
KLF13 Regulates the Activity of the GH-Induced JAK/STAT Signaling by Targeting Genes Involved in the Pathway
 , and
, and 
Abstract
1. Introduction
2. Results
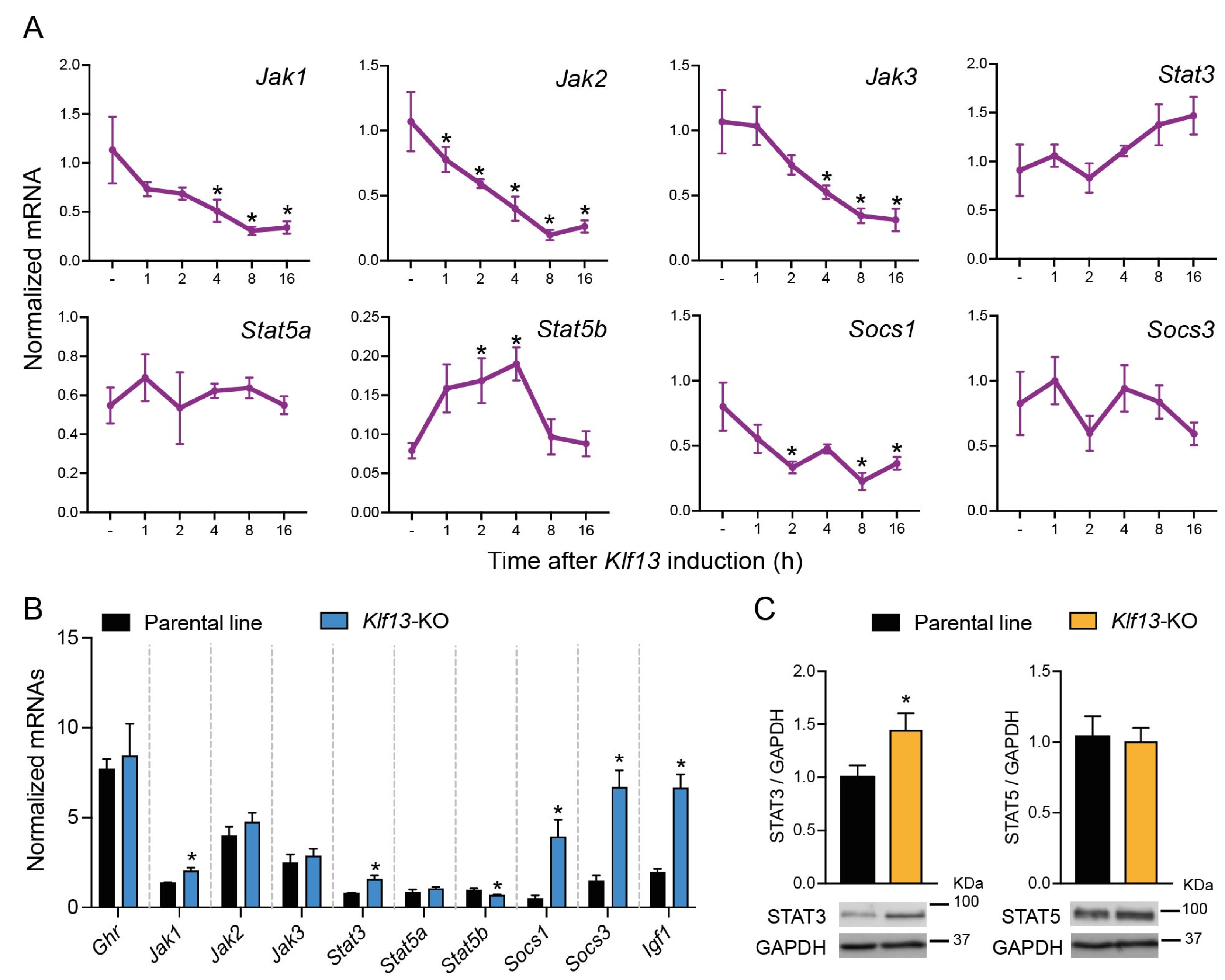
2.1. KLF13 Regulates Several Genes Involved in the JAK/STAT Pathway in HT22 Cells
2.2. The Expression of Several Genes Involved in the JAK/STAT Pathway Is Dysregulated in KLF13-Deficient HT22 Cells
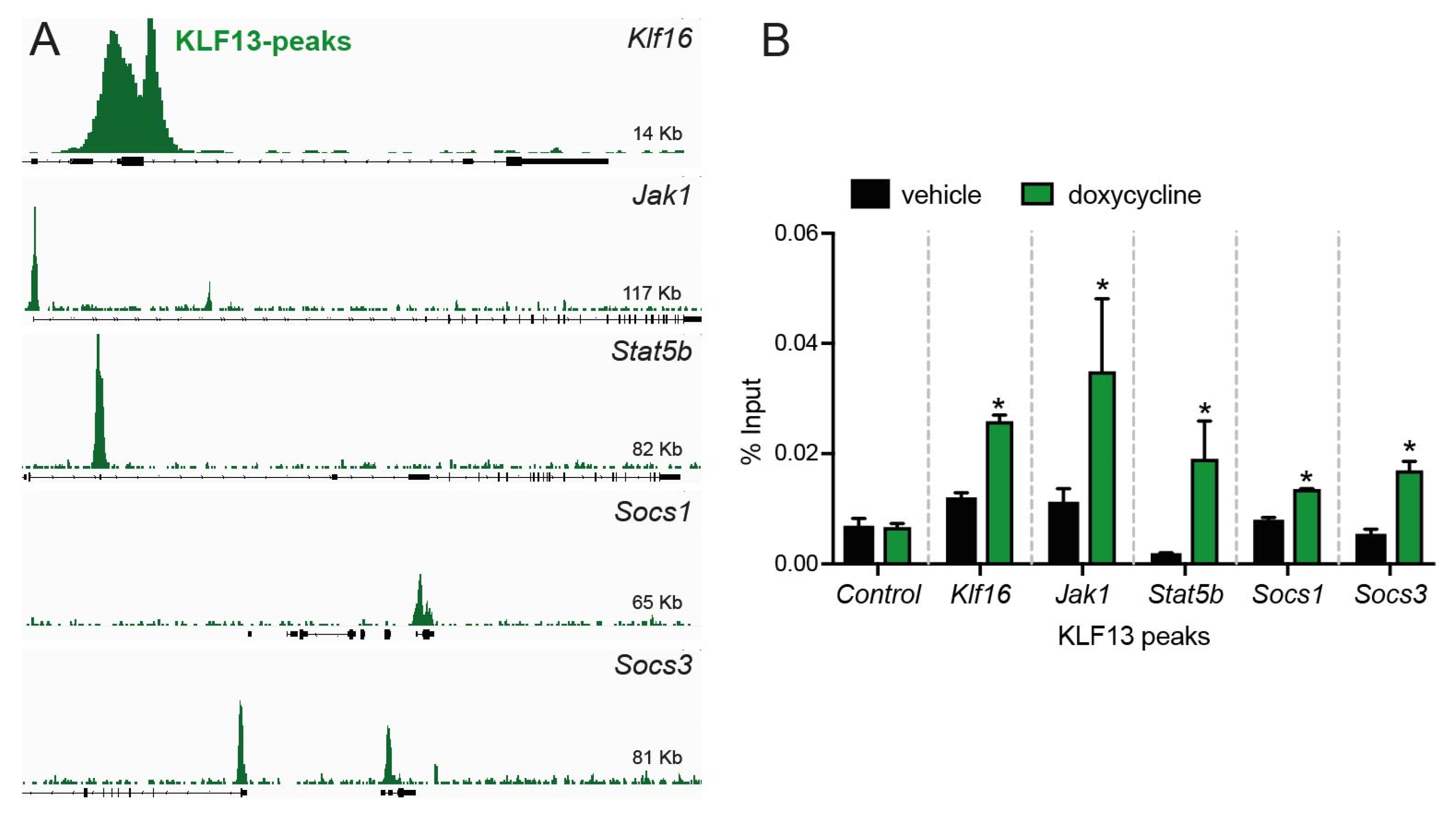
2.3. KLF13 Associates in the Chromatin with Promoters of Jak1, Stat5b, Socs1, and Socs3 Genes in HT22 Cells
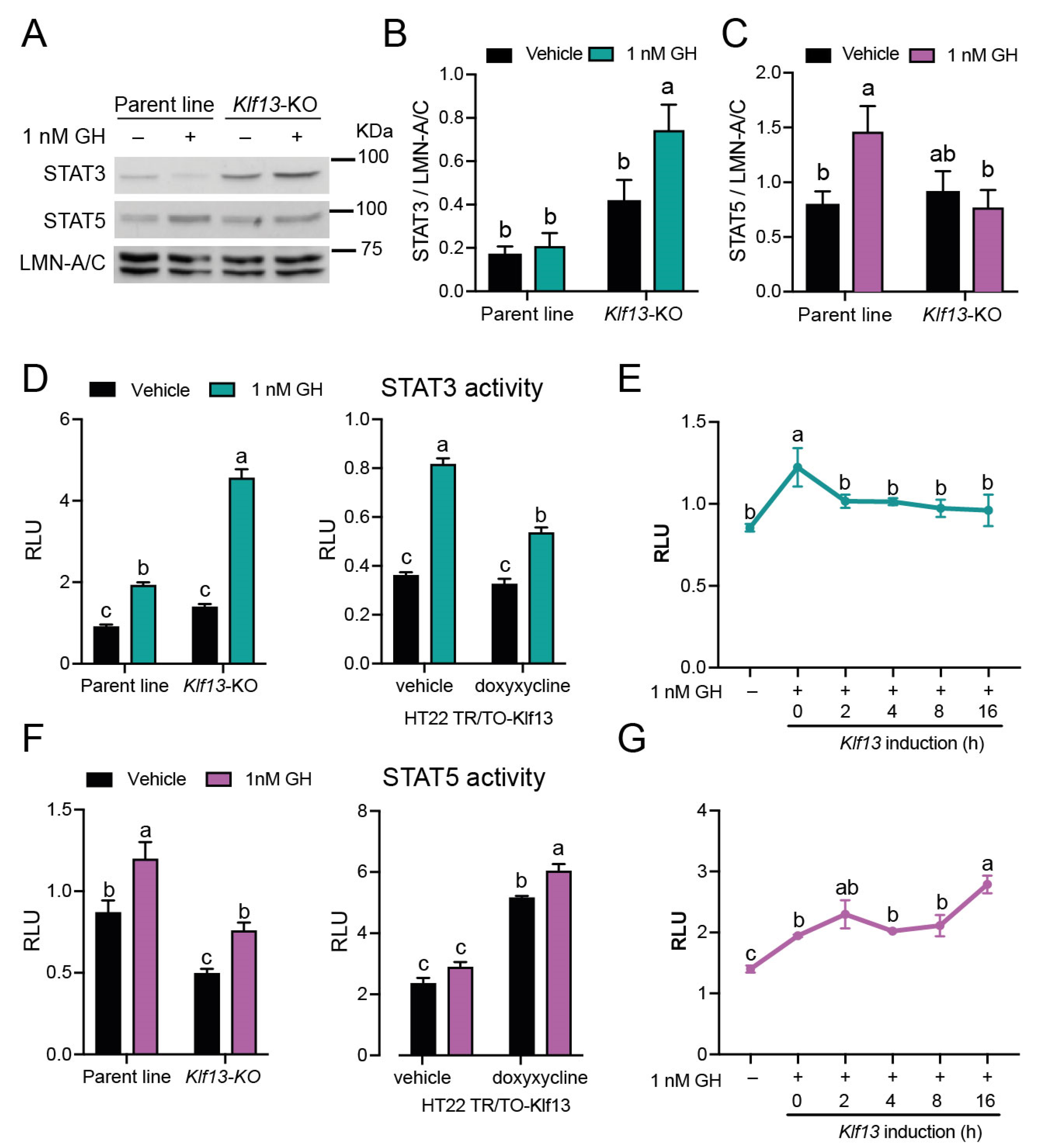
2.4. KLF13 Differentially Regulates GH-Dependent Activity of the STAT3 and STAT5 Branches in the JAK/STAT Pathway
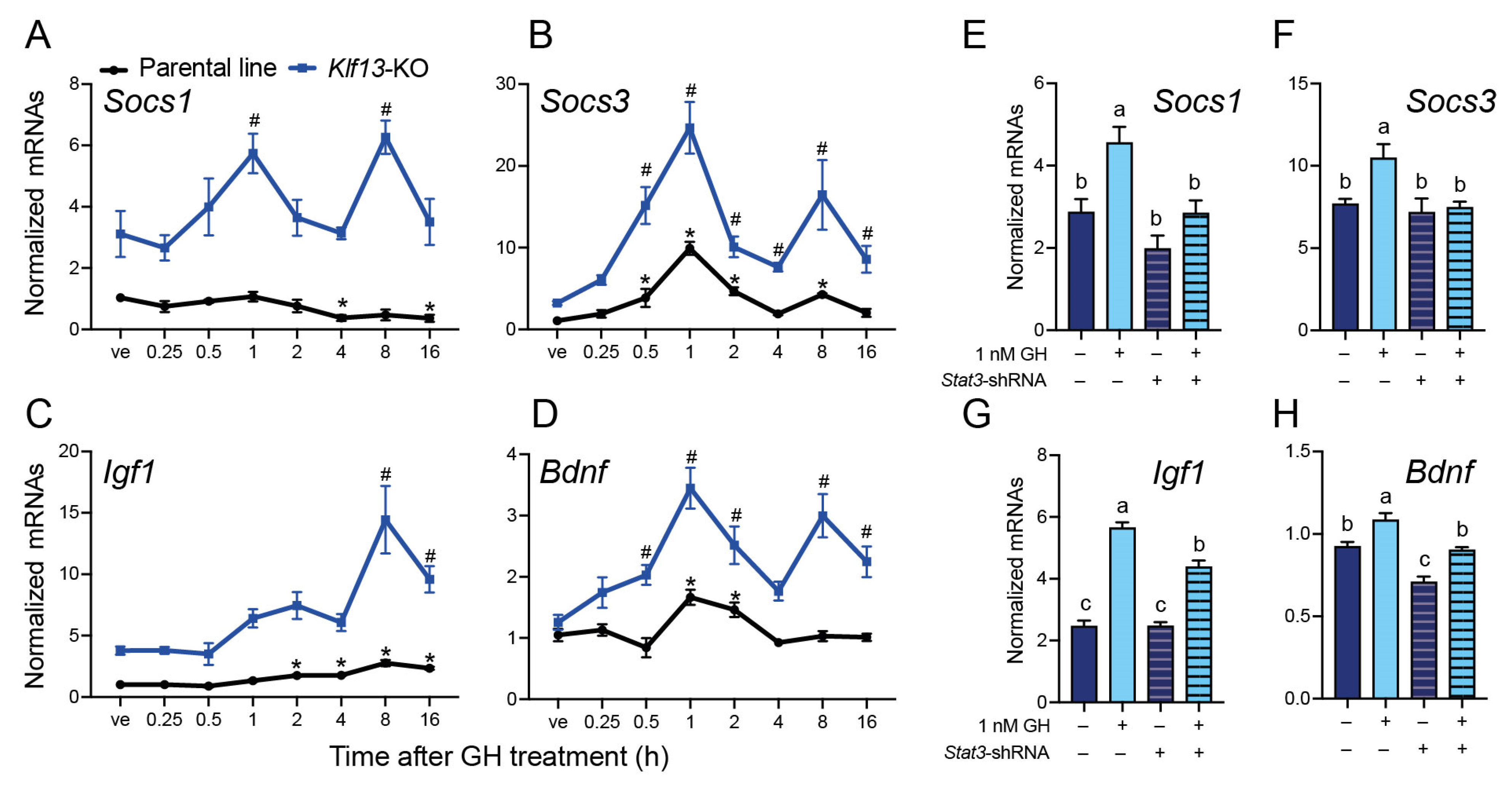
2.5. The GH-Induced Expression of the JAK/STAT Output Genes Socs1, Socs3, Igf1, and Bdnf Is Enhanced in KLF13-Deficient HT22
2.6. STAT3 Mediates the Enhanced GH-Induced Expression of the JAK/STAT Output Genes in KLF13-Deficient HT22 Cells
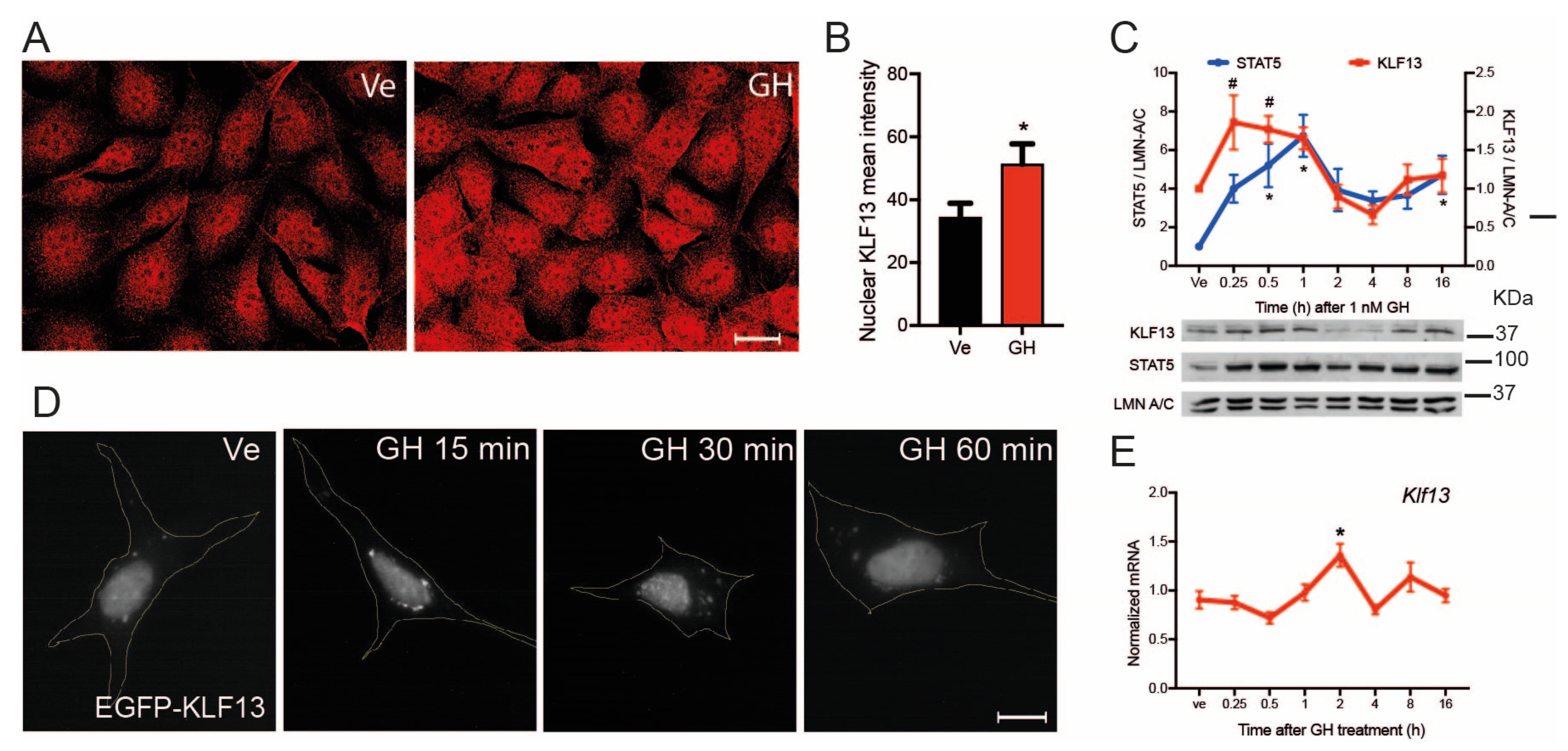
2.7. GH Induces Klf13 Expression and KLF13 Synthesis but Not Its Nuclear Translocation
3. Discussion
4. Materials and Methods
4.1. HT22 Cell Cultures
4.2. Plasmids
4.3. Experimental Design
4.4. RNA Extraction and RT-qPCR
4.5. KLF13-Peaks Visualization
4.6. Chromatin Extraction and Immunoprecipitation
4.7. Dual Luciferase Promoter-Reporter Assays
4.8. SDS-PAGE and Western-Blot Analysis
4.9. Immunofluorescent Staining
4.10. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pei, J.; Grishin, N.V. A New Family of Predicted Krüppel-like Factor Genes and Pseudogenes in Placental Mammals. PLoS ONE 2013, 8, e811109. [Google Scholar] [CrossRef]
- Knoedler, J.R.; Denver, R.J. Krüppel-like Factors Are Effectors of Nuclear Receptor Signaling. Gen. Comp. Endocrinol. 2014, 203, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Moore, D.L.; Apara, A.; Goldberg, J.L. Krüppel-like Transcription Factors in the Nervous System: Novel Players in Neurite Outgrowth and Axon Regeneration. Mol. Cell. Neurosci. 2011, 47, 233–243. [Google Scholar] [CrossRef] [PubMed]
- Yin, K.J.; Hamblin, M.; Fan, Y.; Zhang, J.; Chen, Y.E. Krüppel-like Factors in the Central Nervous System: Novel Mediators in Stroke. Metab. Brain Dis. 2015, 30, 401–410. [Google Scholar] [CrossRef]
- Kennedy, C.L.M.; Price, E.M.; Mifsud, K.R.; Salatino, S.; Sharma, E.; Engledow, S.; Broxholme, J.; Goss, H.M.; Reul, J.M.H.M. Genomic Regulation of Krüppel-like-Factor Family Members by Corticosteroid Receptors in the Rat Brain. Neurobiol. Stress 2023, 23, 100532. [Google Scholar] [CrossRef] [PubMed]
- McConnell, B.B.; Yang, V.W. Mammalian Krüppel-Like Factors in Health and Diseases. Physiol. Rev. 2010, 90, 1337–1381. [Google Scholar] [CrossRef]
- Moore, D.L.; Blackmore, M.G.; Hu, Y.; Kaestner, K.H.; Bixby, J.L.; Lemmon, V.P.; Goldberg, J.L. KLF Family Members Regulate Intrinsic Axon Regeneration Ability. Science 2009, 326, 298–301. [Google Scholar] [CrossRef]
- Ávila-Mendoza, J.; Subramani, A.; Denver, R.J. Krüppel-Like Factors 9 and 13 Block Axon Growth by Transcriptional Repression of Key Components of the cAMP Signaling Pathway. Front. Mol. Neurosci. 2020, 13, 602638. [Google Scholar] [CrossRef]
- Bonett, R.M.; Hu, F.; Bagamasbad, P.; Denver, R.J. Stressor and Glucocorticoid-Dependent Induction of the Immediate Early Gene Krüppel-like Factor 9: Implications for Neural Development and Plasticity. Endocrinology 2009, 150, 1757–1765. [Google Scholar] [CrossRef]
- Ávila-Mendoza, J.; Subramani, A.; Sifuentes, C.J.; Denver, R.J. Molecular Mechanisms for Krüppel-Like Factor 13 Actions in Hippocampal Neurons. Mol. Neurobiol. 2020, 57, 3785–3802. [Google Scholar] [CrossRef]
- Knoedler, J.R.; Subramani, A.; Denver, R.J. The Krüppel-like Factor 9 Cistrome in Mouse Hippocampal Neurons Reveals Predominant Transcriptional Repression via Proximal Promoter Binding. BMC Genom. 2017, 18, 299. [Google Scholar] [CrossRef] [PubMed]
- Bialkowska, A.B.; Yang, V.W.; Mallipattu, S.K. Krüppel-like Factors in Mammalian Stem Cells and Development. Development 2017, 144, 737–754. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Li, J.; Fu, M.; Zhao, X.; Wang, W. The JAK/STAT Signaling Pathway: From Bench to Clinic. Signal Transduct. Target. Ther. 2021, 6, 402. [Google Scholar] [CrossRef]
- Awasthi, N.; Liongue, C.; Ward, A.C. STAT Proteins: A Kaleidoscope of Canonical and Non-Canonical Functions in Immunity and Cancer. J. Hematol. Oncol. 2021, 14, 198. [Google Scholar] [CrossRef] [PubMed]
- Nicolas, C.S.; Amici, M.; Bortolotto, Z.A.; Doherty, A.; Csaba, Z.; Fafouri, A.; Dournaud, P.; Gressens, P.; Collingridge, G.L.; Peineau, S. The Role of JAK-STAT Signaling within the CNS. JAK-STAT 2013, 2, e22925. [Google Scholar] [CrossRef]
- Aittomäki, S.; Pesu, M. Therapeutic Targeting of the JAK/STAT Pathway. Basic Clin. Pharmacol. Toxicol. 2014, 114, 18–23. [Google Scholar] [CrossRef]
- Chhabra, Y.; Lee, C.M.M.; Müller, A.F.; Brooks, A.J. GHR Signaling: Receptor Activation and Degradation Mechanisms. Mol. Cell. Endocrinol. 2021, 520, 111075. [Google Scholar] [CrossRef]
- Bergan-Roller, H.E.; Sheridan, M.A. The Growth Hormone Signaling System: Insights into Coordinating the Anabolic and Catabolic Actions of Growth Hormone. Gen. Comp. Endocrinol. 2018, 258, 119–133. [Google Scholar] [CrossRef]
- Kiu, H.; Nicholson, S.E. Biology and Significance of the JAK/STAT Signaling Pathways. Growth Factors 2012, 30, 88–106. [Google Scholar] [CrossRef]
- Ahmed, S.F.; Farquharson, C. The Effect of GH and IGF1 on Linear Growth and Skeletal Development and Their Modulation by SOCS Proteins. J. Endocrinol. 2010, 206, 249–259. [Google Scholar] [CrossRef]
- Barclay, J.L.; Anderson, S.T.; Waters, M.J.; Curlewis, J.D. Regulation of Suppressor of Cytokine Signaling 3 (SOC3) by Growth Hormone in pro-B Cells. Mol. Endocrinol. 2007, 21, 2503–2515. [Google Scholar] [CrossRef]
- Ranke, M.B.; Wit, J.M. Growth Hormone—Past, Present and Future. Nat. Rev. Endocrinol. 2018, 14, 285–300. [Google Scholar] [CrossRef]
- Arámburo, C.; Alba-Betancourt, C.; Luna, M.; Harvey, S. Expression and Function of Growth Hormone in the Nervous System: A Brief Review. Gen. Comp. Endocrinol. 2014, 203, 35–42. [Google Scholar] [CrossRef]
- Bianchi, V.E.; Locatelli, V.; Rizzi, L. Neurotrophic and Neuroregenerative Effects of GH/IGF1. Int. J. Mol. Sci. 2017, 18, 2441. [Google Scholar] [CrossRef] [PubMed]
- Arce, V.M.; Devesa, P.; Devesa, J. Role of Growth Hormone (GH) in the Treatment on Neural Diseases: From Neuroprotection to Neural Repair. Neurosci. Res. 2013, 76, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Åberg, N.D.; Brywe, K.G.; Isgaard, J. Aspects of Growth Hormone and Insulin-like Growth Factor-I Related to Neuroprotection, Regeneration, and Functional Plasticity in the Adult Brain. Sci. World J. 2006, 6, 53–80. [Google Scholar] [CrossRef] [PubMed]
- Alba-Betancourt, C.; Luna-Acosta, J.L.; Ramírez-Martínez, C.E.; Avila-González, D.; Granados-Ávalos, E.; Carranza, M.; Martínez-Coria, H.; Arámburo, C.; Luna, M. Neuro-Protective Effects of Growth Hormone (GH) after Hypoxia-Ischemia Injury in Embryonic Chicken Cerebellum. Gen. Comp. Endocrinol. 2013, 183, 17–31. [Google Scholar] [CrossRef]
- Olivares-Hernández, J.D.; Carranza, M.; Balderas-Márquez, J.E.; Epardo, D.; Baltazar-Lara, R.; Ávila-Mendoza, J.; Martínez-Moreno, C.G.; Luna, M.; Arámburo, C. Neuroprotective and Regenerative Effects of Growth Hormone (GH) in the Embryonic Chicken Cerebral Pallium Exposed to Hypoxic-Ischemic (HI) Injury. Int. J. Mol. Sci. 2022, 23, 9054. [Google Scholar] [CrossRef]
- Olivares-Hernández, J.D.; Balderas-Márquez, J.E.; Carranza, M.; Luna, M.; Martínez-Moreno, C.G.; Arámburo, C. Growth Hormone (GH) Enhances Endogenous Mechanisms of Neuroprotection and Neuroplasticity after Oxygen and Glucose Deprivation Injury (OGD) and Reoxygenation (OGD/R) in Chicken Hippocampal Cell Cultures. Neural Plast. 2021, 2021, 9990166. [Google Scholar] [CrossRef]
- Martinez-Moreno, C.G.; Fleming, T.; Carranza, M.; Ávila-Mendoza, J.; Luna, M.; Harvey, S.; Arámburo, C. Growth Hormone Protects against Kainate Excitotoxicity and Induces BDNF and NT3 Expression in Chicken Neuroretinal Cells. Exp. Eye Res. 2018, 166, 1–12. [Google Scholar] [CrossRef]
- Heredia, M.; Rodríguez, N.; Robledo, V.S.; Criado, J.M.; de la Fuente, A.; Devesa, J.; Devesa, P.; Riolobos, A.S. Factors Involved in the Functional Motor Recovery of Rats with Cortical Ablation after GH and Rehabilitation Treatment: Cortical Cell Proliferation and Nestin and Actin Expression in the Striatum and Thalamus. Int. J. Mol. Sci. 2019, 20, 5770. [Google Scholar] [CrossRef]
- Sanchez-Bezanilla, S.; David Åberg, N.; Crock, P.; Walker, F.R.; Nilsson, M.; Isgaard, J.; Ong, L.K. Growth Hormone Promotes Motor Function after Experimental Stroke and Enhances Recovery-Promoting Mechanisms within the Peri-Infarct Area. Int. J. Mol. Sci. 2020, 21, 606. [Google Scholar] [CrossRef]
- Baltazar-Lara, R.; Ávila-Mendoza, J.; Martínez-Moreno, C.G.; Carranza, M.; Pech-Pool, S.; Vázquez-Martínez, O.; Díaz-Muñoz, M.; Luna, M.; Arámburo, C. Neuroprotective Effects of Growth Hormone (GH) and Insulin-like Growth Factor Type 1 (IGF-1) after Hypoxic-Ischemic Injury in Chicken Cerebellar Cell Cultures. Int. J. Mol. Sci. 2021, 22, 256. [Google Scholar] [CrossRef]
- Bjørbæk, C.; Buchholz, R.M.; Davis, S.M.; Bates, S.H.; Pierroz, D.D.; Gu, H.; Neel, B.G.; Myers, M.G.; Flier, J.S. Divergent Roles of SHP-2 in ERK Activation by Leptin Receptors. J. Biol. Chem. 2001, 276, 4747–4755. [Google Scholar] [CrossRef] [PubMed]
- Cui, M.Y.; Hu, C.K.; Pelletier, C.; Dziuba, A.; Slupski, R.H.; Li, C.; Denver, R.J. Ancient Origins and Evolutionary Conservation of Intracellular and Neural Signaling Pathways Engaged by the Leptin Receptor. Endocrinology 2014, 155, 4202–4214. [Google Scholar] [CrossRef] [PubMed]
- Knoedler, J.R.; Ávila-Mendoza, J.; Subramani, A.; Denver, R.J. The Paralogous Krüppel-like Factors 9 and 13 Regulate the Mammalian Cellular Circadian Clock Output Gene Dbp. J. Biol. Rhythm. 2020, 35, 257–274. [Google Scholar] [CrossRef]
- Heard, M.E.; Pabona, J.M.P.; Clayberger, C.; Krensky, A.M.; Simmen, F.A.; Simmen, R.C.M. The Reproductive Phenotype of Mice Null for Transcription Factor Krüppel-like Factor 13 Suggests Compensatory Function of Family Member Krüppel-like Factor 9 in the Peri-Implantation Uterus. Biol. Reprod. 2012, 87, 115. [Google Scholar] [CrossRef]
- Lomberk, G.; Urrutia, R. The Family Feud: Turning off Sp1 by Sp1-like KLF Proteins. Biochem. J. 2005, 392, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Kaczynski, J.; Zhang, J.S.; Ellenrieder, V.; Conley, A.; Duenes, T.; Kester, H.; Van der Burg, B.; Urrutia, R. The Sp1-like Protein BTEB3 Inhibits Transcription via the Basic Transcription Element Box by Interacting with MSin3A and HDAC-1 Co-Repressors and Competing with Sp1. J. Biol. Chem. 2001, 276, 36749–36756. [Google Scholar] [CrossRef]
- Manstein, V.; Yang, C.; Richter, D.; Delis, N.; Vafaizadeh, V.; Groner, B. Resistance of Cancer Cells to Targeted Therapies through the Activation of Compensating Signaling Loops. Curr. Signal Transduct. Ther. 2013, 8, 193–202. [Google Scholar] [CrossRef]
- Hosui, A.; Hennighausen, L. Genomic Dissection of the Cytokine-Controlled STAT5 Signaling Network in Liver. Physiol. Genom. 2008, 34, 135–143. [Google Scholar] [CrossRef]
- Zhang, W.; Hong, S.; Maniar, K.P.; Cheng, S.; Jie, C.; Rademaker, A.W.; Krensky, A.M.; Clayberger, C. KLF13 Regulates the Differentiation-Dependent Human Papillomavirus Life Cycle in Keratinocytes through STAT5 and IL-8. Oncogene 2016, 35, 5565–5575. [Google Scholar] [CrossRef]
- Li, Y.; Shi, X.; Li, J.; Zhang, M.; Yu, B. Knockdown of KLF11 Attenuates Hypoxia/Reoxygenation Injury via JAK2/STAT3 Signaling in H9c2. Apoptosis 2017, 22, 510–518. [Google Scholar] [CrossRef] [PubMed]
- Schindler, C.; Levy, D.E.; Decker, T. JAK-STAT Signaling: From Interferons to Cytokines. J. Biol. Chem. 2007, 282, 20059–20063. [Google Scholar] [CrossRef]
- Fleming, T.; Martínez-Moreno, C.G.; Mora, J.; Aizouki, M.; Luna, M.; Arámburo, C.; Harvey, S. Internalization and Synaptogenic Effect of GH in Retinal Ganglion Cells (RGCs). Gen. Comp. Endocrinol. 2016, 234, 151–160. [Google Scholar] [CrossRef]
- Martínez-Moreno, C.G.; Ávila-Mendoza, J.; Wu, Y.; Arellanes-Licea, E.d.C.; Louie, M.; Luna, M.; Arámburo, C.; Harvey, S. Neuroprotection by GH against Excitotoxic-Induced Cell Death in Retinal Ganglion Cells. Gen. Comp. Endocrinol. 2016, 234, 68–80. [Google Scholar] [CrossRef]
- Baltazar-Lara, R.; Mora-Zenil, J.; Carranza, M.; Ávila-Mendoza, J.; Martínez-Moreno, C.G.; Arámburo, C.; Luna, M. Growth Hormone (GH) Crosses the Blood-Brain Barrier (BBB) and Induces Neuroprotective Effects in the Embryonic Chicken Cerebellum after a Hypoxic Injury. Int. J. Mol. Sci. 2022, 23, 11546. [Google Scholar] [CrossRef] [PubMed]
- Fleming, T.; Balderas-Márquez, J.E.; Epardo, D.; Ávila-Mendoza, J.; Carranza, M.; Luna, M.; Harvey, S.; Arámburo, C.; Martínez-Moreno, C.G. Growth Hormone Neuroprotection against Kainate Excitotoxicity in the Retina Is Mediated by Notch/PTEN/ Akt Signaling. Investig. Ophthalmol. Vis. Sci. 2019, 60, 4532–4547. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Moreno, C.G.; Epardo, D.; Balderas-Márquez, J.E.; Fleming, T.; Carranza, M.; Luna, M.; Harvey, S.; Arámburo, C. Regenerative Effect of Growth Hormone (GH) in the Retina after Kainic Acid Excitotoxic Damage. Int. J. Mol. Sci. 2019, 20, 4433. [Google Scholar] [CrossRef]
- Rahaman, S.O.; Harbor, P.C.; Chernova, O.; Barnett, G.H.; Vogelbaum, M.A.; Haque, S.J. Inhibition of Constitutively Active Stat3 Suppresses Proliferation and Induces Apoptosis in Glioblastoma Multiforme Cells. Oncogene 2002, 21, 8404–8413. [Google Scholar] [CrossRef]
- Abou-Ghazal, M.; Yng, D.S.; Qiao, W.; Reina-Ortiz, C.; Wei, J.; Kong, L.Y.; Fuller, G.N.; Hiraoka, N.; Priebe, W.; Sawaya, R.; et al. The Incidence, Correlation with Tumor-Infiltrating Inflammation, and Prognosis of Phosphorylated STAT3 Expression in Human Gliomas. Clin. Cancer Res. 2008, 14, 8228–8235. [Google Scholar] [CrossRef]
- Lo, H.W.; Cao, X.; Zhu, H.; Ali-Osman, F. Constitutively Activated STAT3 Frequently Coexpresses with Epidermal Growth Factor Receptor in High-Grade Gliomas and Targeting STAT3 Sensitizes Them to Iressa and Alkylators. Clin. Cancer Res. 2008, 14, 6042–6054. [Google Scholar] [CrossRef]
- Zhou, M.; McPherson, L.; Feng, D.; Song, A.; Dong, C.; Lyu, S.-C.; Zhou, L.; Shi, X.; Ahn, Y.-T.; Wang, D.; et al. Krüppel-like Transcription Factor 13 Regulates T Lymphocyte Survival in Vivo. J. Immunol. 2007, 178, 5496–5504. [Google Scholar] [CrossRef]
- Jing, D.; Bhadri, V.A.; Beck, D.; Thoms, J.A.I.; Yakob, N.A.; Wong, J.W.H.; Knezevic, K.; Pimanda, J.E.; Lock, R.B. Opposing Regulation of BIM and BCL2 Controls Glucocorticoid-Induced Apoptosis of Pediatric Acute Lymphoblastic Leukemia Cells. Blood 2015, 125, 273–283. [Google Scholar] [CrossRef]
- Hu, Y.; Zhang, M.; Tian, N.; Li, D.; Wu, F.; Hu, P.; Wang, Z.; Wang, L.; Hao, W.; Kang, J.; et al. The Antibiotic Clofoctol Suppresses Glioma Stem Cell Proliferation by Activating KLF13. J. Clin. Investig. 2019, 129, 3072–3085. [Google Scholar] [CrossRef]
- Wu, R.; Yun, Q.; Zhang, J.; Bao, J. Downregulation of KLF13 through DNMT1-Mediated Hypermethylation Promotes Glioma Cell Proliferation and Invasion. OncoTargets Ther. 2019, 12, 1509–1520. [Google Scholar] [CrossRef]
- Luo, X.; Ribeiro, M.; Bray, E.R.; Lee, D.H.; Yungher, B.J.; Mehta, S.T.; Thakor, K.A.; Diaz, F.; Lee, J.K.; Moraes, C.T.; et al. Enhanced Transcriptional Activity and Mitochondrial Localization of STAT3 Co-Induce Axon Regrowth in the Adult Central Nervous System. Cell Rep. 2016, 15, 398–410. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Li, M.; Zhang, N.; Sun, W.; Wang, H.; Wei, W. Mechanism of MiR-218-5p in Autophagy, Apoptosis and Oxidative Stress in Rheumatoid Arthritis Synovial Fibroblasts Is Mediated by KLF9 and JAK/STAT3 Pathways. J. Investig. Med. 2021, 69, 824–832. [Google Scholar] [CrossRef] [PubMed]
- Ben-Yaakov, K.; Dagan, S.Y.; Segal-Ruder, Y.; Shalem, O.; Vuppalanchi, D.; Willis, D.E.; Yudin, D.; Rishal, I.; Rother, F.; Bader, M.; et al. Axonal Transcription Factors Signal Retrogradely in Lesioned Peripheral Nerve. EMBO J. 2012, 31, 1350–1363. [Google Scholar] [CrossRef] [PubMed]
- Bareyre, F.M.; Garzorz, N.; Lang, C.; Misgeld, T.; Büning, H.; Kerschensteiner, M. In Vivo Imaging Reveals a Phase-Specific Role of STAT3 during Central and Peripheral Nervous System Axon Regeneration. Proc. Natl. Acad. Sci. USA 2011, 108, 6282–6287. [Google Scholar] [CrossRef]
- Benowitz, L.I.; He, Z.; Goldberg, J.L. Reaching the Brain: Advances in Optic Nerve Regeneration. Exp. Neurol. 2017, 287, 365–373. [Google Scholar] [CrossRef]
- Baldwin, K.T.; Giger, R.J. Insights into the Physiological Role of CNS Regeneration Inhibitors. Front. Mol. Neurosci. 2015, 8, 23. [Google Scholar] [CrossRef]
- Zhang, Y.; Williams, P.R.; Jacobi, A.; Wang, C.; Goel, A.; Hirano, A.A.; Brecha, N.C.; Kerschensteiner, D.; He, Z. Elevating Growth Factor Responsiveness and Axon Regeneration by Modulating Presynaptic Inputs. Neuron 2019, 103, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, B.H.; Koshland, D.E. Induction and Expression of Long- and Short-Term Neurosecretory Potentiation in a Neural Cell Line. Neuron 1990, 5, 875–880. [Google Scholar] [CrossRef]
- Davis, J.B.; Maher, P. Protein Kinase C Activation Inhibits Glutamate-Induced Cytotoxicity in a Neuronal Cell Line. Brain Res. 1994, 652, 169–173. [Google Scholar] [CrossRef]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative Genomics Viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [PubMed]
- Denver, R.J.; Williamson, K.E. Identification of a Thyroid Hormone Response Element in the Mouse Krüppel-like Factor 9 Gene to Explain Its Postnatal Expression in the Brain. Endocrinology 2009, 150, 3935–3943. [Google Scholar] [CrossRef] [PubMed]
- Gade, P.; Kalvakolanu, D.V. Chromatin Immunoprecipitation Assay as a Tool for Analyzing Transcription Factor Activity. Methods Mol. Biol. 2012, 809, 85–104. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An Open-Source Platform for Biological-Image Analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| To Create the Vectors | Forward (5′–3′) | Reverse (5′–3′) |
|---|---|---|
| shStat3 | AAAATCGATGGGTGAAATTGACCAGCAATACGAATATTGCTGGTCAATTTCACCCTTTTTTTCTAGAAAA | TTTTCTAGAAAAAAAGGGTGAAATTGACCAGCAATATTCGTATTGCTGGTCAATTTCACCCATCGATTTT |
| shNc | AAAATCGATGCGCGATAGCGCTAATAATTTCGAAAAATTATTAGCGCTATCGCGCTTTTTTTCTAGAAAA | TTTTCTAGAAAAAAAGCGCGATAGCGCTAATAATTTTTCGAAATTATTAGCGCTATCGCGCATCGATTTT |
| pTOEgfp-Klf13 | TTTAAACTTAAGCTTGGTACCACCATGGCGCCCATGGCAG | TTTAAACTTAAGCTTGGTACCACCATGGCGCCCATGGCAG |
| For RT-qPCR | ||
| Gadph-mRNA | TGTGTCCGTCGTGGATCTGA | CTTCACCACCTTCTTGATGTCACT |
| Ppia-mRNA | GGTTCCTCCTTTCACAGAAT | AATTTCTCTCCGTAGATGGAC |
| Jak1-mRNA | CAAGTCTAGTGACCCTGGCA | CAGATTTCCCAGAGCGTGGT |
| Jak2-mRNA | TTGGGCAAGCTGAAGGAGAG | CATGCCTGGTTGACTCGTCT |
| Jak3-mRNA | GAACCTGGGTCACGGTTCTT | GCGGGTAGGATACTTGGCTC |
| Stat3-mRNA | TGGATGCGACCAACATCCTG | CAATGGTATTGCTGCAGGTCG |
| Stat5a-mRNA | CACTCCTGTACTTGGTTCGTCA | CCAGGTCAAACTCGCCATCT |
| Stat5b-mRNA | GTACTACACACCGGTCCCCT | ATGCATTTGCAAACTCGGGG |
| Socs1-mRNA | GATTCTGCGTGCCGCTCTC | CGGGGAGATCGCATTGTCG |
| Socs3-mRNA | CTACGCATCCAGTGTGAGGG | TGAGTACACAGTCGAAGCGG |
| Ghr-mRNA | AAGTACAGCGAGTTCAGCGA | GGACTGGGGGTAAAATCAGCA |
| Igf1-mRNA | TGGATGCTCTTCAGTTCGTG | GTGGGGCACAGTACATCTCC |
| Bdnf-mRNA | GCTCACACTCCACTGCCCAT | TCCCTGACCCATGCCAGAAGA |
| For ChIP-qPCR | ||
| Klf16-Intron | ACTAAACTCCACCCCACAAC | TCTTTCAAACACTCCCTCGC |
| Klf16-promoter | GTACGCACTACCCTCACCAG | GGTGGGCGTAACTCTCAAAG |
| Jak1-promoter | GAGCTGACCAGGGGTGAAC | TCCGCCGACATCCTGTTTAT |
| Stat5b-promoter | TTCTAGACAGCAGGAGCACG | TTCTAGACAGCAGGAGCACG |
| Socs1-promoter | AGCTCGAAAAGGCAGTCGAA | AGCTCGAAAAGGCAGTCGAA |
| Socs3-promoter | GGCAGTTCCAGGAATCGGG | GGCAGTTCCAGGAATCGGG |
| Target | Host/Type | Dilution | Assay | Source | Cat No. |
|---|---|---|---|---|---|
| GAPDH | Rabbit/polyclonal | 1:2000 | WB | Cell Signaling | 14C10 |
| LMN A/C | Goat/polyclonal | 1:2000 | WB | Santa Cruz Biotechnology | SC-6215 |
| Total STAT3 | Rabbit/polyclonal | 1:1000 | WB | Cell Signaling | 12640S |
| Total STAT5 | Rabbit/polyclonal | 1:1000 | WB | Cell Signaling | 94105S |
| KLF13 | Rabbit/polyclonal | 1:2000 | WB | Custom | Avila-Mendoza et al., 2020 [10] |
| Rabbit IgG | Goat/HRP-conjugated | 1:5000 | WB | Invitrogen | 65–6120 |
| Goat IgG | Rabbit/HRP-conjugated | 1:5000 | WB | Thermo Fisher Scientific | 81–1620 |
| KLF13 | Rabbit/polyclonal | 1:1000 | IHC | Custom | Avila-Mendoza et al., 2020 [10] |
| Rabbit IgG | Goat/Alexa 594-conjugated | 1:1000 | IHC | Invitrogen | A-11012 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ávila-Mendoza, J.; Delgado-Rueda, K.; Urban-Sosa, V.A.; Carranza, M.; Luna, M.; Martínez-Moreno, C.G.; Arámburo, C. KLF13 Regulates the Activity of the GH-Induced JAK/STAT Signaling by Targeting Genes Involved in the Pathway. Int. J. Mol. Sci. 2023, 24, 11187. https://doi.org/10.3390/ijms241311187
Ávila-Mendoza J, Delgado-Rueda K, Urban-Sosa VA, Carranza M, Luna M, Martínez-Moreno CG, Arámburo C. KLF13 Regulates the Activity of the GH-Induced JAK/STAT Signaling by Targeting Genes Involved in the Pathway. International Journal of Molecular Sciences. 2023; 24(13):11187. https://doi.org/10.3390/ijms241311187
Chicago/Turabian StyleÁvila-Mendoza, José, Karen Delgado-Rueda, Valeria A. Urban-Sosa, Martha Carranza, Maricela Luna, Carlos G. Martínez-Moreno, and Carlos Arámburo. 2023. "KLF13 Regulates the Activity of the GH-Induced JAK/STAT Signaling by Targeting Genes Involved in the Pathway" International Journal of Molecular Sciences 24, no. 13: 11187. https://doi.org/10.3390/ijms241311187
APA StyleÁvila-Mendoza, J., Delgado-Rueda, K., Urban-Sosa, V. A., Carranza, M., Luna, M., Martínez-Moreno, C. G., & Arámburo, C. (2023). KLF13 Regulates the Activity of the GH-Induced JAK/STAT Signaling by Targeting Genes Involved in the Pathway. International Journal of Molecular Sciences, 24(13), 11187. https://doi.org/10.3390/ijms241311187