Values
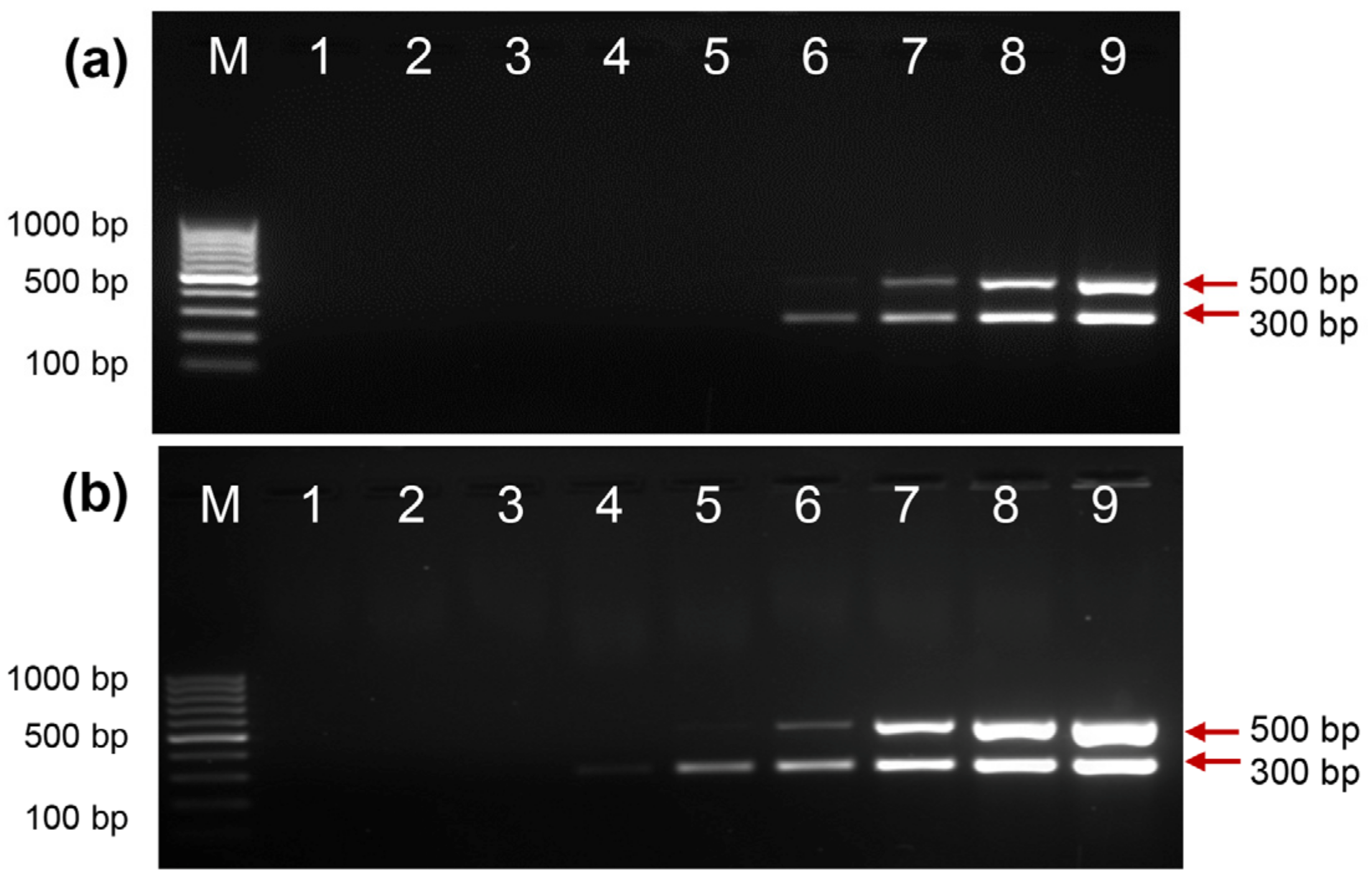
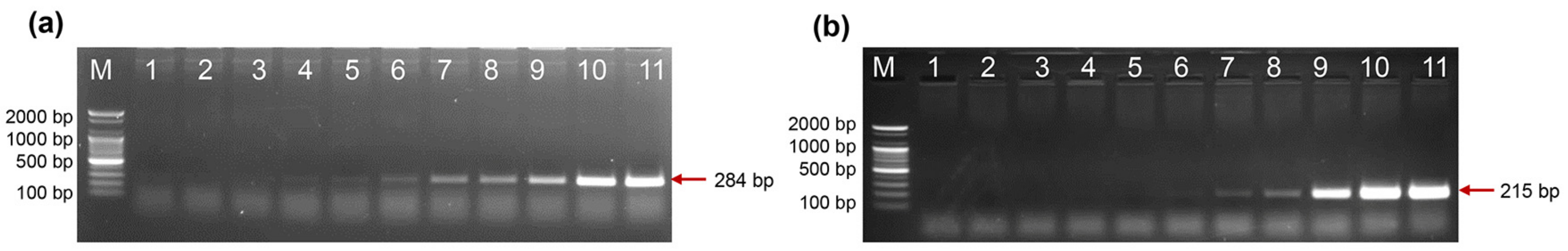
2.6. Direct PCR Detection of Salmonella and Shigella in Whole Blood
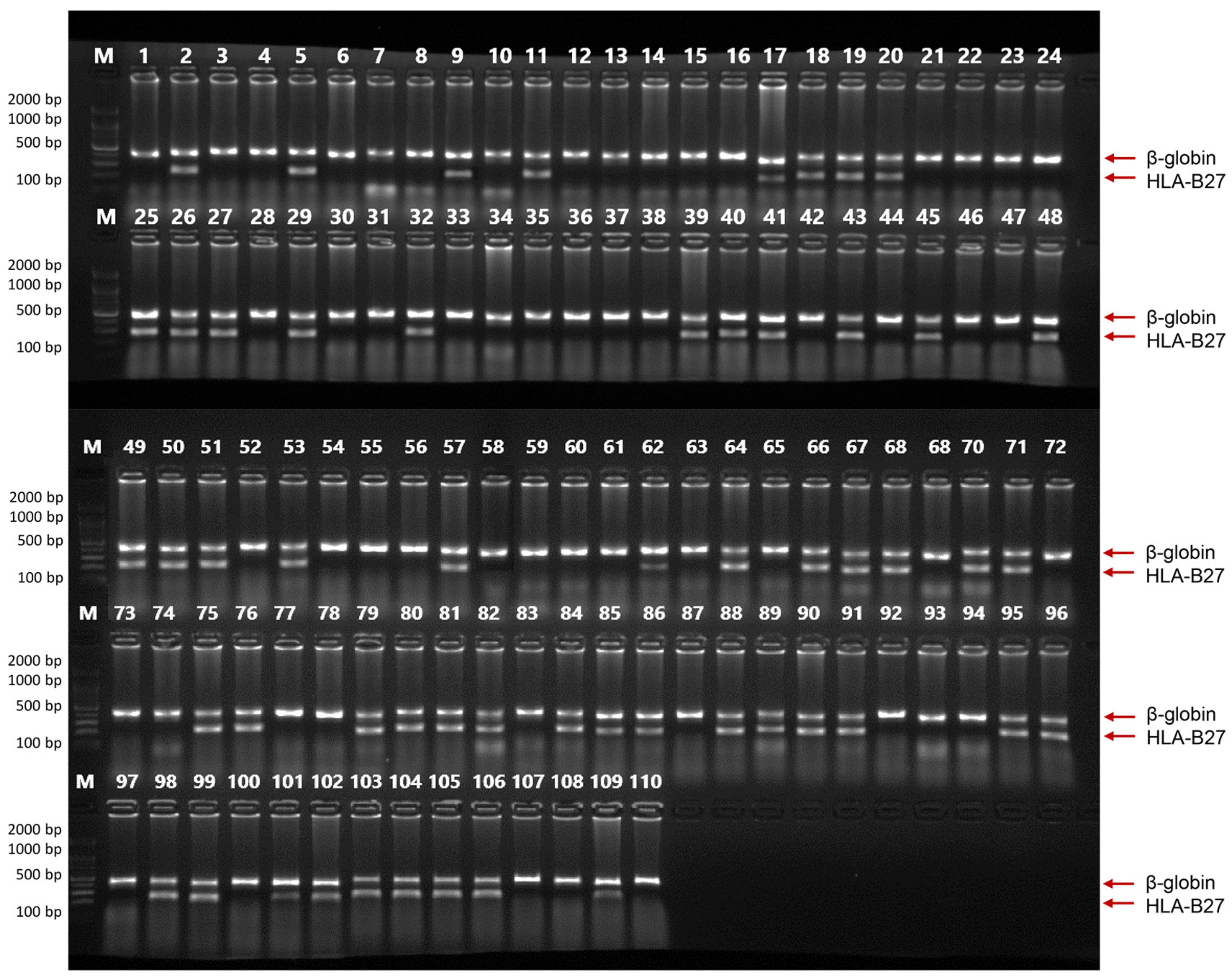
2.7. HLA-B27 Genotyping Using Clinical Samples
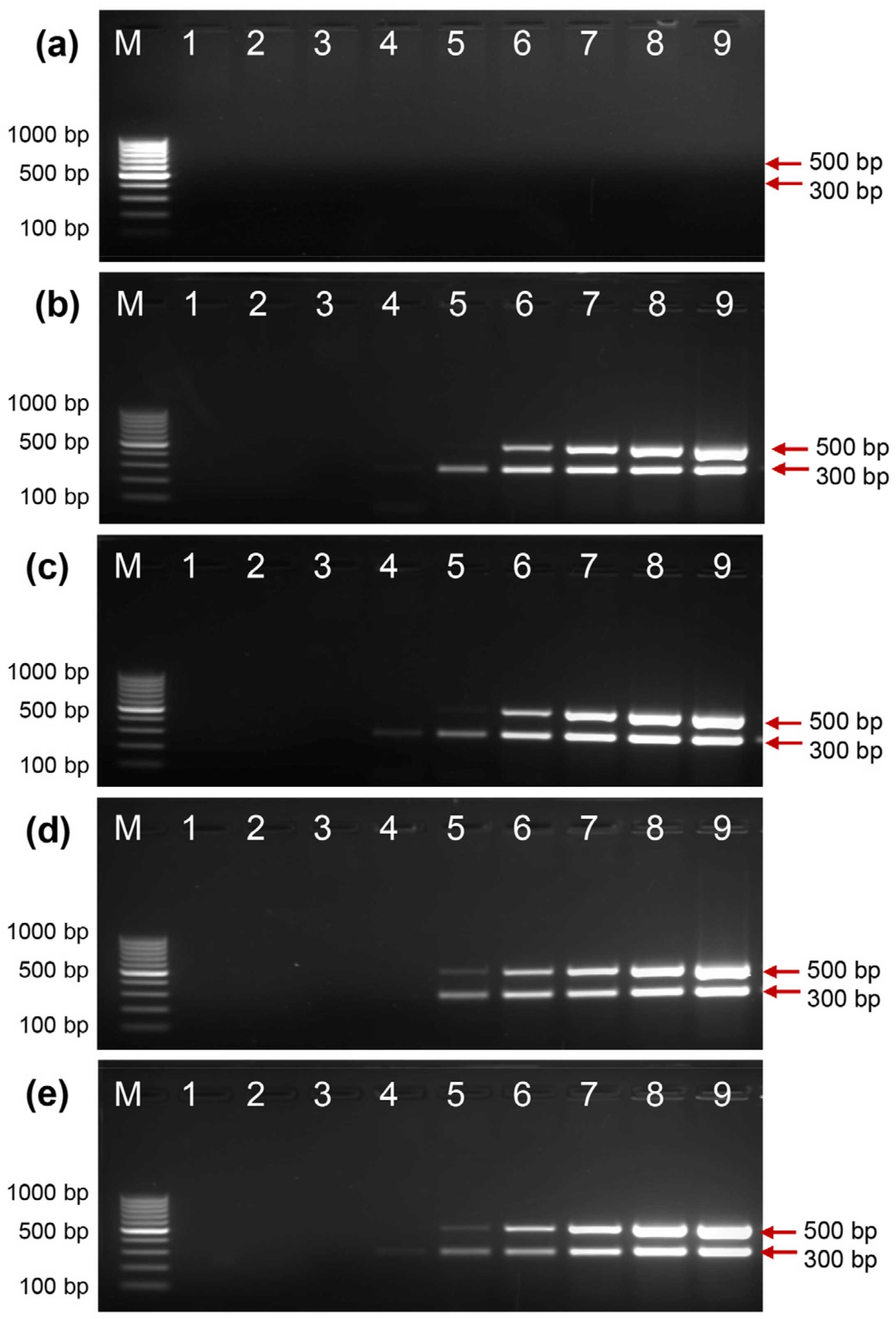
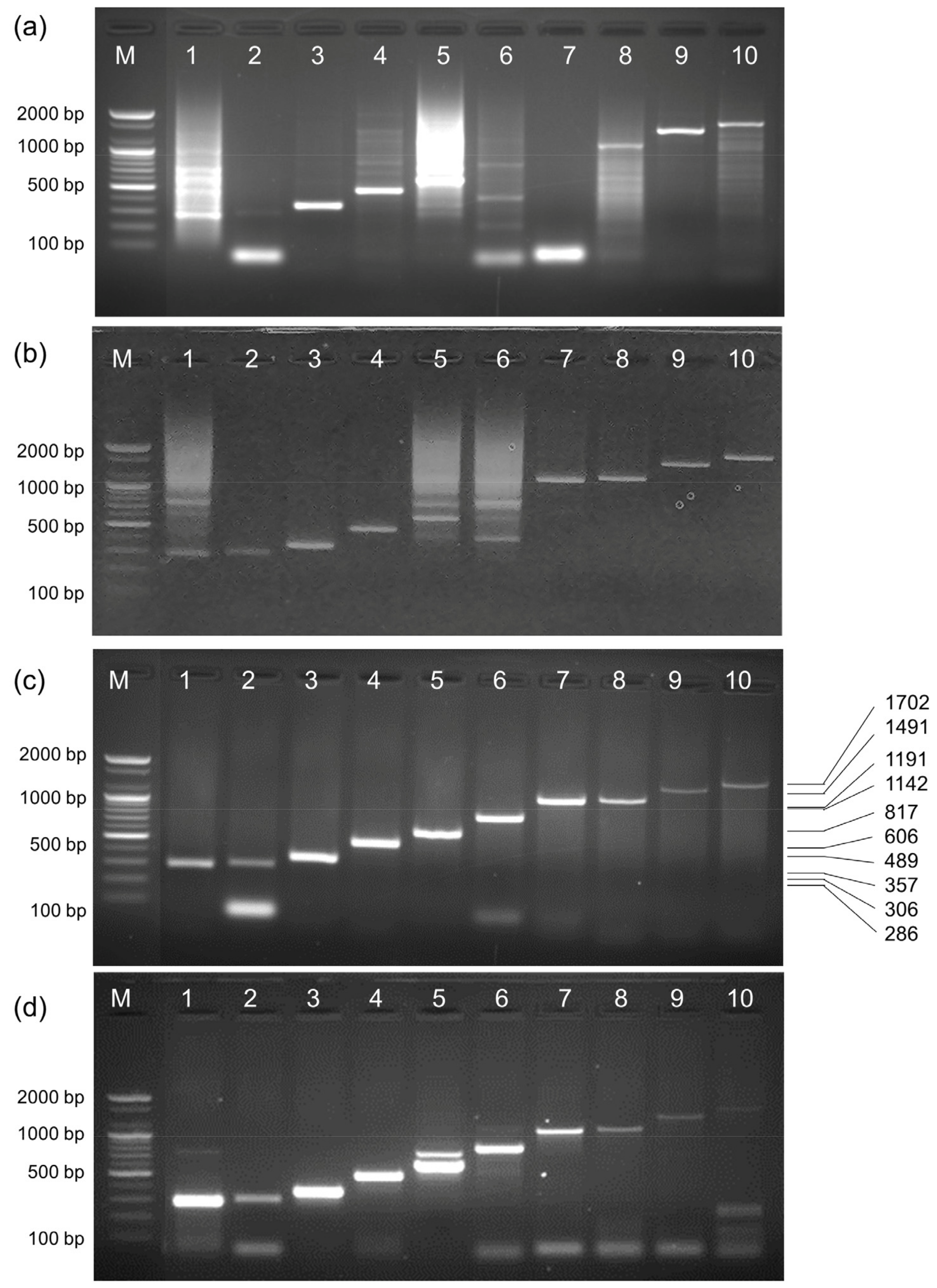
2.8. Direct Allele-Specific PCR Using DNA and Whole Blood Samples
2.9. Activity of EcoliTaq in Hot-Start PCR
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Preparation of EcoliTaq
4.3. PCR Amplification for Assay of EcoliTaq Activity
4.4. Detection of Salmonella spp. and Shigella spp. in Whole Blood Samples
4.5. HLA-B27 Genotyping Using Clinical Samples
4.6. ASPCR
4.7. Hot-Start PCR
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Saiki, R.K.; Scharf, S.; Faloona, F.; Mullis, K.B.; Horn, G.T.; Erlich, H.A.; Arnheim, N. Enzymatic Amplification of β-Globin Genomic Sequences and Restriction Site Analysis for Diagnosis of Sickle Cell Anemia. Science 1985, 230, 1350–1354. [Google Scholar] [CrossRef] [PubMed]
- Harzandi, N.; Jamshidi, S.; Dezfulian, M.; Bahonar, A.; Bakhtiari, A.; Banihashemi, K. Molecular Detection and Speciation of Campylobacter Species in Children With Gastroenteritis Using Polymerase Chain Reaction in Bahonar Hospital of Karaj City. Int. J. Enteric Pathog. 2015, 3, 4–7. [Google Scholar] [CrossRef]
- Bermingham, N.; Luettich, K. Polymerase Chain Reaction and Its Applications. Curr. Diagn. Pathol. 2003, 9, 159–164. [Google Scholar] [CrossRef]
- Roayaei, M.; Galehdari, H. Cloning and Expression of Thermus Aquaticus DNA Polymerase in Escherichia coli. Jundishapur J. Microbiol. 2008, 1, 1–5. [Google Scholar]
- Bloos, F.; Sachse, S.; Kortgen, A.; Pletz, M.W.; Lehmann, M.; Straube, E.; Riedemann, N.C.; Reinhart, K.; Bauer, M. Evaluation of a Polymerase Chain Reaction Assay for Pathogen Detection in Septic Patients under Routine Condition: An Observational Study. PLoS ONE 2012, 7, e46003. [Google Scholar] [CrossRef] [PubMed]
- Singh, I.; Swarup, V.; Shakya, S.; Goyal, V.; Faruq, M.; Srivastava, A.K. Single-Step Blood Direct PCR: A Robust and Rapid Method to Diagnose Triplet Repeat Disorders. J. Neurol. Sci. 2017, 379, 49–54. [Google Scholar] [CrossRef]
- Wang, J.T.; Wang, T.H.; Sheu, J.C.; Lin, S.M.; Lin, J.T.; Chen, D.S. Effects of Anticoagulants and Storage of Blood Samples on Efficacy of the Polymerase Chain Reaction Assay for Hepatitis C Virus. J. Clin. Microbiol. 1992, 30, 750–753. [Google Scholar] [CrossRef]
- Yokota, M.; Tatsumi, N.; Nathalang, O.; Yamada, T.; Tsuda, I. Effects of Heparin on Polymerase Chain Reaction for Blood White Cells. J. Clin. Lab. Anal. 1999, 13, 133–140. [Google Scholar] [CrossRef]
- Akane, A.; Matsubara, K.; Nakamura, H.; Takahashi, S.; Kimura, K. Identification of the Heme Compound Copurified with Deoxyribonucleic Acid (DNA) from Bloodstains, a Major Inhibitor of Polymerase Chain Reaction (PCR) Amplification. J. Forensic Sci. 1994, 39, 362–372. [Google Scholar] [CrossRef]
- Ali, N.; Rampazzo, R.d.C.P.; Costa, A.D.T.; Krieger, M.A. Current Nucleic Acid Extraction Methods and Their Implications to Point-of-Care Diagnostics. BioMed Res. Int. 2017, 2017, 9306564. [Google Scholar] [CrossRef]
- D’aquila, R.T.; Bechtel, L.J.; Videler, J.A.; Eron, J.J.; Gorczyca, P.; Kaplan, J.C. Maximizing Sensitivity and Specificity of PCR by Pre-Amplification Heating. Nucleic Acids Res. 1991, 19, 3749. [Google Scholar] [CrossRef]
- Birch, D.E.; Kolmodin, L.; Wong, J.; Zangenberg, G.A.; Zoccoli, M.A.; McKinney, N.; Young, K.K.Y. Simplified Hot Start PCR. Nature 1996, 381, 445–446. [Google Scholar] [CrossRef]
- Moretti, T.; Koons, B.; Budowle, B. Enhancement of PCR Amplification Yield and Specificity Using AmpliTaq Gold(TM) DNA Polymerase. Biotechniques 1998, 25, 716–722. [Google Scholar]
- Sharkey, D.J.; Scalice, E.R.; Christy, K.G.; Atwood, S.M.; Daiss, J.L. Antibodies as Thermolabile Switches: High Temperature Triggering for the Polymerase Chain Reaction. Bio/Technology 1994, 12, 506–509. [Google Scholar] [CrossRef]
- Kellogg, D.E.; Rybalkin, I.; Chen, S.; Mukhamedova, N.; Vlasik, T.; Siebert, P.D.; Chenchik, A. TaqStart Antibody(TM): “Hot Start” PCR Facilitated by a Neutralizing Monoclonal Antibody Directed against Taq DNA Polymerase. Biotechniques 1994, 16, 1134–1137. [Google Scholar]
- Mizuguchi, H.; Nakatsuji, M.; Fujiwara, S.; Takagi, M.; Imanaka, T. Characterization and Application to Hot Start PCR of Neutralizing Monoclonal Antibodies against KOD DNA Polymerase. J. Biochem. 1999, 126, 762–768. [Google Scholar] [CrossRef]
- Dang, C.; Jayasena, S.D. Oligonucleotide Inhibitors OfTaqDNA Polymerase Facilitate Detection of Low Copy Number Targets by PCR. J. Mol. Biol. 1996, 264, 268–278. [Google Scholar] [CrossRef]
- Lin, Y.; Jayasena, S.D. Inhibition of Multiple Thermostable DNA Polymerases by a Heterodimeric Aptamer. J. Mol. Biol. 1997, 271, 100–111. [Google Scholar] [CrossRef]
- Kaijalainen, S.; Karhunen, P.J.; Lalu, K.; Lindström, K. An Alternative Hot Start Technique for PCR in Small Volumes Using Beads of Wax-Embedded Reaction Components Dried in Trehalose. Nucleic Acids Res. 1993, 21, 2959–2960. [Google Scholar] [CrossRef]
- Barnes, W.M.; Rowlyk, K.R. Magnesium Precipitate Hot Start Method for PCR. Mol. Cell. Probes 2002, 16, 167–171. [Google Scholar] [CrossRef]
- Kaboev, O.K. PCR Hot Start Using Primers with the Structure of Molecular Beacons (Hairpin-like Structure). Nucleic Acids Res. 2000, 28, e94. [Google Scholar] [CrossRef] [PubMed]
- Ailenberg, M.; Silverman, M. Controlled Hot Start and Improved Specificity in Carrying Out PCR Utilizing Touch-Up and Loop Incorporated Primers (TULIPS). Biotechniques 2000, 29, 1018–1024. [Google Scholar] [CrossRef] [PubMed]
- Kermekchiev, M.B.; Tzekov, A.; Barnes, W.M. Cold-Sensitive Mutants of Taq DNA Polymerase Provide a Hot Start for PCR. Nucleic Acids Res. 2003, 31, 6139–6147. [Google Scholar] [CrossRef]
- Horáková, H.; Polakovičová, I.; Shaik, G.M.; Eitler, J.; Bugajev, V.; Dráberová, L.; Dráber, P. 1,2-Propanediol-Trehalose Mixture as a Potent Quantitative Real-Time PCR Enhancer. BMC Biotechnol. 2011, 11, 41. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Park, G.; Yang, Y.G.; Lee, S.G.; Kim, S.W. Rapid ABO Genotyping Using Whole Blood without DNA Purification. Ann. Lab. Med. 2009, 29, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Bu, Y.; Huang, H.; Zhou, G. Direct Polymerase Chain Reaction (PCR) from Human Whole Blood and Filter-Paper-Dried Blood by Using a PCR Buffer with a Higher PH. Anal. Biochem. 2008, 375, 370–372. [Google Scholar] [CrossRef] [PubMed]
- Bachmann, B.; Lüke, W.; Hunsmann, G. Improvement of PCR Amplified DNA Sequencing with the Aid of Detergents. Nucleic Acids Res. 1990, 18, 1309. [Google Scholar] [CrossRef] [PubMed]
- Arachea, B.T.; Sun, Z.; Potente, N.; Malik, R.; Isailovic, D.; Viola, R.E. Detergent Selection for Enhanced Extraction of Membrane Proteins. Protein Expr. Purif. 2012, 86, 12–20. [Google Scholar] [CrossRef]
- Carninci, P.; Nishiyama, Y.; Westover, A.; Itoh, M.; Nagaoka, S.; Sasaki, N.; Okazaki, Y.; Muramatsu, M.; Hayashizaki, Y. Thermostabilization and Thermoactivation of Thermolabile Enzymes by Trehalose and Its Application for the Synthesis of Full Length CDNA. Proc. Natl. Acad. Sci. USA 1998, 95, 520–524. [Google Scholar] [CrossRef]
- Spiess, A.N.; Mueller, N.; Ivell, R. Trehalose Is a Potent PCR Enhancer: Lowering of DNA Melting Temperature and Thermal Stabilization of Taq Polymerase by the Disaccharide Trehalose. Clin. Chem. 2004, 50, 1256–1259. [Google Scholar] [CrossRef]
- Zhang, Z.; Kermekchiev, M.B.; Barnes, W.M. Direct DNA Amplification from Crude Clinical Samples Using a PCR Enhancer Cocktail and Novel Mutants of Taq. J. Mol. Diagn. 2010, 12, 152–161. [Google Scholar] [CrossRef]
- Ishino, S.; Ishino, Y. DNA Polymerases as Useful Reagents for Biotechnology—The History of Developmental Research in the Field. Front. Microbiol. 2014, 5, 465. [Google Scholar] [CrossRef]
- Yang, Y.G.; Kim, J.Y.; Song, Y.H.; Kim, D.S. A Novel Buffer System, AnyDirect, Can Improve Polymerase Chain Reaction from Whole Blood without DNA Isolation. Clin. Chim. Acta 2007, 380, 112–117. [Google Scholar] [CrossRef]
- Mercier, B.; Gaucher, C.; Feugeas, O.; Mazurier, C. Direct PCR from Whole Blood, without DNA Extraction. Nucleic Acids Res. 1990, 18, 5908–5909. [Google Scholar] [CrossRef]
- Śpibida, M.; Krawczyk, B.; Olszewski, M.; Kur, J. Modified DNA Polymerases for PCR Troubleshooting. J. Appl. Genet. 2017, 58, 133–142. [Google Scholar] [CrossRef]
- Corless, C.E.; Guiver, M.; Borrow, R.; Edwards-Jones, V.; Kaczmarski, E.B.; Fox, A.J. Contamination and Sensitivity Issues with a Real-Time Universal 16S RRNA PCR. J. Clin. Microbiol. 2000, 38, 1747–1752. [Google Scholar] [CrossRef]
- Eom, S.H.; Wang, J.; Steitz, T.A. Structure of Taq Polymerase with DNA at the Polymerase Active Site. Nature 1996, 382, 278–281. [Google Scholar] [CrossRef]
- Rahn, K.; De Grandis, S.A.; Clarke, R.C.; McEwen, S.A.; Galán, J.E.; Ginocchio, C.; Curtiss, R.; Gyles, C.L. Amplification of an InvA Gene Sequence of Salmonella Typhimurium by Polymerase Chain Reaction as a Specific Method of Detection of Salmonella. Mol. Cell. Probes 1992, 6, 271–279. [Google Scholar] [CrossRef]
- Villalobo, E.; Torres, A. PCR for Detection of Shigella Spp. in Mayonnaise. Appl. Environ. Microbiol. 1998, 64, 1242–1245. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PCR Reaction | Fragment Size (bp) | Primer Pair | Allele Specificity |
|---|---|---|---|
| 1 | 205 | 261G: 5′-GCAGTAGGAAGGATGTCCTCGTGtTG-3′ | A101, A102, B101 |
| int6: 5′-AGACCTCAATGTCCACAGTCACTCG-3′ | |||
| 2 | 381 | 467C: 5′-CCACTACTATGTCTTCACCGACCAtCC-3′ | A101, O01, O02 |
| 803G: 5′-CACCGACCCCCCGAAGAtCC-3′ | |||
| 3 | 164 | 297A: 5′-CCATTGTCTGGGAGGGCcCA-3′ | A101, A102, O01 |
| int6: 5′-AGACCTCAATGTCCACAGTCACTCG-3′ | |||
| 4 | 381 | 467C: 5′-CCACTACTATGTCTTCACCGACCAtCC-3′ | B101 |
| 803C: 5′-CACCGACCCCCCGAAGAtCG-3′ | |||
| 5 | 205 | 261A: 5′-GCAGTAGGAAGGATGTCCTCGTGtTA-3′ | O01, O02 |
| int6: 5′-AGACCTCAATGTCCACAGTCACTCG-3′ | |||
| 6 | 381 | 467T: 5′-CCACTACTATGTCTTCACCGACCAtCT-3′ | A102 |
| 803G: 5′-CACCGACCCCCCGAAGAtCC-3′ | |||
| 7 | 164 | 297G: 5′-CCATTGTCTGGGAGGGCcCG-3′ | B101, O02 |
| int6: 5′-AGACCTCAATGTCCACAGTCACTCG-3′ |
| PCR Reaction | Fragment Size (bp) | Primer Sequence |
|---|---|---|
| 1 | 286 | 5′-GGCGACAGAGCGAGATTCCA-3′ |
| 5′-GGGTCAGCGGCAAGCAGAGG-3′ | ||
| 2 | 306 | 5′-GAGGTTCCTACAGGCACCTGCCCAG-3′ |
| 5′-CAAAATCACCCCTCACAGTACTCTG-3′ | ||
| 3 | 357 | 5′-GACAAGGGTGGTTGGGAGTAGATG-3′ |
| 5′-AGAGGAGCTGGTGTTGTTGG-3′ | ||
| 4 | 489 | 5′-TGTTCACTTGTGCCCTGACT-3′ |
| 5′-GGAGGGCCACTGACAACCA-3′ | ||
| 5 | 606 | 5′-GGCGACAGAGCGAGATTCCA-3′ |
| 5′-CACAAACACGCACCTCAAAG-3′ | ||
| 6 | 817 | 5′-GGCGACAGAGCGAGATTCCA-3′ 5′-AGAGGAGCTGGTGTTGTTGG-3′ |
| 7 | 1142 | 5′-GCTTTATCTGTTCACTTGTGCCC-3′ 5′-TGTGCAGGGTGGCAAGTGGC-3′ |
| 8 | 1191 | 5′-TGTTCACTTGTGCCCTGACT-3′ 5′-GGGTCAGCGGCAAGCAGAGG-3′ |
| 9 | 1491 | 5′-TGTTCACTTGTGCCCTGACT-3′ 5′-CACAAACACGCACCTCAAAG-3′ |
| 10 | 1702 | 5′-TGTTCACTTGTGCCCTGACT-3′ |
| 5′-AGAGGAGCTGGTGTTGTTGG-3′ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, S.J.; Park, S.-Y.; Lee, K.-H.; Lee, M.-W.; Yu, C.-Y.; Maeng, J.; Kim, H.-D.; Kim, S.W. Development of a Simple Direct and Hot-Start PCR Using Escherichia coli-Expressing Taq DNA Polymerase. Int. J. Mol. Sci. 2023, 24, 11405. https://doi.org/10.3390/ijms241411405
Lee SJ, Park S-Y, Lee K-H, Lee M-W, Yu C-Y, Maeng J, Kim H-D, Kim SW. Development of a Simple Direct and Hot-Start PCR Using Escherichia coli-Expressing Taq DNA Polymerase. International Journal of Molecular Sciences. 2023; 24(14):11405. https://doi.org/10.3390/ijms241411405
Chicago/Turabian StyleLee, Sun Ju, Sang-Yong Park, Kwang-Ho Lee, Min-Woo Lee, Chae-Yeon Yu, Jaeyoung Maeng, Hyeong-Dong Kim, and Suhng Wook Kim. 2023. "Development of a Simple Direct and Hot-Start PCR Using Escherichia coli-Expressing Taq DNA Polymerase" International Journal of Molecular Sciences 24, no. 14: 11405. https://doi.org/10.3390/ijms241411405
APA StyleLee, S. J., Park, S.-Y., Lee, K.-H., Lee, M.-W., Yu, C.-Y., Maeng, J., Kim, H.-D., & Kim, S. W. (2023). Development of a Simple Direct and Hot-Start PCR Using Escherichia coli-Expressing Taq DNA Polymerase. International Journal of Molecular Sciences, 24(14), 11405. https://doi.org/10.3390/ijms241411405