
The Complex Relationship between Hypoxia Signaling, Mitochondrial Dysfunction and Inflammation in Calcific Aortic Valve Disease: Insights from the Molecular Mechanisms to Therapeutic Approaches

 , ,
, ,  ,
,  and
and 
Abstract
1. Introduction
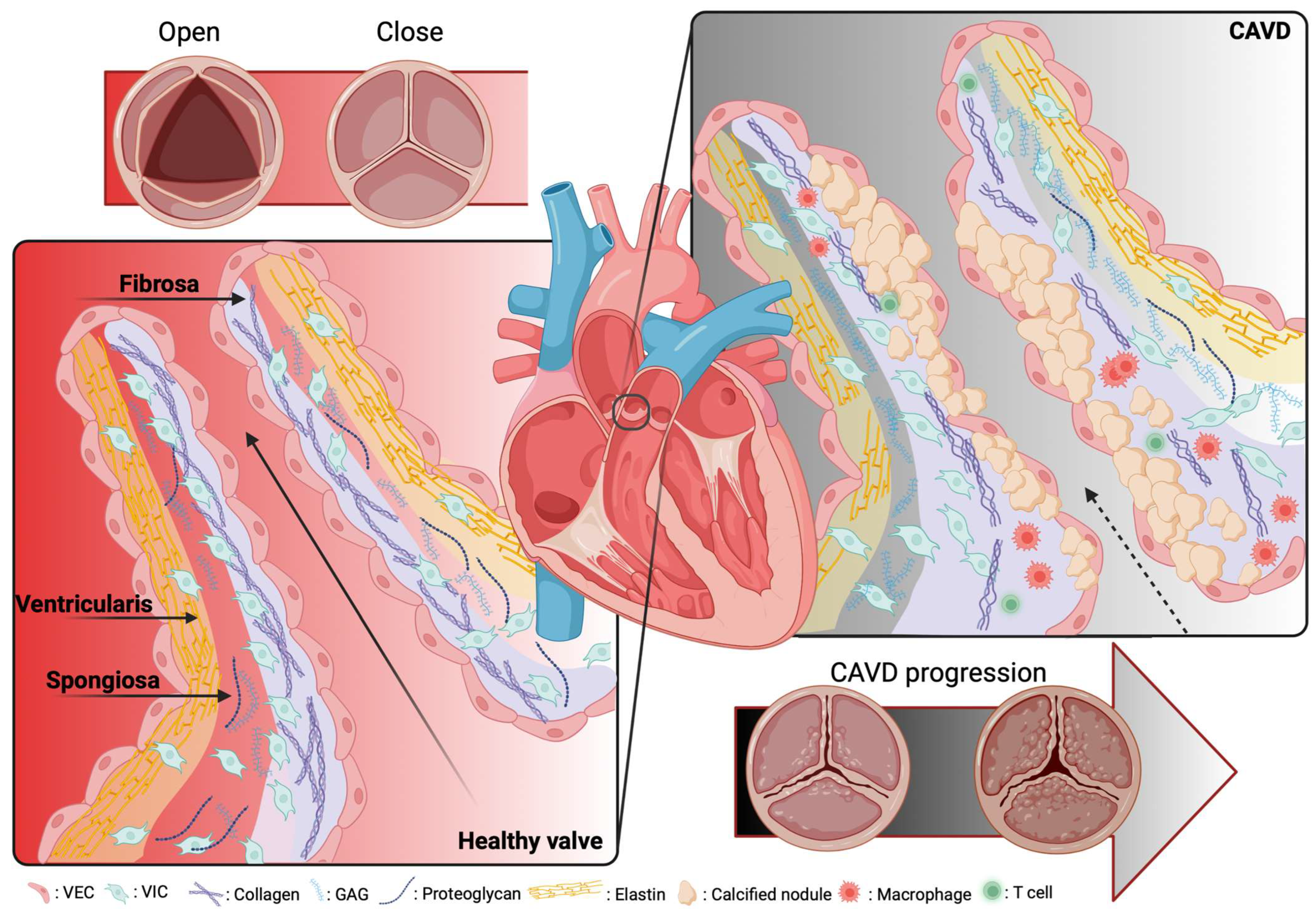
2. Overview of the Calcific Aortic Valve Disease
Epidemiology and Histological Structure
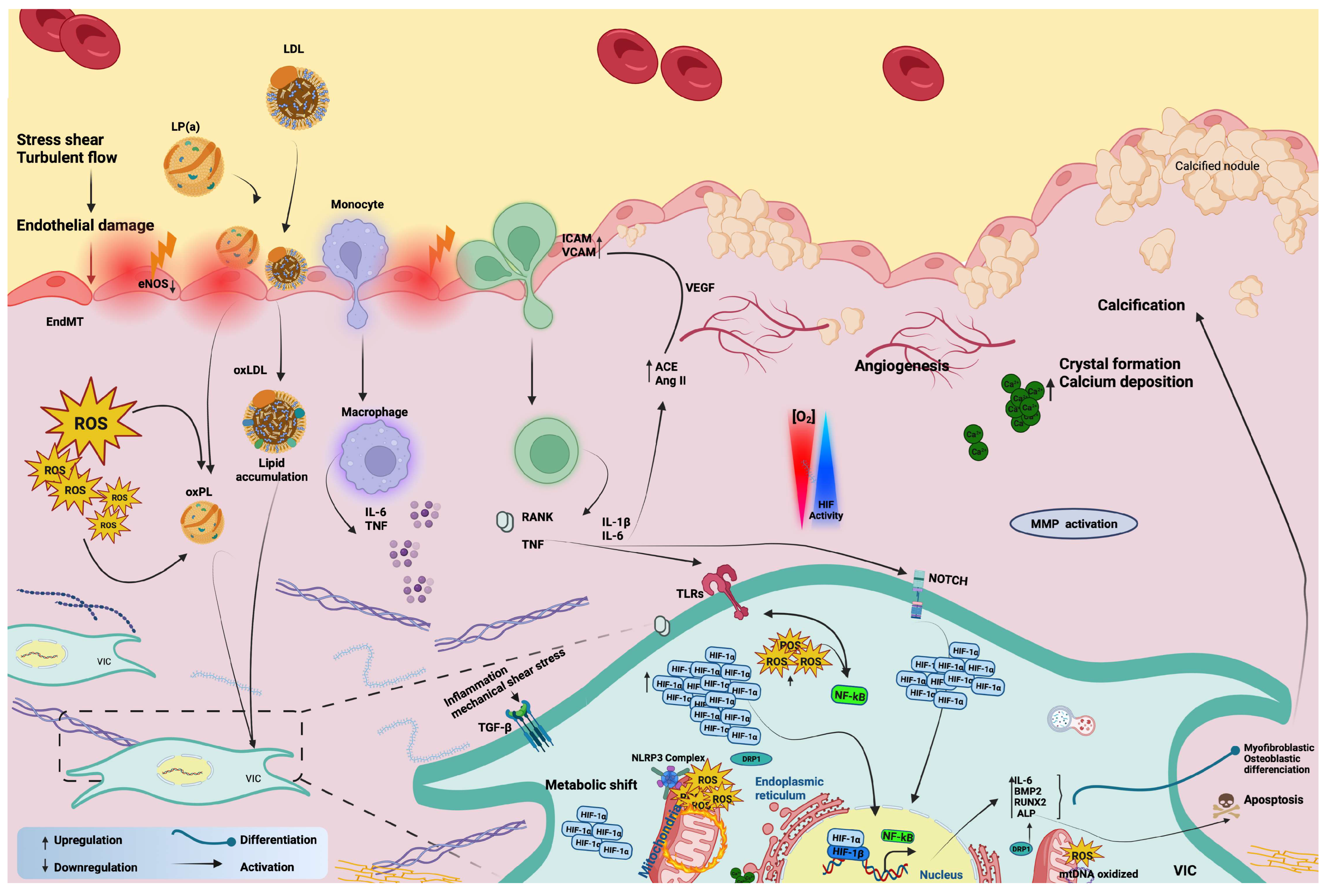
3. The Complex Interplay of Hypoxia Signaling, Mitochondrial Dysfunction, and Inflammation
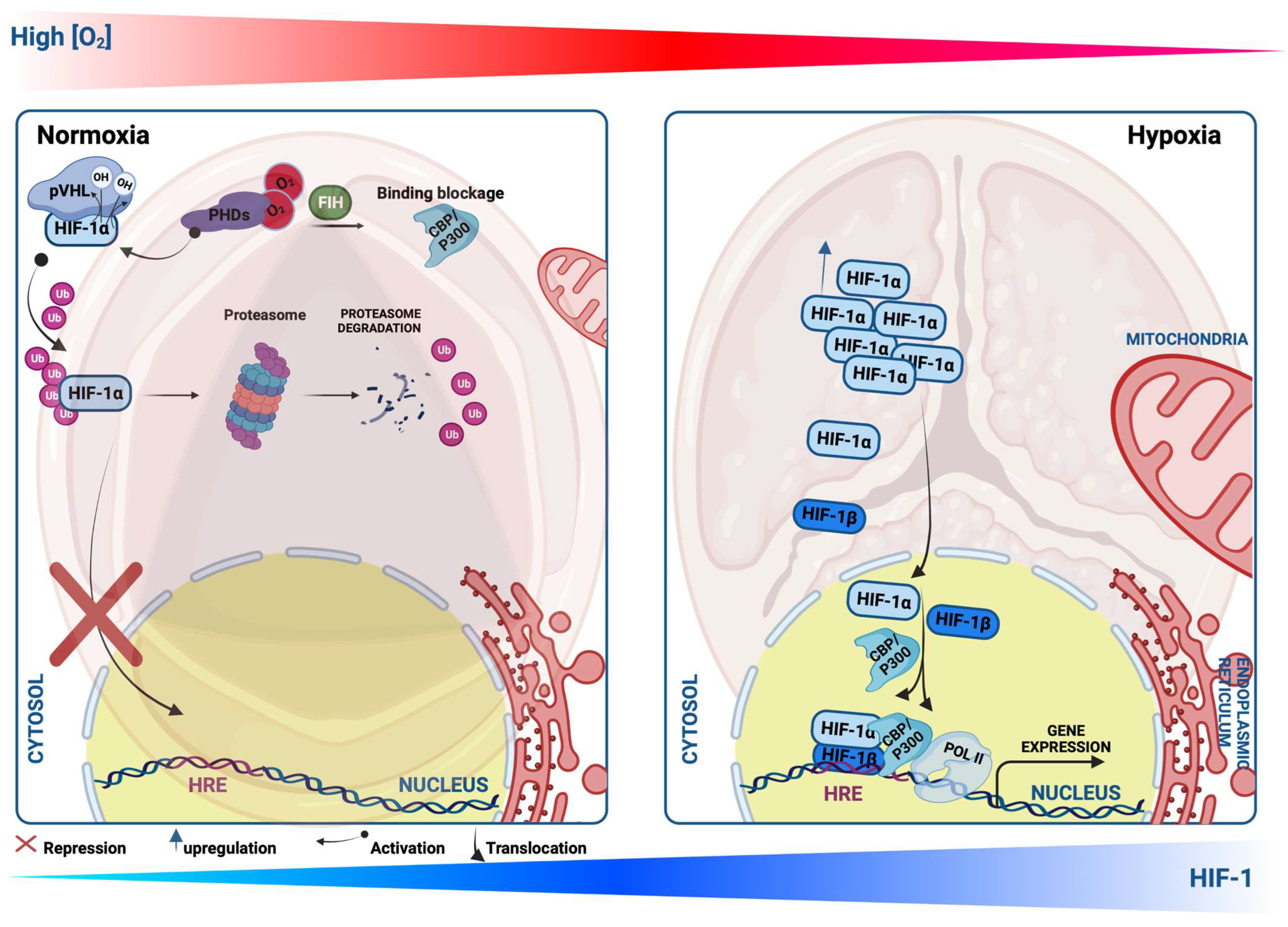
3.1. Hypoxia Signaling and Molecular Regulation of HIF-1
3.2. Hypoxia Signaling and Inflammation in CAVD
3.3. Mitochondrial Impairment in CAVD
3.4. Relationship between Inflammation and Mitochondria in CAVD
4. Novel Insights on the Therapeutic Strategies of CAVS

{kind=link}
{kind=link}
{kind=link}
| Therapeutic Target | Treatment | Mechanism of Action and Effect | References |
|---|---|---|---|
| HIF-1α inhibitors | Dimethylloxetane | Downregulates osteogenic transcription factors (RUNX2 and BMP-2), reduces vascular calcification | [168,205] |
| PX-478 | Downregulates porcine AV (PAV) calcification, and significantly blocks the deposition of calcium, thereby alleviating CAVD | [58,167] | |
| EZN-2968 | Possible to mitigate the calcification | [172] | |
| Estrogen drug therapy | - | Alleviates HIF-1α expression and vascular calcification, inhibits NFκB signaling and RNKL, blocks p53 activity and NADPH oxidase, as well as inflammation. Activates antioxidant enzymes including in mitochondria and lysosomes, as well as the cytosol | [168,169,170,206,207,208,209] |
| Angiogenesis repressor | S1P | Halts neo-vessels formation via sFlt1 activation | [65] |
| RAAS inhibitors (ACE-I therapy Ang-II blockers) | Fimasartan and losartan | Reduces CAV and a slowdown in mild CAVS development. Improves hemodynamic and left ventricular (LV) hypertrophy in patients with moderate and severe CAVS | [179,180,181,182], https://clinicaltrials.gov/show/nct01589380, https://clinicaltrials.gov/ct2/show/NCT03666351 (accessed on 30 March 2023) |
| Exportin-1 (XPO1) inhibitor | KPT-330 | Mitigates calcific nodule formation via CCAAT/enhancing-binding protein (C/EBPβ) signaling pathway, protecting against CAVS | [187] |
| Melatonine (N-acetyl-5-methoxytryptamine) | Melatonine | Improves CAV in VICs via RNA CircRIC3/miR-204-5p/DPP4 signaling and targets MT1/NF-κB/RUNX2 signaling, reducing VIC calcification-mediated osteogenic stimulation. Inhibits calcification via the AMPK/DRP1 pathway in VSMCs | [194,195,196] |
| CROT inhibitor | - | Blocks mitochondrial fragmentation, restores mitochondrial proteome changes in the osteogenic environment, and promotes fatty acid metabolism, and reduces fibrocalcification | [197] |
| PCSK9 inhibitors | Evolocumab | Reduces CAVS events | [143,202] |
| PTPB inhibitor | MSI-1436 | Prevents AV fibrocalcification, protects mitochondrial biogenesis, and performances via OPA1 | [116] |
| MMP9 inhibitor | MMP-9i | Attenuates mitochondrial impairments, represses oxidative stress, and VIC calcification | [120] |
| Autophagy activator | Rapamycin | Reverts calcification through cell death downregulation and restores calcium dysregulation in CAVS patients | [46] |
5. Conclusions and Future Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 18F-FDG | 18F-Flurodeoxyglucose |
| ACE-I | Angiotensin-converting enzyme |
| Ang-II | Angiotensin-II |
| APC/C | Complex/Cyclosome |
| AS | aortic stenosis |
| AV | Aortic valve |
| BMP-2 | Bone morphogenetic protein-2 |
| C/EBPβ | CCAAT/enhancing-binding protein |
| CACNA1C | Calcium voltage-gated channel subunit alpha 1 C |
| CAVD | Calcific aortic valve disease |
| CAVS | Calcific aortic valve stenosis |
| CBP | CREB-binding protein |
| CCL19 | C-C motif chemokine ligand 19 |
| CD11 | Surface marker of M1 |
| CLPP | Caseinolytic Mitochondrial Matrix Peptidase Proteolytic Subunit |
| CROT | Carnitine O-octanoyltransferase |
| CTB | Cytotrophoblast |
| CVD | Cardiovascular disease |
| DRP1 | Dynamin-related protein 1 |
| ECM | Extracellular matrix |
| EPAS-1 | Endothelial PAS domain protein-1 |
| EPO | Erythropoietin |
| ERK1/2 | Extracellular signal-regulated kinase |
| FAT/CD36 | Fatty acid translocase |
| FOURIER | Further Cardiovascular Outcomes Research with PCSK9 Inhibition in Subjects with Elevated Risk |
| Glut 1, 4 | Glucose transporter 1 and 4 |
| HAVEC | Human aortic endothelial cells |
| HC-OA | Human osteoarthritic |
| HF | Heart failure |
| HIF-1α | Hypoxia-inducible factor 1 alpha subunit |
| HIFs | Hypoxia-inducible transcription factors |
| HMOX1 | Heme Oxygenase 1 |
| HTRA2 | HtrA Serine Peptidase 2 |
| HUVEC | Human umbilical endothelial cells |
| ICAM-1 | Intracellular adhesion molecule-1 |
| IFN-γ | Interferon-γ |
| IHD | ischemic heart disease |
| IL- | Interleukin |
| iNOS | Inducible nitric oxide synthase |
| LDH-A | Lactate dehydrogenase-A |
| LDL | Low-density lipoprotein |
| Lp(a) | Lipoprotein a |
| LV | Left ventricular |
| MCP-1 | Monocyte chemoattractant protein-1 |
| MMP9 | Matrix metalloproteinase 9 expression |
| MMPs | Matrix metalloproteinases |
| MMP-9i | Matrix metalloproteinase 9 inhibitor |
| MT1 | Melatonin receptor |
| mtDNA | Mitochondrial DNA |
| mtROS | Mitochondrial ROS |
| mtUPR | Mitochondrial unfolded protein response |
| N-acetyl-5-methoxytryptamine | Melatonine |
| NF-κB | Nuclear factor kappa B |
| NLRC4 | Pyrin domain-containing protein |
| NOS | Nitric oxide synthase |
| O2 | Oxygen |
| OPA1 | Optic atrophy 1 |
| OXPHOS | Oxidative phosphorylation |
| PAV | Porcine AV |
| PCSK9 | Proprotein convertase subtilisin/kexin type 9 |
| PDK | Dehydrogenase kinase isoform 1 |
| PET | Positron emission tomography |
| PGK1 | Phosphoglycerate kinase-1 |
| PHD1, -2, -3 | Propyl-hydroxylase-1, -2, and -3 enzyme activity |
| PI3K/Akt | P hosphatidylinositol 3-kinase (PI3K)-Akt (protein kinase B) |
| PKM2 | M2 isoform of pyruvate kinase |
| PTP1B | Protein tyrosine phosphatase 1B |
| pVHL | von Hippel–Lindau |
| RANKL | Receptor-activator of nuclear factor κB ligand |
| SCG2 | Secretogranin II |
| sFlt1 | Soluble fms-like tyrosine kinase 1 |
| SMC | Smooth muscle cells |
| SOD2 | Superoxide dismutase 2 |
| STAT3 | Signal Transducer and Activator of Transcription 3 |
| TG | Triglyceride levels |
| TGF-β | Transforming growth factor-beta |
| TLR | Toll-like receptor |
| UBE2C | Ubiquitin E2 ligase C |
| VECs | Valvular endothelial cells |
| VEGF | Vascular endothelial growth factor |
| VICs | valvular interstitial cells |
| VSMC | Vascular smooth muscle cells |
| XPO1 | Exportin-1 |
References
- Aisagbonhi, O.; Rai, M.; Ryzhov, S.; Atria, N.; Feoktistov, I.; Hatzopoulos, A.K. Experimental myocardial infarction triggers canonical Wnt signaling and endothelial-to-mesenchymal transition. Dis. Model. Mech. 2011, 4, 469–483. [Google Scholar] [CrossRef]
- Akahori, H.; Tsujino, T.; Naito, Y.; Sawada, H.; Sugahara, M.; Fukui, M.; Ohyanagi, M.; Mitsuno, M.; Miyamoto, Y.; Masuyama, T. Nuclear factor-kappaB-hypoxia-inducible factor-2 pathway in aortic valve stenosis. J. Heart Valve Dis. 2014, 23, 558–566. [Google Scholar]
- Andersson, C.; Abdulla, J. Is the use of renin-angiotensin system inhibitors in patients with aortic valve stenosis safe and of prognostic benefit? A systematic review and meta-analysis. Eur. Heart J. Cardiovasc. Pharmacother. 2017, 3, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Angelo, L.S.; Kurzrock, R. Vascular endothelial growth factor and its relationship to inflammatory mediators. Clin. Cancer Res. 2007, 13, 2825–2830. [Google Scholar] [CrossRef] [PubMed]
- Bai, C.G.; Liu, X.H.; Liu, W.Q.; Ma, D.L. Regional expression of the hypoxia-inducible factor (HIF) system and association with cardiomyocyte cell cycle re-entry after myocardial infarction in rats. Heart Vessels 2008, 23, 193–200. [Google Scholar] [CrossRef]
- Bergmark, B.A.; O’Donoghue, M.L.; Murphy, S.A.; Kuder, J.F.; Ezhov, M.V.; Ceska, R.; Gouni-Berthold, I.; Jensen, H.K.; Tokgozoglu, S.L.; Mach, F.; et al. An Exploratory Analysis of Proprotein Convertase Subtilisin/Kexin Type 9 Inhibition and Aortic Stenosis in the FOURIER Trial. JAMA Cardiol. 2020, 5, 709–713. [Google Scholar] [CrossRef]
- Bertazzo, S.; Gentleman, E.; Cloyd, K.L.; Chester, A.H.; Yacoub, M.H.; Stevens, M.M. Nano-analytical electron microscopy reveals fundamental insights into human cardiovascular tissue calcification. Nat. Mater. 2013, 12, 576–583. [Google Scholar] [CrossRef]
- Bonora, M.; Giorgi, C.; Pinton, P. Molecular mechanisms and consequences of mitochondrial permeability transition. Nat. Rev. Mol. Cell Biol. 2022, 23, 266–285. [Google Scholar] [CrossRef]
- Bonora, M.; Pinton, P. A New Current for the Mitochondrial Permeability Transition. Trends Biochem. Sci. 2019, 44, 559–561. [Google Scholar] [CrossRef] [PubMed]
- Borchi, E.; Bargelli, V.; Stillitano, F.; Giordano, C.; Sebastiani, M.; Nassi, P.A.; d’Amati, G.; Cerbai, E.; Nediani, C. Enhanced ROS production by NADPH oxidase is correlated to changes in antioxidant enzyme activity in human heart failure. Biochim. Biophys. Acta 2010, 1802, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Bouchareb, R.; Boulanger, M.C.; Fournier, D.; Pibarot, P.; Messaddeq, Y.; Mathieu, P. Mechanical strain induces the production of spheroid mineralized microparticles in the aortic valve through a RhoA/ROCK-dependent mechanism. J. Mol. Cell. Cardiol. 2014, 67, 49–59. [Google Scholar] [CrossRef]
- Bouhamida, E.; Morciano, G.; Perrone, M.; Kahsay, A.E.; Della Sala, M.; Wieckowski, M.R.; Fiorica, F.; Pinton, P.; Giorgi, C.; Patergnani, S. The Interplay of Hypoxia Signaling on Mitochondrial Dysfunction and Inflammation in Cardiovascular Diseases and Cancer: From Molecular Mechanisms to Therapeutic Approaches. Biology 2022, 11, 300. [Google Scholar] [CrossRef] [PubMed]
- Bull, S.; Loudon, M.; Francis, J.M.; Joseph, J.; Gerry, S.; Karamitsos, T.D.; Prendergast, B.D.; Banning, A.P.; Neubauer, S.; Myerson, S.G. A prospective, double-blind, randomized controlled trial of the angiotensin-converting enzyme inhibitor Ramipril In Aortic Stenosis (RIAS trial). Eur. Heart J. Cardiovasc. Imaging 2015, 16, 834–841. [Google Scholar] [CrossRef]
- Butcher, J.T.; Nerem, R.M. Valvular endothelial cells regulate the phenotype of interstitial cells in co-culture: Effects of steady shear stress. Tissue Eng. 2006, 12, 905–915. [Google Scholar] [CrossRef] [PubMed]
- Butcher, J.T.; Penrod, A.M.; Garcia, A.J.; Nerem, R.M. Unique morphology and focal adhesion development of valvular endothelial cells in static and fluid flow environments. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1429–1434. [Google Scholar] [CrossRef]
- Byon, C.H.; Javed, A.; Dai, Q.; Kappes, J.C.; Clemens, T.L.; Darley-Usmar, V.M.; McDonald, J.M.; Chen, Y. Oxidative stress induces vascular calcification through modulation of the osteogenic transcription factor Runx2 by AKT signaling. J. Biol. Chem. 2008, 283, 15319–15327. [Google Scholar] [CrossRef] [PubMed]
- Carracedo, M.; Back, M. Fetuin A in aortic stenosis and valve calcification: Not crystal clear. Int. J. Cardiol. 2018, 265, 77–78. [Google Scholar] [CrossRef] [PubMed]
- Castardo-de-Paula, J.C.; de Campos, B.H.; Amorim, E.D.T.; da Silva, R.V.; de Farias, C.C.; Higachi, L.; Pinge-Filho, P.; Barbosa, D.S.; Martins-Pinge, M.C. Cardiovascular risk and the effect of nitric oxide synthase inhibition in female rats: The role of estrogen. Exp. Gerontol. 2017, 97, 38–48. [Google Scholar] [CrossRef]
- Castellazzi, M.; Patergnani, S.; Donadio, M.; Giorgi, C.; Bonora, M.; Fainardi, E.; Casetta, I.; Granieri, E.; Pugliatti, M.; Pinton, P. Correlation between auto/mitophagic processes and magnetic resonance imaging activity in multiple sclerosis patients. J. Neuroinflamm. 2019, 16, 131. [Google Scholar] [CrossRef]
- Chen, J.; Lin, Y.; Sun, Z. Inhibition of miR-101-3p prevents human aortic valve interstitial cell calcification through regulation of CDH11/SOX9 expression. Mol. Med. 2023, 29, 24. [Google Scholar] [CrossRef]
- Chen, J.H.; Simmons, C.A. Cell-matrix interactions in the pathobiology of calcific aortic valve disease: Critical roles for matricellular, matricrine, and matrix mechanics cues. Circ. Res. 2011, 108, 1510–1524. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.R.; Liu, H.B.; Chen, Y.D.; Sha, Y.; Ma, Q.; Zhu, P.J.; Mu, Y. Melatonin Attenuates Myocardial Ischemia/Reperfusion Injury by Inhibiting Autophagy Via an AMPK/mTOR Signaling Pathway. Cell. Physiol. Biochem. 2018, 47, 2067–2076. [Google Scholar] [CrossRef]
- Chen, W.R.; Zhou, Y.J.; Sha, Y.; Wu, X.P.; Yang, J.Q.; Liu, F. Melatonin attenuates vascular calcification by inhibiting mitochondria fission via an AMPK/Drp1 signalling pathway. J. Cell. Mol. Med. 2020, 24, 6043–6054. [Google Scholar] [CrossRef]
- Chignalia, A.Z.; Oliveira, M.A.; Debbas, V.; Dull, R.O.; Laurindo, F.R.; Touyz, R.M.; Carvalho, M.H.; Fortes, Z.B.; Tostes, R.C. Testosterone induces leucocyte migration by NADPH oxidase-driven ROS- and COX2-dependent mechanisms. Clin. Sci. 2015, 129, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Chockalingam, A.; Venkatesan, S.; Subramaniam, T.; Jagannathan, V.; Elangovan, S.; Alagesan, R.; Gnanavelu, G.; Dorairajan, S.; Krishna, B.P.; Chockalingam, V.; et al. Safety and efficacy of angiotensin-converting enzyme inhibitors in symptomatic severe aortic stenosis: Symptomatic Cardiac Obstruction-Pilot Study of Enalapril in Aortic Stenosis (SCOPE-AS). Am. Heart J. 2004, 147, E19. [Google Scholar] [CrossRef]
- Chowdhury, A.; Aich, A.; Jain, G.; Wozny, K.; Luchtenborg, C.; Hartmann, M.; Bernhard, O.; Balleiniger, M.; Alfar, E.A.; Zieseniss, A.; et al. Defective Mitochondrial Cardiolipin Remodeling Dampens HIF-1alpha Expression in Hypoxia. Cell Rep. 2018, 25, 561–570.e566. [Google Scholar] [CrossRef]
- Coffey, S.; Cox, B.; Williams, M.J. The prevalence, incidence, progression, and risks of aortic valve sclerosis: A systematic review and meta-analysis. J. Am. Coll. Cardiol. 2014, 63, 2852–2861. [Google Scholar] [CrossRef] [PubMed]
- Crouzier, L.; Danese, A.; Yasui, Y.; Richard, E.M.; Lievens, J.C.; Patergnani, S.; Couly, S.; Diez, C.; Denus, M.; Cubedo, N.; et al. Activation of the sigma-1 receptor chaperone alleviates symptoms of Wolfram syndrome in preclinical models. Sci. Transl. Med. 2022, 14, eabh3763. [Google Scholar] [CrossRef]
- Cui, J.; Li, Z.; Zhuang, S.; Qi, S.; Li, L.; Zhou, J.; Zhang, W.; Zhao, Y. Melatonin alleviates inflammation-induced apoptosis in human umbilical vein endothelial cells via suppression of Ca2+-XO-ROS-Drp1-mitochondrial fission axis by activation of AMPK/SERCA2a pathway. Cell Stress Chaperones 2018, 23, 281–293. [Google Scholar] [CrossRef]
- Dachs, G.U.; Tozer, G.M. Hypoxia modulated gene expression: Angiogenesis, metastasis and therapeutic exploitation. Eur. J. Cancer 2000, 36, 1649–1660. [Google Scholar] [CrossRef]
- Dalsgaard, M.; Iversen, K.; Kjaergaard, J.; Grande, P.; Goetze, J.P.; Clemmensen, P.; Hassager, C. Short-term hemodynamic effect of angiotensin-converting enzyme inhibition in patients with severe aortic stenosis: A placebo-controlled, randomized study. Am. Heart J. 2014, 167, 226–234. [Google Scholar] [CrossRef]
- Danese, A.; Patergnani, S.; Maresca, A.; Peron, C.; Raimondi, A.; Caporali, L.; Marchi, S.; La Morgia, C.; Del Dotto, V.; Zanna, C.; et al. Pathological mitophagy disrupts mitochondrial homeostasis in Leber’s hereditary optic neuropathy. Cell Rep. 2022, 40, 111124. [Google Scholar] [CrossRef]
- Dhalla, A.K.; Hill, M.F.; Singal, P.K. Role of oxidative stress in transition of hypertrophy to heart failure. J. Am. Coll. Cardiol. 1996, 28, 506–514. [Google Scholar] [CrossRef]
- Di Minno, A.; Zanobini, M.; Myasoedova, V.A.; Valerio, V.; Songia, P.; Saccocci, M.; Di Minno, M.N.D.; Tremoli, E.; Poggio, P. Could circulating fetuin A be a biomarker of aortic valve stenosis? Int. J. Cardiol. 2017, 249, 426–430. [Google Scholar] [CrossRef]
- Dinarello, C.A.; Nold-Petry, C.; Nold, M.; Fujita, M.; Li, S.; Kim, S.; Bufler, P. Suppression of innate inflammation and immunity by interleukin-37. Eur. J. Immunol. 2016, 46, 1067–1081. [Google Scholar] [CrossRef]
- Ding, M.; Feng, N.; Tang, D.; Feng, J.; Li, Z.; Jia, M.; Liu, Z.; Gu, X.; Wang, Y.; Fu, F.; et al. Melatonin prevents Drp1-mediated mitochondrial fission in diabetic hearts through SIRT1-PGC1alpha pathway. J. Pineal Res. 2018, 65, e12491. [Google Scholar] [CrossRef]
- GBD 2019 Risk Factors Collaborators. Global burden of 369 diseases and injuries in 204 countries and territories, 1990–2019: A systematic analysis for the Global Burden of Disease Study 2019. Lancet 2020, 396, 1204–1222. [Google Scholar] [CrossRef]
- Dodd, M.S.; Sousa Fialho, M.D.L.; Montes Aparicio, C.N.; Kerr, M.; Timm, K.N.; Griffin, J.L.; Luiken, J.; Glatz, J.F.C.; Tyler, D.J.; Heather, L.C. Fatty Acids Prevent Hypoxia-Inducible Factor-1alpha Signaling Through Decreased Succinate in Diabetes. JACC Basic Transl. Sci. 2018, 3, 485–498. [Google Scholar] [CrossRef] [PubMed]
- Duan, C.; Cao, Z.; Tang, F.; Jian, Z.; Liang, C.; Liu, H.; Xiao, Y.; Liu, L.; Ma, R. miRNA-mRNA crosstalk in myocardial ischemia induced by calcified aortic valve stenosis. Aging 2019, 11, 448–466. [Google Scholar] [CrossRef] [PubMed]
- Dutta, P.; Kodigepalli, K.M.; LaHaye, S.; Thompson, J.W.; Rains, S.; Nagel, C.; Thatcher, K.; Hinton, R.B.; Lincoln, J. KPT-330 Prevents Aortic Valve Calcification via a Novel C/EBPbeta Signaling Pathway. Circ. Res. 2021, 128, 1300–1316. [Google Scholar] [CrossRef]
- Dweck, M.R.; Boon, N.A.; Newby, D.E. Calcific aortic stenosis: A disease of the valve and the myocardium. J. Am. Coll. Cardiol. 2012, 60, 1854–1863. [Google Scholar] [CrossRef] [PubMed]
- Dweck, M.R.; Jones, C.; Joshi, N.V.; Fletcher, A.M.; Richardson, H.; White, A.; Marsden, M.; Pessotto, R.; Clark, J.C.; Wallace, W.A.; et al. Assessment of valvular calcification and inflammation by positron emission tomography in patients with aortic stenosis. Circulation 2012, 125, 76–86. [Google Scholar] [CrossRef] [PubMed]
- Dweck, M.R.; Khaw, H.J.; Sng, G.K.; Luo, E.L.; Baird, A.; Williams, M.C.; Makiello, P.; Mirsadraee, S.; Joshi, N.V.; van Beek, E.J.; et al. Aortic stenosis, atherosclerosis, and skeletal bone: Is there a common link with calcification and inflammation? Eur. Heart J. 2013, 34, 1567–1574. [Google Scholar] [CrossRef] [PubMed]
- El Husseini, D.; Boulanger, M.C.; Mahmut, A.; Bouchareb, R.; Laflamme, M.H.; Fournier, D.; Pibarot, P.; Bosse, Y.; Mathieu, P. P2Y2 receptor represses IL-6 expression by valve interstitial cells through Akt: Implication for calcific aortic valve disease. J. Mol. Cell. Cardiol. 2014, 72, 146–156. [Google Scholar] [CrossRef]
- Engberding, N.; Spiekermann, S.; Schaefer, A.; Heineke, A.; Wiencke, A.; Muller, M.; Fuchs, M.; Hilfiker-Kleiner, D.; Hornig, B.; Drexler, H.; et al. Allopurinol attenuates left ventricular remodeling and dysfunction after experimental myocardial infarction: A new action for an old drug? Circulation 2004, 110, 2175–2179. [Google Scholar] [CrossRef]
- Fang, C.; Dai, L.; Wang, C.; Fan, C.; Yu, Y.; Yang, L.; Deng, H.; Zhou, Z. Secretogranin II impairs tumor growth and angiogenesis by promoting degradation of hypoxia-inducible factor-1alpha in colorectal cancer. Mol. Oncol. 2021, 15, 3513–3526. [Google Scholar] [CrossRef]
- GBD 2019 Risk Factors Collaborators. Global burden of 87 risk factors in 204 countries and territories, 1990–2019: A systematic analysis for the Global Burden of Disease Study 2019. Lancet 2020, 396, 1223–1249. [Google Scholar] [CrossRef]
- Farrar, E.J.; Huntley, G.D.; Butcher, J. Endothelial-derived oxidative stress drives myofibroblastic activation and calcification of the aortic valve. PLoS ONE 2015, 10, e0123257. [Google Scholar] [CrossRef]
- Feldhoff, L.M.; Rueda, C.M.; Moreno-Fernandez, M.E.; Sauer, J.; Jackson, C.M.; Chougnet, C.A.; Rupp, J. IL-1beta induced HIF-1alpha inhibits the differentiation of human FOXP3+ T cells. Sci. Rep. 2017, 7, 465. [Google Scholar] [CrossRef]
- Fernandez-Pisonero, I.; Lopez, J.; Onecha, E.; Duenas, A.I.; Maeso, P.; Crespo, M.S.; San Roman, J.A.; Garcia-Rodriguez, C. Synergy between sphingosine 1-phosphate and lipopolysaccharide signaling promotes an inflammatory, angiogenic and osteogenic response in human aortic valve interstitial cells. PLoS ONE 2014, 9, e109081. [Google Scholar] [CrossRef]
- Fernandez Esmerats, J.; Villa-Roel, N.; Kumar, S.; Gu, L.; Salim, M.T.; Ohh, M.; Taylor, W.R.; Nerem, R.M.; Yoganathan, A.P.; Jo, H. Disturbed Flow Increases UBE2C (Ubiquitin E2 Ligase C) via Loss of miR-483-3p, Inducing Aortic Valve Calcification by the pVHL (von Hippel-Lindau Protein) and HIF-1alpha (Hypoxia-Inducible Factor-1alpha) Pathway in Endothelial Cells. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 467–481. [Google Scholar] [CrossRef]
- Ferrara, N.; Gerber, H.P.; LeCouter, J. The biology of VEGF and its receptors. Nat. Med. 2003, 9, 669–676. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, S.; Pesce, M. The Complex Interplay of Inflammation, Metabolism, Epigenetics, and Sex in Calcific Disease of the Aortic Valve. Front. Cardiovasc. Med. 2021, 8, 791646. [Google Scholar] [CrossRef] [PubMed]
- Ferreira-Gonzalez, I.; Pinar-Sopena, J.; Ribera, A.; Marsal, J.R.; Cascant, P.; Gonzalez-Alujas, T.; Evangelista, A.; Brotons, C.; Moral, I.; Permanyer-Miralda, G.; et al. Prevalence of calcific aortic valve disease in the elderly and associated risk factors: A population-based study in a Mediterranean area. Eur. J. Prev. Cardiol. 2013, 20, 1022–1030. [Google Scholar] [CrossRef] [PubMed]
- Forsythe, J.A.; Jiang, B.H.; Iyer, N.V.; Agani, F.; Leung, S.W.; Koos, R.D.; Semenza, G.L. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol. Cell. Biol. 1996, 16, 4604–4613. [Google Scholar] [CrossRef]
- Gendron, N.; Rosa, M.; Blandinieres, A.; Sottejeau, Y.; Rossi, E.; Van Belle, E.; Idelcadi, S.; Lecourt, S.; Vincentelli, A.; Cras, A.; et al. Human Aortic Valve Interstitial Cells Display Proangiogenic Properties During Calcific Aortic Valve Disease. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 415–429. [Google Scholar] [CrossRef] [PubMed]
- Ghaisas, N.K.; Foley, J.B.; O’Briain, D.S.; Crean, P.; Kelleher, D.; Walsh, M. Adhesion molecules in nonrheumatic aortic valve disease: Endothelial expression, serum levels and effects of valve replacement. J. Am. Coll. Cardiol. 2000, 36, 2257–2262. [Google Scholar] [CrossRef] [PubMed]
- Gil-Gil, M.J.; Mesia, C.; Rey, M.; Bruna, J. Bevacizumab for the treatment of glioblastoma. Clin. Med. Insights Oncol. 2013, 7, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Giorgi, C.; Bouhamida, E.; Danese, A.; Previati, M.; Pinton, P.; Patergnani, S. Relevance of Autophagy and Mitophagy Dynamics and Markers in Neurodegenerative Diseases. Biomedicines 2021, 9, 149. [Google Scholar] [CrossRef]
- Giorgi, C.; Danese, A.; Missiroli, S.; Patergnani, S.; Pinton, P. Calcium Dynamics as a Machine for Decoding Signals. Trends Cell Biol. 2018, 28, 258–273. [Google Scholar] [CrossRef]
- Giorgi, C.; Marchi, S.; Pinton, P. The machineries, regulation and cellular functions of mitochondrial calcium. Nat. Rev. Mol. Cell Biol. 2018, 19, 713–730. [Google Scholar] [CrossRef] [PubMed]
- Gravina, G.L.; Mancini, A.; Sanita, P.; Vitale, F.; Marampon, F.; Ventura, L.; Landesman, Y.; McCauley, D.; Kauffman, M.; Shacham, S.; et al. KPT-330, a potent and selective exportin-1 (XPO-1) inhibitor, shows antitumor effects modulating the expression of cyclin D1 and survivin [corrected] in prostate cancer models. BMC Cancer 2015, 15, 941. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, H.Z.E.; Zhao, G.; Shah, A.M.; Zhang, M. Role of oxidative stress in calcific aortic valve disease and its therapeutic implications. Cardiovasc. Res. 2022, 118, 1433–1451. [Google Scholar] [CrossRef] [PubMed]
- Greene, A.W.; Grenier, K.; Aguileta, M.A.; Muise, S.; Farazifard, R.; Haque, M.E.; McBride, H.M.; Park, D.S.; Fon, E.A. Mitochondrial processing peptidase regulates PINK1 processing, import and Parkin recruitment. EMBO Rep. 2012, 13, 378–385. [Google Scholar] [CrossRef]
- Guauque-Olarte, S.; Messika-Zeitoun, D.; Droit, A.; Lamontagne, M.; Tremblay-Marchand, J.; Lavoie-Charland, E.; Gaudreault, N.; Arsenault, B.J.; Dube, M.P.; Tardif, J.C.; et al. Calcium Signaling Pathway Genes RUNX2 and CACNA1C Are Associated With Calcific Aortic Valve Disease. Circ. Cardiovasc. Genet. 2015, 8, 812–822. [Google Scholar] [CrossRef]
- Hakuno, D.; Kimura, N.; Yoshioka, M.; Fukuda, K. Molecular mechanisms underlying the onset of degenerative aortic valve disease. J. Mol. Med. 2009, 87, 17–24. [Google Scholar] [CrossRef]
- Head, S.J.; Celik, M.; Kappetein, A.P. Mechanical versus bioprosthetic aortic valve replacement. Eur. Heart J. 2017, 38, 2183–2191. [Google Scholar] [CrossRef]
- Heather, L.C.; Howell, N.J.; Emmanuel, Y.; Cole, M.A.; Frenneaux, M.P.; Pagano, D.; Clarke, K. Changes in cardiac substrate transporters and metabolic proteins mirror the metabolic shift in patients with aortic stenosis. PLoS ONE 2011, 6, e26326. [Google Scholar] [CrossRef]
- Hutcheson, J.D.; Aikawa, E.; Merryman, W.D. Potential drug targets for calcific aortic valve disease. Nat. Rev. Cardiol. 2014, 11, 218–231. [Google Scholar] [CrossRef]
- Isoda, K.; Matsuki, T.; Kondo, H.; Iwakura, Y.; Ohsuzu, F. Deficiency of interleukin-1 receptor antagonist induces aortic valve disease in BALB/c mice. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 708–715. [Google Scholar] [CrossRef]
- Jansen, F.; Xiang, X.; Werner, N. Role and function of extracellular vesicles in calcific aortic valve disease. Eur. Heart J. 2017, 38, 2714–2716. [Google Scholar] [CrossRef] [PubMed]
- Jaskiewicz, M.; Moszynska, A.; Kroliczewski, J.; Cabaj, A.; Bartoszewska, S.; Charzynska, A.; Gebert, M.; Dabrowski, M.; Collawn, J.F.; Bartoszewski, R. The transition from HIF-1 to HIF-2 during prolonged hypoxia results from reactivation of PHDs and HIF1A mRNA instability. Cell Mol. Biol. Lett. 2022, 27, 109. [Google Scholar] [CrossRef] [PubMed]
- Jeong, W.; Rapisarda, A.; Park, S.R.; Kinders, R.J.; Chen, A.; Melillo, G.; Turkbey, B.; Steinberg, S.M.; Choyke, P.; Doroshow, J.H.; et al. Pilot trial of EZN-2968, an antisense oligonucleotide inhibitor of hypoxia-inducible factor-1 alpha (HIF-1alpha), in patients with refractory solid tumors. Cancer Chemother. Pharmacol. 2014, 73, 343–348. [Google Scholar] [CrossRef]
- Jersmann, H.P.; Dransfield, I.; Hart, S.P. Fetuin/alpha2-HS glycoprotein enhances phagocytosis of apoptotic cells and macropinocytosis by human macrophages. Clin. Sci. 2003, 105, 273–278. [Google Scholar] [CrossRef]
- Jiang, B.H.; Rue, E.; Wang, G.L.; Roe, R.; Semenza, G.L. Dimerization, DNA binding, and transactivation properties of hypoxia-inducible factor 1. J. Biol. Chem. 1996, 271, 17771–17778. [Google Scholar] [CrossRef]
- Jiang, X.; Tian, W.; Tu, A.B.; Pasupneti, S.; Shuffle, E.; Dahms, P.; Zhang, P.; Cai, H.; Dinh, T.T.; Liu, B.; et al. Endothelial Hypoxia-Inducible Factor-2alpha Is Required for the Maintenance of Airway Microvasculature. Circulation 2019, 139, 502–517. [Google Scholar] [CrossRef]
- Jimenez-Candil, J.; Bermejo, J.; Yotti, R.; Cortina, C.; Moreno, M.; Cantalapiedra, J.L.; Garcia-Fernandez, M.A. Effects of angiotensin converting enzyme inhibitors in hypertensive patients with aortic valve stenosis: A drug withdrawal study. Heart 2005, 91, 1311–1318. [Google Scholar] [CrossRef]
- Jung, Y.J.; Isaacs, J.S.; Lee, S.; Trepel, J.; Neckers, L. IL-1beta-mediated up-regulation of HIF-1alpha via an NFkappaB/COX-2 pathway identifies HIF-1 as a critical link between inflammation and oncogenesis. FASEB J. 2003, 17, 2115–2117. [Google Scholar] [CrossRef] [PubMed]
- Kaden, J.J.; Dempfle, C.E.; Grobholz, R.; Fischer, C.S.; Vocke, D.C.; Kilic, R.; Sarikoc, A.; Pinol, R.; Hagl, S.; Lang, S.; et al. Inflammatory regulation of extracellular matrix remodeling in calcific aortic valve stenosis. Cardiovasc. Pathol. 2005, 14, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Kaden, J.J.; Dempfle, C.E.; Grobholz, R.; Tran, H.T.; Kilic, R.; Sarikoc, A.; Brueckmann, M.; Vahl, C.; Hagl, S.; Haase, K.K.; et al. Interleukin-1 beta promotes matrix metalloproteinase expression and cell proliferation in calcific aortic valve stenosis. Atherosclerosis 2003, 170, 205–211. [Google Scholar] [CrossRef]
- Kim, J.W.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006, 3, 177–185. [Google Scholar] [CrossRef]
- Kobayashi, K.; Maeda, K.; Takefuji, M.; Kikuchi, R.; Morishita, Y.; Hirashima, M.; Murohara, T. Dynamics of angiogenesis in ischemic areas of the infarcted heart. Sci. Rep. 2017, 7, 7156. [Google Scholar] [CrossRef] [PubMed]
- Kokudo, T.; Suzuki, Y.; Yoshimatsu, Y.; Yamazaki, T.; Watabe, T.; Miyazono, K. Snail is required for TGFbeta-induced endothelial-mesenchymal transition of embryonic stem cell-derived endothelial cells. J. Cell Sci. 2008, 121, 3317–3324. [Google Scholar] [CrossRef] [PubMed]
- Koos, R.; Brandenburg, V.; Mahnken, A.H.; Muhlenbruch, G.; Stanzel, S.; Gunther, R.W.; Floege, J.; Jahnen-Dechent, W.; Kelm, M.; Kuhl, H.P. Association of fetuin-A levels with the progression of aortic valve calcification in non-dialyzed patients. Eur. Heart J. 2009, 30, 2054–2061. [Google Scholar] [CrossRef]
- Kornicka-Garbowska, K.; Bourebaba, L.; Rocken, M.; Marycz, K. Inhibition of protein tyrosine phosphatase improves mitochondrial bioenergetics and dynamics, reduces oxidative stress, and enhances adipogenic differentiation potential in metabolically impaired progenitor stem cells. Cell Commun. Signal. 2021, 19, 106. [Google Scholar] [CrossRef]
- Kronenberg, F.; Mora, S.; Stroes, E.S.G.; Ference, B.A.; Arsenault, B.J.; Berglund, L.; Dweck, M.R.; Koschinsky, M.; Lambert, G.; Mach, F.; et al. Lipoprotein(a) in atherosclerotic cardiovascular disease and aortic stenosis: A European Atherosclerosis Society consensus statement. Eur. Heart J. 2022, 43, 3925–3946. [Google Scholar] [CrossRef]
- Lagace, T.A.; Curtis, D.E.; Garuti, R.; McNutt, M.C.; Park, S.W.; Prather, H.B.; Anderson, N.N.; Ho, Y.K.; Hammer, R.E.; Horton, J.D. Secreted PCSK9 decreases the number of LDL receptors in hepatocytes and in livers of parabiotic mice. J. Clin. Investig. 2006, 116, 2995–3005. [Google Scholar] [CrossRef]
- Leopold, J.A. PCSK9 and Calcific Aortic Valve Stenosis: Moving Beyond Lipids. JACC Basic Transl. Sci. 2020, 5, 662–664. [Google Scholar] [CrossRef]
- Lewis, C.T.A.; Mascall, K.S.; Wilson, H.M.; Murray, F.; Kerr, K.M.; Gibson, G.; Buchan, K.; Small, G.R.; Nixon, G.F. An endogenous inhibitor of angiogenesis downregulated by hypoxia in human aortic valve stenosis promotes disease pathogenesis. J. Mol. Cell. Cardiol. 2023, 174, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Qiao, W.; Zhang, W.; Li, F.; Shi, J.; Dong, N. The shift of macrophages toward M1 phenotype promotes aortic valvular calcification. J. Thorac. Cardiovasc. Surg. 2017, 153, 1318–1327.e1311. [Google Scholar] [CrossRef]
- Li, S.J.; Cheng, W.L.; Kao, Y.H.; Chung, C.C.; Trang, N.N.; Chen, Y.J. Melatonin Inhibits NF-kappaB/CREB/Runx2 Signaling and Alleviates Aortic Valve Calcification. Front. Cardiovasc. Med. 2022, 9, 885293. [Google Scholar] [CrossRef]
- Li, X.Z.; Xiong, Z.C.; Zhang, S.L.; Hao, Q.Y.; Gao, M.; Wang, J.F.; Gao, J.W.; Liu, P.M. Potential ferroptosis key genes in calcific aortic valve disease. Front. Cardiovasc. Med. 2022, 9, 916841. [Google Scholar] [CrossRef] [PubMed]
- Liberman, M.; Bassi, E.; Martinatti, M.K.; Lario, F.C.; Wosniak, J., Jr.; Pomerantzeff, P.M.; Laurindo, F.R. Oxidant generation predominates around calcifying foci and enhances progression of aortic valve calcification. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 463–470. [Google Scholar] [CrossRef] [PubMed]
- Lindman, B.R.; Clavel, M.A.; Mathieu, P.; Iung, B.; Lancellotti, P.; Otto, C.M.; Pibarot, P. Calcific aortic stenosis. Nat. Rev. Dis. Primers 2016, 2, 16006. [Google Scholar] [CrossRef]
- Liu, C.; Liu, R.; Cao, Z.; Guo, Q.; Huang, H.; Liu, L.; Xiao, Y.; Duan, C.; Ma, R. Identification of MMP9 as a Novel Biomarker to Mitochondrial Metabolism Disorder and Oxidative Stress in Calcific Aortic Valve Stenosis. Oxidat. Med. Cell. Longev. 2022, 2022, 3858871. [Google Scholar] [CrossRef]
- Liu, D.; Iruthayanathan, M.; Homan, L.L.; Wang, Y.; Yang, L.; Wang, Y.; Dillon, J.S. Dehydroepiandrosterone stimulates endothelial proliferation and angiogenesis through extracellular signal-regulated kinase 1/2-mediated mechanisms. Endocrinology 2008, 149, 889–898. [Google Scholar] [CrossRef]
- Liu, F.; Chen, J.; Hu, W.; Gao, C.; Zeng, Z.; Cheng, S.; Yu, K.; Qian, Y.; Xu, D.; Zhu, G.; et al. PTP1B Inhibition Improves Mitochondrial Dynamics to Alleviate Calcific Aortic Valve Disease Via Regulating OPA1 Homeostasis. JACC Basic Transl. Sci. 2022, 7, 697–712. [Google Scholar] [CrossRef]
- Liu, W.; Xin, H.; Eckert, D.T.; Brown, J.A.; Gnarra, J.R. Hypoxia and cell cycle regulation of the von Hippel-Lindau tumor suppressor. Oncogene 2011, 30, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Losenno, K.L.; Goodman, R.L.; Chu, M.W. Bicuspid aortic valve disease and ascending aortic aneurysms: Gaps in knowledge. Cardiol. Res. Pract. 2012, 2012, 145202. [Google Scholar] [CrossRef]
- Luo, M.J.; Rao, S.S.; Tan, Y.J.; Yin, H.; Hu, X.K.; Zhang, Y.; Liu, Y.W.; Yue, T.; Chen, L.J.; Li, L.; et al. Fasting before or after wound injury accelerates wound healing through the activation of pro-angiogenic SMOC1 and SCG2. Theranostics 2020, 10, 3779–3792. [Google Scholar] [CrossRef]
- Luo, W.; Hu, H.; Chang, R.; Zhong, J.; Knabel, M.; O’Meally, R.; Cole, R.N.; Pandey, A.; Semenza, G.L. Pyruvate kinase M2 is a PHD3-stimulated coactivator for hypoxia-inducible factor 1. Cell 2011, 145, 732–744. [Google Scholar] [CrossRef] [PubMed]
- Ma, F.; Li, Y.; Jia, L.; Han, Y.; Cheng, J.; Li, H.; Qi, Y.; Du, J. Macrophage-stimulated cardiac fibroblast production of IL-6 is essential for TGF beta/Smad activation and cardiac fibrosis induced by angiotensin II. PLoS ONE 2012, 7, e35144. [Google Scholar]
- Maeda, K.; Ma, X.; Chalajour, F.; Hanley, F.L.; Riemer, R.K. Critical Role of Coaptive Strain in Aortic Valve Leaflet Homeostasis: Use of a Novel Flow Culture Bioreactor to Explore Heart Valve Mechanobiology. J. Am. Heart Assoc. 2016, 5, e003506. [Google Scholar] [CrossRef] [PubMed]
- Maemura, K.; Hsieh, C.M.; Jain, M.K.; Fukumoto, S.; Layne, M.D.; Liu, Y.; Kourembanas, S.; Yet, S.F.; Perrella, M.A.; Lee, M.E. Generation of a dominant-negative mutant of endothelial PAS domain protein 1 by deletion of a potent C-terminal transactivation domain. J. Biol. Chem. 1999, 274, 31565–31570. [Google Scholar] [CrossRef] [PubMed]
- Marchi, S.; Giorgi, C.; Galluzzi, L.; Pinton, P. Ca2+ Fluxes and Cancer. Mol. Cell 2020, 78, 1055–1069. [Google Scholar] [CrossRef]
- Masri, A.; Svensson, L.G.; Griffin, B.P.; Desai, M.Y. Contemporary natural history of bicuspid aortic valve disease: A systematic review. Heart 2017, 103, 1323–1330. [Google Scholar] [CrossRef]
- Matsui, M.; Bouchareb, R.; Storto, M.; Hussain, Y.; Gregg, A.; Marx, S.O.; Pitt, G.S. Increased Ca2+ influx through CaV1.2 drives aortic valve calcification. JCI Insight 2022, 7, e155569. [Google Scholar] [CrossRef]
- Mazzone, A.; Epistolato, M.C.; De Caterina, R.; Storti, S.; Vittorini, S.; Sbrana, S.; Gianetti, J.; Bevilacqua, S.; Glauber, M.; Biagini, A.; et al. Neoangiogenesis, T-lymphocyte infiltration, and heat shock protein-60 are biological hallmarks of an immunomediated inflammatory process in end-stage calcified aortic valve stenosis. J. Am. Coll. Cardiol. 2004, 43, 1670–1676. [Google Scholar] [CrossRef]
- McClelland, G.B.; Brooks, G.A. Changes in MCT 1, MCT 4, and LDH expression are tissue specific in rats after long-term hypobaric hypoxia. J. Appl. Physiol. 2002, 92, 1573–1584. [Google Scholar] [CrossRef]
- Melillo, G.; Musso, T.; Sica, A.; Taylor, L.S.; Cox, G.W.; Varesio, L. A hypoxia-responsive element mediates a novel pathway of activation of the inducible nitric oxide synthase promoter. J. Exp. Med. 1995, 182, 1683–1693. [Google Scholar] [CrossRef]
- Meng, X.; Ao, L.; Song, Y.; Babu, A.; Yang, X.; Wang, M.; Weyant, M.J.; Dinarello, C.A.; Cleveland, J.C., Jr.; Fullerton, D.A. Expression of functional Toll-like receptors 2 and 4 in human aortic valve interstitial cells: Potential roles in aortic valve inflammation and stenosis. Am. J. Physiol. Cell Physiol. 2008, 294, C29–C35. [Google Scholar] [CrossRef] [PubMed]
- Metry, G.; Stenvinkel, P.; Qureshi, A.R.; Carrero, J.J.; Yilmaz, M.I.; Barany, P.; Snaedal, S.; Heimburger, O.; Lindholm, B.; Suliman, M.E. Low serum fetuin-A concentration predicts poor outcome only in the presence of inflammation in prevalent haemodialysis patients. Eur. J. Clin. Investig. 2008, 38, 804–811. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.D.; Chu, Y.; Brooks, R.M.; Richenbacher, W.E.; Pena-Silva, R.; Heistad, D.D. Dysregulation of antioxidant mechanisms contributes to increased oxidative stress in calcific aortic valvular stenosis in humans. J. Am. Coll. Cardiol. 2008, 52, 843–850. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.D.; Weiss, R.M.; Heistad, D.D. Calcific aortic valve stenosis: Methods, models, and mechanisms. Circ. Res. 2011, 108, 1392–1412. [Google Scholar] [CrossRef] [PubMed]
- Missiroli, S.; Bonora, M.; Patergnani, S.; Poletti, F.; Perrone, M.; Gafa, R.; Magri, E.; Raimondi, A.; Lanza, G.; Tacchetti, C.; et al. PML at Mitochondria-Associated Membranes Is Critical for the Repression of Autophagy and Cancer Development. Cell Rep. 2016, 16, 2415–2427. [Google Scholar] [CrossRef]
- Missiroli, S.; Perrone, M.; Gafa, R.; Nicoli, F.; Bonora, M.; Morciano, G.; Boncompagni, C.; Marchi, S.; Lebiedzinska-Arciszewska, M.; Vezzani, B.; et al. PML at mitochondria-associated membranes governs a trimeric complex with NLRP3 and P2X7R that modulates the tumor immune microenvironment. Cell Death Differ. 2023, 30, 429–441. [Google Scholar] [CrossRef]
- Moorlag, S.; Roring, R.J.; Joosten, L.A.B.; Netea, M.G. The role of the interleukin-1 family in trained immunity. Immunol. Rev. 2018, 281, 28–39. [Google Scholar] [CrossRef]
- Morciano, G.; Patergnani, S.; Pedriali, G.; Cimaglia, P.; Mikus, E.; Calvi, S.; Albertini, A.; Giorgi, C.; Campo, G.; Ferrari, R.; et al. Impairment of mitophagy and autophagy accompanies calcific aortic valve stenosis favouring cell death and the severity of disease. Cardiovasc. Res. 2022, 118, 2548–2559. [Google Scholar] [CrossRef]
- Mosch, J.; Gleissner, C.A.; Body, S.; Aikawa, E. Histopathological assessment of calcification and inflammation of calcific aortic valves from patients with and without diabetes mellitus. Histol. Histopathol. 2017, 32, 293–306. [Google Scholar]
- Nadlonek, N.; Lee, J.H.; Reece, T.B.; Weyant, M.J.; Cleveland, J.C., Jr.; Meng, X.; Fullerton, D.A. Interleukin-1 Beta induces an inflammatory phenotype in human aortic valve interstitial cells through nuclear factor kappa Beta. Ann. Thorac. Surg. 2013, 96, 155–162. [Google Scholar] [CrossRef]
- Nagy, E.; Lei, Y.; Martinez-Martinez, E.; Body, S.C.; Schlotter, F.; Creager, M.; Assmann, A.; Khabbaz, K.; Libby, P.; Hansson, G.K.; et al. Interferon-gamma Released by Activated CD8+ T Lymphocytes Impairs the Calcium Resorption Potential of Osteoclasts in Calcified Human Aortic Valves. Am. J. Pathol. 2017, 187, 1413–1425. [Google Scholar] [CrossRef]
- Naldini, A.; Filippi, I.; Miglietta, D.; Moschetta, M.; Giavazzi, R.; Carraro, F. Interleukin-1beta regulates the migratory potential of MDAMB231 breast cancer cells through the hypoxia-inducible factor-1alpha. Eur. J. Cancer 2010, 46, 3400–3408. [Google Scholar] [CrossRef] [PubMed]
- Negri, A.L. Role of prolyl hydroxylase/HIF-1 signaling in vascular calcification. Clin. Kidney J. 2023, 16, 205–209. [Google Scholar] [CrossRef] [PubMed]
- Ng, M.Y.W.; Wai, T.; Simonsen, A. Quality control of the mitochondrion. Dev. Cell 2021, 56, 881–905. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.D.; Schwarzer, M.; Schrepper, A.; Amorim, P.A.; Blum, D.; Hain, C.; Faerber, G.; Haendeler, J.; Altschmied, J.; Doenst, T. Increased Protein Tyrosine Phosphatase 1B (PTP1B) Activity and Cardiac Insulin Resistance Precede Mitochondrial and Contractile Dysfunction in Pressure-Overloaded Hearts. J. Am. Heart Assoc. 2018, 7, e008865. [Google Scholar] [CrossRef]
- Nkomo, V.T.; Gardin, J.M.; Skelton, T.N.; Gottdiener, J.S.; Scott, C.G.; Enriquez-Sarano, M. Burden of valvular heart diseases: A population-based study. Lancet 2006, 368, 1005–1011. [Google Scholar] [CrossRef]
- Nsaibia, M.J.; Mahmut, A.; Boulanger, M.C.; Arsenault, B.J.; Bouchareb, R.; Simard, S.; Witztum, J.L.; Clavel, M.A.; Pibarot, P.; Bosse, Y.; et al. Autotaxin interacts with lipoprotein(a) and oxidized phospholipids in predicting the risk of calcific aortic valve stenosis in patients with coronary artery disease. J. Internal Med. 2016, 280, 509–517. [Google Scholar] [CrossRef]
- O’Brien, K.D.; Zhao, X.Q.; Shavelle, D.M.; Caulfield, M.T.; Letterer, R.A.; Kapadia, S.R.; Probstfield, J.L.; Otto, C.M. Hemodynamic effects of the angiotensin-converting enzyme inhibitor, ramipril, in patients with mild to moderate aortic stenosis and preserved left ventricular function. J. Investig. Med. 2004, 52, 185–191. [Google Scholar] [CrossRef]
- Okui, T.; Iwashita, M.; Rogers, M.A.; Halu, A.; Atkins, S.K.; Kuraoka, S.; Abdelhamid, I.; Higashi, H.; Ramsaroop, A.; Aikawa, M.; et al. CROT (Carnitine O-Octanoyltransferase) Is a Novel Contributing Factor in Vascular Calcification via Promoting Fatty Acid Metabolism and Mitochondrial Dysfunction. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 755–768. [Google Scholar] [CrossRef]
- Osako, M.K.; Nakagami, H.; Koibuchi, N.; Shimizu, H.; Nakagami, F.; Koriyama, H.; Shimamura, M.; Miyake, T.; Rakugi, H.; Morishita, R. Estrogen inhibits vascular calcification via vascular RANKL system: Common mechanism of osteoporosis and vascular calcification. Circ. Res. 2010, 107, 466–475. [Google Scholar] [CrossRef]
- Osnabrugge, R.L.; Mylotte, D.; Head, S.J.; Van Mieghem, N.M.; Nkomo, V.T.; LeReun, C.M.; Bogers, A.J.; Piazza, N.; Kappetein, A.P. Aortic stenosis in the elderly: Disease prevalence and number of candidates for transcatheter aortic valve replacement: A meta-analysis and modeling study. J. Am. Coll. Cardiol. 2013, 62, 1002–1012. [Google Scholar] [CrossRef] [PubMed]
- Otto, C.M.; Burwash, I.G.; Legget, M.E.; Munt, B.I.; Fujioka, M.; Healy, N.L.; Kraft, C.D.; Miyake-Hull, C.Y.; Schwaegler, R.G. Prospective study of asymptomatic valvular aortic stenosis. Clinical, echocardiographic, and exercise predictors of outcome. Circulation 1997, 95, 2262–2270. [Google Scholar] [CrossRef]
- Otto, C.M.; Kuusisto, J.; Reichenbach, D.D.; Gown, A.M.; O’Brien, K.D. Characterization of the early lesion of ‘degenerative’ valvular aortic stenosis. Histological and immunohistochemical studies. Circulation 1994, 90, 844–853. [Google Scholar] [CrossRef] [PubMed]
- Packer, M. Mutual Antagonism of Hypoxia-Inducible Factor Isoforms in Cardiac, Vascular, and Renal Disorders. JACC Basic Transl. Sci. 2020, 5, 961–968. [Google Scholar] [CrossRef] [PubMed]
- Parra-Izquierdo, I.; Castanos-Mollor, I.; Lopez, J.; Gomez, C.; San Roman, J.A.; Sanchez Crespo, M.; Garcia-Rodriguez, C. Calcification Induced by Type I Interferon in Human Aortic Valve Interstitial Cells Is Larger in Males and Blunted by a Janus Kinase Inhibitor. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 2148–2159. [Google Scholar] [CrossRef] [PubMed]
- Parra-Izquierdo, I.; Castanos-Mollor, I.; Lopez, J.; Gomez, C.; San Roman, J.A.; Sanchez Crespo, M.; Garcia-Rodriguez, C. Lipopolysaccharide and interferon-gamma team up to activate HIF-1alpha via STAT1 in normoxia and exhibit sex differences in human aortic valve interstitial cells. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 2168–2179. [Google Scholar] [CrossRef] [PubMed]
- Patergnani, S.; Bonora, M.; Ingusci, S.; Previati, M.; Marchi, S.; Zucchini, S.; Perrone, M.; Wieckowski, M.R.; Castellazzi, M.; Pugliatti, M.; et al. Antipsychotic drugs counteract autophagy and mitophagy in multiple sclerosis. Proc. Natl. Acad. Sci. USA 2021, 118, e2020078118. [Google Scholar] [CrossRef]
- Patergnani, S.; Bouhamida, E.; Leo, S.; Pinton, P.; Rimessi, A. Mitochondrial Oxidative Stress and “Mito-Inflammation”: Actors in the Diseases. Biomedicines 2021, 9, 216. [Google Scholar] [CrossRef]
- Pedriali, G.; Morciano, G.; Patergnani, S.; Cimaglia, P.; Morelli, C.; Mikus, E.; Ferrari, R.; Gasbarro, V.; Giorgi, C.; Wieckowski, M.R.; et al. Aortic Valve Stenosis and Mitochondrial Dysfunctions: Clinical and Molecular Perspectives. Int. J. Mol. Sci. 2020, 21, 4899. [Google Scholar] [CrossRef]
- Perrotta, I.; Moraca, F.M.; Sciangula, A.; Aquila, S.; Mazzulla, S. HIF-1alpha and VEGF: Immunohistochemical Profile and Possible Function in Human Aortic Valve Stenosis. Ultrastruct. Pathol. 2015, 39, 198–206. [Google Scholar] [CrossRef]
- Pickles, S.; Vigie, P.; Youle, R.J. Mitophagy and Quality Control Mechanisms in Mitochondrial Maintenance. Curr. Biol. 2018, 28, R170–R185. [Google Scholar] [CrossRef]
- Qian, D.; Lin, H.Y.; Wang, H.M.; Zhang, X.; Liu, D.L.; Li, Q.L.; Zhu, C. Normoxic induction of the hypoxic-inducible factor-1 alpha by interleukin-1 beta involves the extracellular signal-regulated kinase 1/2 pathway in normal human cytotrophoblast cells. Biol. Reprod. 2004, 70, 1822–1827. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.; Liang, S.; Sanchez-Lopez, E.; He, F.; Shalapour, S.; Lin, X.J.; Wong, J.; Ding, S.; Seki, E.; Schnabl, B.; et al. New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature 2018, 560, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Rajamannan, N.M.; Nealis, T.B.; Subramaniam, M.; Pandya, S.; Stock, S.R.; Ignatiev, C.I.; Sebo, T.J.; Rosengart, T.K.; Edwards, W.D.; McCarthy, P.M.; et al. Calcified rheumatic valve neoangiogenesis is associated with vascular endothelial growth factor expression and osteoblast-like bone formation. Circulation 2005, 111, 3296–3301. [Google Scholar] [CrossRef]
- Rajamannan, N.M.; Subramaniam, M.; Rickard, D.; Stock, S.R.; Donovan, J.; Springett, M.; Orszulak, T.; Fullerton, D.A.; Tajik, A.J.; Bonow, R.O.; et al. Human aortic valve calcification is associated with an osteoblast phenotype. Circulation 2003, 107, 2181–2184. [Google Scholar] [CrossRef]
- Richards, J.; El-Hamamsy, I.; Chen, S.; Sarang, Z.; Sarathchandra, P.; Yacoub, M.H.; Chester, A.H.; Butcher, J.T. Side-specific endothelial-dependent regulation of aortic valve calcification: Interplay of hemodynamics and nitric oxide signaling. Am. J. Pathol. 2013, 182, 1922–1931. [Google Scholar] [CrossRef] [PubMed]
- Roberts, W.C.; Ko, J.M. Frequency by decades of unicuspid, bicuspid, and tricuspid aortic valves in adults having isolated aortic valve replacement for aortic stenosis, with or without associated aortic regurgitation. Circulation 2005, 111, 920–925. [Google Scholar] [CrossRef]
- Rogers, M.A.; Maldonado, N.; Hutcheson, J.D.; Goettsch, C.; Goto, S.; Yamada, I.; Faits, T.; Sesaki, H.; Aikawa, M.; Aikawa, E. Dynamin-Related Protein 1 Inhibition Attenuates Cardiovascular Calcification in the Presence of Oxidative Stress. Circ. Res. 2017, 121, 220–233. [Google Scholar] [CrossRef]
- Roos, C.M.; Zhang, B.; Hagler, M.A.; Verzosa, G.C.; Huang, R.; Oehler, E.A.; Arghami, A.; Miller, J.D. Effects of Altering Mitochondrial Antioxidant Capacity on Molecular and Phenotypic Drivers of Fibrocalcific Aortic Valve Stenosis. Front. Cardiovasc. Med. 2021, 8, 694881. [Google Scholar] [CrossRef]
- Rudloff, S.; Jahnen-Dechent, W.; Huynh-Do, U. Tissue chaperoning-the expanded functions of fetuin-A beyond inhibition of systemic calcification. Pflugers Arch. 2022, 474, 949–962. [Google Scholar] [CrossRef]
- Rudloff, S.; Janot, M.; Rodriguez, S.; Dessalle, K.; Jahnen-Dechent, W.; Huynh-Do, U. Fetuin-A is a HIF target that safeguards tissue integrity during hypoxic stress. Nat. Commun. 2021, 12, 549. [Google Scholar] [CrossRef]
- Sabatine, M.S.; Giugliano, R.P.; Keech, A.C.; Honarpour, N.; Wiviott, S.D.; Murphy, S.A.; Kuder, J.F.; Wang, H.; Liu, T.; Wasserman, S.M.; et al. Evolocumab and Clinical Outcomes in Patients with Cardiovascular Disease. N. Engl. J. Med. 2017, 376, 1713–1722. [Google Scholar] [CrossRef]
- Sahoo, S.; Meijles, D.N.; Pagano, P.J. NADPH oxidases: Key modulators in aging and age-related cardiovascular diseases? Clin. Sci. 2016, 130, 317–335. [Google Scholar] [CrossRef]
- Sainger, R.; Grau, J.B.; Branchetti, E.; Poggio, P.; Lai, E.; Koka, E.; Vernick, W.J.; Gorman, R.C.; Bavaria, J.E.; Ferrari, G. Comparison of transesophageal echocardiographic analysis and circulating biomarker expression profile in calcific aortic valve disease. J. Heart Valve Dis. 2013, 22, 156–165. [Google Scholar]
- Salhiyyah, K.; Sarathchandra, P.; Latif, N.; Yacoub, M.H.; Chester, A.H. Hypoxia-mediated regulation of the secretory properties of mitral valve interstitial cells. Am. J. Physiol. Heart Circ. Physiol. 2017, 313, H14–H23. [Google Scholar] [CrossRef] [PubMed]
- Salim, M.T.; Villa-Roel, N.; Vogel, B.; Jo, H.; Yoganathan, A.P. HIF1A inhibitor PX-478 reduces pathological stretch-induced calcification and collagen turnover in aortic valve. Front. Cardiovasc. Med. 2022, 9, 1002067. [Google Scholar] [CrossRef]
- Sanchez-Duffhues, G.; Garcia de Vinuesa, A.; van de Pol, V.; Geerts, M.E.; de Vries, M.R.; Janson, S.G.; van Dam, H.; Lindeman, J.H.; Goumans, M.J.; Ten Dijke, P. Inflammation induces endothelial-to-mesenchymal transition and promotes vascular calcification through downregulation of BMPR2. J. Pathol. 2019, 247, 333–346. [Google Scholar] [CrossRef]
- Sanjuan-Pla, A.; Cervera, A.M.; Apostolova, N.; Garcia-Bou, R.; Victor, V.M.; Murphy, M.P.; McCreath, K.J. A targeted antioxidant reveals the importance of mitochondrial reactive oxygen species in the hypoxic signaling of HIF-1alpha. FEBS Lett. 2005, 579, 2669–2674. [Google Scholar] [CrossRef] [PubMed]
- Sapp, M.C.; Krishnamurthy, V.K.; Puperi, D.S.; Bhatnagar, S.; Fatora, G.; Mutyala, N.; Grande-Allen, K.J. Differential cell-matrix responses in hypoxia-stimulated aortic versus mitral valves. J. R. Soc. Interface 2016, 13, 20160449. [Google Scholar] [CrossRef]
- Sasagawa, T.; Nagamatsu, T.; Morita, K.; Mimura, N.; Iriyama, T.; Fujii, T.; Shibuya, M. HIF-2alpha, but not HIF-1alpha, mediates hypoxia-induced up-regulation of Flt-1 gene expression in placental trophoblasts. Sci. Rep. 2018, 8, 17375. [Google Scholar] [CrossRef] [PubMed]
- Schlotter, F.; de Freitas, R.C.C.; Rogers, M.A.; Blaser, M.C.; Wu, P.J.; Higashi, H.; Halu, A.; Iqbal, F.; Andraski, A.B.; Rodia, C.N.; et al. ApoC-III is a novel inducer of calcification in human aortic valves. J. Biol. Chem. 2021, 296, 100193. [Google Scholar] [CrossRef]
- Schoen, F.J. Evolving concepts of cardiac valve dynamics: The continuum of development, functional structure, pathobiology, and tissue engineering. Circulation 2008, 118, 1864–1880. [Google Scholar] [CrossRef]
- Schoen, F.J.; Levy, R.J. Calcification of tissue heart valve substitutes: Progress toward understanding and prevention. Ann. Thorac. Surg. 2005, 79, 1072–1080. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Hypoxia-inducible factor 1: Regulator of mitochondrial metabolism and mediator of ischemic preconditioning. Biochim. Biophys. Acta 2011, 1813, 1263–1268. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L.; Wang, G.L. A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol. Cell. Biol. 1992, 12, 5447–5454. [Google Scholar] [PubMed]
- Shin, M.; Beane, T.J.; Quillien, A.; Male, I.; Zhu, L.J.; Lawson, N.D. Vegfa signals through ERK to promote angiogenesis, but not artery differentiation. Development 2016, 143, 3796–3805. [Google Scholar] [CrossRef] [PubMed]
- Shworak, N.W. Angiogenic modulators in valve development and disease: Does valvular disease recapitulate developmental signaling pathways? Curr. Opin. Cardiol. 2004, 19, 140–146. [Google Scholar] [CrossRef]
- Simon, M.C.; Liu, L.; Barnhart, B.C.; Young, R.M. Hypoxia-induced signaling in the cardiovascular system. Annu. Rev. Physiol. 2008, 70, 51–71. [Google Scholar] [CrossRef]
- Souilhol, C.; Harmsen, M.C.; Evans, P.C.; Krenning, G. Endothelial-mesenchymal transition in atherosclerosis. Cardiovasc. Res. 2018, 114, 565–577. [Google Scholar] [CrossRef]
- Stiehl, D.P.; Jelkmann, W.; Wenger, R.H.; Hellwig-Burgel, T. Normoxic induction of the hypoxia-inducible factor 1alpha by insulin and interleukin-1beta involves the phosphatidylinositol 3-kinase pathway. FEBS Lett. 2002, 512, 157–162. [Google Scholar] [CrossRef]
- Sun, Y.; Wang, W.; Tang, Y.; Wang, D.; Li, L.; Na, M.; Jiang, G.; Li, Q.; Chen, S.; Zhou, J. Microarray profiling and functional analysis of differentially expressed plasma exosomal circular RNAs in Graves’ disease. Biol. Res. 2020, 53, 32. [Google Scholar] [CrossRef] [PubMed]
- Swaminathan, G.; Krishnamurthy, V.K.; Sridhar, S.; Robson, D.C.; Ning, Y.; Grande-Allen, K.J. Hypoxia Stimulates Synthesis of Neutrophil Gelatinase-Associated Lipocalin in Aortic Valve Disease. Front. Cardiovasc. Med. 2019, 6, 156. [Google Scholar] [CrossRef] [PubMed]
- Szweras, M.; Liu, D.; Partridge, E.A.; Pawling, J.; Sukhu, B.; Clokie, C.; Jahnen-Dechent, W.; Tenenbaum, H.C.; Swallow, C.J.; Grynpas, M.D.; et al. alpha 2-HS glycoprotein/fetuin, a transforming growth factor-beta/bone morphogenetic protein antagonist, regulates postnatal bone growth and remodeling. J. Biol. Chem. 2002, 277, 19991–19997. [Google Scholar] [CrossRef] [PubMed]
- Tan, D.X.; Manchester, L.C.; Qin, L.; Reiter, R.J. Melatonin: A Mitochondrial Targeting Molecule Involving Mitochondrial Protection and Dynamics. Int. J. Mol. Sci. 2016, 17, 2124. [Google Scholar] [CrossRef] [PubMed]
- Thanassoulis, G. Lipoprotein (a) in calcific aortic valve disease: From genomics to novel drug target for aortic stenosis. J. Lipid Res. 2016, 57, 917–924. [Google Scholar] [CrossRef]
- Tian, H.; McKnight, S.L.; Russell, D.W. Endothelial PAS domain protein 1 (EPAS1), a transcription factor selectively expressed in endothelial cells. Genes Dev. 1997, 11, 72–82. [Google Scholar] [CrossRef]
- Tian, X.; Zhou, N.; Yuan, J.; Lu, L.; Zhang, Q.; Wei, M.; Zou, Y.; Yuan, L. Heat shock transcription factor 1 regulates exercise-induced myocardial angiogenesis after pressure overload via HIF-1alpha/VEGF pathway. J. Cell. Mol. Med. 2020, 24, 2178–2188. [Google Scholar] [CrossRef]
- Tuder, R.M.; Flook, B.E.; Voelkel, N.F. Increased gene expression for VEGF and the VEGF receptors KDR/Flk and Flt in lungs exposed to acute or to chronic hypoxia. Modulation of gene expression by nitric oxide. J. Clin. Investig. 1995, 95, 1798–1807. [Google Scholar] [CrossRef]
- Verstraete, A.; Herregods, M.C.; Verbrugghe, P.; Lamberigts, M.; Vanassche, T.; Meyns, B.; Oosterlinck, W.; Rega, F.; Adriaenssens, T.; Van Hoof, L.; et al. Antithrombotic Treatment After Surgical and Transcatheter Heart Valve Repair and Replacement. Front. Cardiovasc. Med. 2021, 8, 702780. [Google Scholar] [CrossRef]
- Viau, A.; El Karoui, K.; Laouari, D.; Burtin, M.; Nguyen, C.; Mori, K.; Pillebout, E.; Berger, T.; Mak, T.W.; Knebelmann, B.; et al. Lipocalin 2 is essential for chronic kidney disease progression in mice and humans. J. Clin. Investig. 2010, 120, 4065–4076. [Google Scholar] [CrossRef]
- Wakabayashi, K.; Tsujino, T.; Naito, Y.; Ezumi, A.; Lee-Kawabata, M.; Nakao, S.; Goda, A.; Sakata, Y.; Yamamoto, K.; Daimon, T.; et al. Administration of angiotensin-converting enzyme inhibitors is associated with slow progression of mild aortic stenosis in Japanese patients. Heart Vessels 2011, 26, 252–257. [Google Scholar] [CrossRef]
- Wang, A.; Rana, S.; Karumanchi, S.A. Preeclampsia: The role of angiogenic factors in its pathogenesis. Physiology 2009, 24, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.L.; Jiang, B.H.; Rue, E.A.; Semenza, G.L. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. USA 1995, 92, 5510–5514. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Klionsky, D.J. Mitochondria removal by autophagy. Autophagy 2011, 7, 297–300. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Fang, D.; Yang, Q.; You, J.; Wang, L.; Wu, J.; Zeng, M.; Luo, M. Interactions between PCSK9 and NLRP3 inflammasome signaling in atherosclerosis. Front. Immunol. 2023, 14, 1126823. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Han, D.; Zhou, T.; Zhang, J.; Liu, C.; Cao, F.; Dong, N. Melatonin ameliorates aortic valve calcification via the regulation of circular RNA CircRIC3/miR-204-5p/DPP4 signaling in valvular interstitial cells. J. Pineal Res. 2020, 69, e12666. [Google Scholar] [CrossRef]
- Weisell, J.; Ohukainen, P.; Napankangas, J.; Ohlmeier, S.; Bergmann, U.; Peltonen, T.; Taskinen, P.; Ruskoaho, H.; Rysa, J. Heat shock protein 90 is downregulated in calcific aortic valve disease. BMC Cardiovasc. Disord. 2019, 19, 306. [Google Scholar] [CrossRef]
- Wu, X.; Zhao, Q.; Chen, Z.; Geng, Y.J.; Zhang, W.; Zhou, Q.; Yang, W.; Liu, Q.; Liu, H. Estrogen inhibits vascular calcification in rats via hypoxia-induced factor-1alpha signaling. Vascular 2020, 28, 465–474. [Google Scholar] [CrossRef]
- Xie, C.; Shen, Y.; Hu, W.; Chen, Z.; Li, Y. Angiotensin II promotes an osteoblast-like phenotype in porcine aortic valve myofibroblasts. Aging Clin. Exp. Res. 2016, 28, 181–187. [Google Scholar] [CrossRef]
- Xu, X.H.; Xu, J.; Xue, L.; Cao, H.L.; Liu, X.; Chen, Y.J. VEGF attenuates development from cardiac hypertrophy to heart failure after aortic stenosis through mitochondrial mediated apoptosis and cardiomyocyte proliferation. J. Cardiothorac. Surg. 2011, 6, 54. [Google Scholar] [CrossRef]
- Yan, H.F.; Liu, Z.Y.; Guan, Z.A.; Guo, C. Deferoxamine ameliorates adipocyte dysfunction by modulating iron metabolism in ob/ob mice. Endocr. Connect. 2018, 7, 604–616. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Fullerton, D.A.; Su, X.; Ao, L.; Cleveland, J.C., Jr.; Meng, X. Pro-osteogenic phenotype of human aortic valve interstitial cells is associated with higher levels of Toll-like receptors 2 and 4 and enhanced expression of bone morphogenetic protein 2. J. Am. Coll. Cardiol. 2009, 53, 491–500. [Google Scholar] [CrossRef] [PubMed]
- Yi, B.; Zeng, W.; Lv, L.; Hua, P. Changing epidemiology of calcific aortic valve disease: 30-year trends of incidence, prevalence, and deaths across 204 countries and territories. Aging 2021, 13, 12710–12732. [Google Scholar] [CrossRef] [PubMed]
- Yip, C.Y.; Chen, J.H.; Zhao, R.; Simmons, C.A. Calcification by valve interstitial cells is regulated by the stiffness of the extracellular matrix. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 936–942. [Google Scholar] [CrossRef] [PubMed]
- Youle, R.J.; van der Bliek, A.M. Mitochondrial fission, fusion, and stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef]
- Yu, B.; Khan, K.; Hamid, Q.; Mardini, A.; Siddique, A.; Aguilar-Gonzalez, L.P.; Makhoul, G.; Alaws, H.; Genest, J.; Thanassoulis, G.; et al. Pathological significance of lipoprotein(a) in aortic valve stenosis. Atherosclerosis 2018, 272, 168–174. [Google Scholar] [CrossRef]
- Yuan, L.; Wang, M.; Liu, T.; Lei, Y.; Miao, Q.; Li, Q.; Wang, H.; Zhang, G.; Hou, Y.; Chang, X. Carbonic Anhydrase 1-Mediated Calcification Is Associated With Atherosclerosis, and Methazolamide Alleviates Its Pathogenesis. Front. Pharmacol. 2019, 10, 766. [Google Scholar] [CrossRef]
- Zeng, Q.; Song, R.; Fullerton, D.A.; Ao, L.; Zhai, Y.; Li, S.; Ballak, D.B.; Cleveland, J.C., Jr.; Reece, T.B.; McKinsey, T.A.; et al. Interleukin-37 suppresses the osteogenic responses of human aortic valve interstitial cells in vitro and alleviates valve lesions in mice. Proc. Natl. Acad. Sci. USA 2017, 114, 1631–1636. [Google Scholar] [CrossRef]
- Zhan, Q.; Zeng, Q.; Song, R.; Zhai, Y.; Xu, D.; Fullerton, D.A.; Dinarello, C.A.; Meng, X. IL-37 suppresses MyD88-mediated inflammatory responses in human aortic valve interstitial cells. Mol. Med. 2017, 23, 83–91. [Google Scholar] [CrossRef]
- Zhao, L.; Yang, N.; Song, Y.; Si, H.; Qin, Q.; Guo, Z. Effect of iron overload on endothelial cell calcification and its mechanism. Ann. Transl. Med. 2021, 9, 1658. [Google Scholar] [CrossRef]
- Zhao, T.; Jin, F.; Xiao, D.; Wang, H.; Huang, C.; Wang, X.; Gao, S.; Liu, J.; Yang, S.; Hao, J. IL-37/ STAT3/ HIF-1alpha negative feedback signaling drives gemcitabine resistance in pancreatic cancer. Theranostics 2020, 10, 4088–4100. [Google Scholar] [CrossRef]
- Zhao, W.; Zhao, D.; Yan, R.; Sun, Y. Cardiac oxidative stress and remodeling following infarction: Role of NADPH oxidase. Cardiovasc. Pathol. 2009, 18, 156–166. [Google Scholar] [CrossRef]
- Zhou, P.; Li, Q.; Su, S.; Dong, W.; Zong, S.; Ma, Q.; Yang, X.; Zuo, D.; Zheng, S.; Meng, X.; et al. Interleukin 37 Suppresses M1 Macrophage Polarization Through Inhibition of the Notch1 and Nuclear Factor Kappa B Pathways. Front. Cell Dev. Biol. 2020, 8, 56. [Google Scholar] [CrossRef] [PubMed]
- Zimna, A.; Kurpisz, M. Hypoxia-Inducible Factor-1 in Physiological and Pathophysiological Angiogenesis: Applications and Therapies. BioMed Res. Int. 2015, 2015, 549412. [Google Scholar] [CrossRef]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [PubMed]
- Smyrnias, I.; Gray, S.P.; Okonko, D.O.; Sawyer, G.; Zoccarato, A.; Catibog, N.; Lopez, B.; Gonzalez, A.; Ravassa, S.; Diez, J.; et al. Cardioprotective Effect of the Mitochondrial Unfolded Protein Response During Chronic Pressure Overload. J. Am. Coll. Cardiol. 2019, 73, 1795–1806. [Google Scholar] [CrossRef] [PubMed]
- Soini, Y.; Salo, T.; Satta, J. Angiogenesis is involved in the pathogenesis of nonrheumatic aortic valve stenosis. Human Pathol. 2003, 34, 756–763. [Google Scholar] [CrossRef]
- Stephens, E.H.; Saltarrelli, J.G., Jr.; Balaoing, L.R.; Baggett, L.S.; Nandi, I.; Anderson, K.M.; Morrisett, J.D.; Reardon, M.J.; Simpson, M.A.; Weigel, P.H.; et al. Hyaluronan turnover and hypoxic brown adipocytic differentiation are co-localized with ossification in calcified human aortic valves. Pathol. Res. Pract. 2012, 208, 642–650. [Google Scholar] [CrossRef]
- Stewart, B.F.; Siscovick, D.; Lind, B.K.; Gardin, J.M.; Gottdiener, J.S.; Smith, V.E.; Kitzman, D.W.; Otto, C.M. Clinical factors associated with calcific aortic valve disease. Cardiovascular Health Study. J. Am. Coll. Cardiol. 1997, 29, 630–634. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bouhamida, E.; Morciano, G.; Pedriali, G.; Ramaccini, D.; Tremoli, E.; Giorgi, C.; Pinton, P.; Patergnani, S. The Complex Relationship between Hypoxia Signaling, Mitochondrial Dysfunction and Inflammation in Calcific Aortic Valve Disease: Insights from the Molecular Mechanisms to Therapeutic Approaches. Int. J. Mol. Sci. 2023, 24, 11105. https://doi.org/10.3390/ijms241311105
Bouhamida E, Morciano G, Pedriali G, Ramaccini D, Tremoli E, Giorgi C, Pinton P, Patergnani S. The Complex Relationship between Hypoxia Signaling, Mitochondrial Dysfunction and Inflammation in Calcific Aortic Valve Disease: Insights from the Molecular Mechanisms to Therapeutic Approaches. International Journal of Molecular Sciences. 2023; 24(13):11105. https://doi.org/10.3390/ijms241311105
Chicago/Turabian StyleBouhamida, Esmaa, Giampaolo Morciano, Gaia Pedriali, Daniela Ramaccini, Elena Tremoli, Carlotta Giorgi, Paolo Pinton, and Simone Patergnani. 2023. "The Complex Relationship between Hypoxia Signaling, Mitochondrial Dysfunction and Inflammation in Calcific Aortic Valve Disease: Insights from the Molecular Mechanisms to Therapeutic Approaches" International Journal of Molecular Sciences 24, no. 13: 11105. https://doi.org/10.3390/ijms241311105
APA StyleBouhamida, E., Morciano, G., Pedriali, G., Ramaccini, D., Tremoli, E., Giorgi, C., Pinton, P., & Patergnani, S. (2023). The Complex Relationship between Hypoxia Signaling, Mitochondrial Dysfunction and Inflammation in Calcific Aortic Valve Disease: Insights from the Molecular Mechanisms to Therapeutic Approaches. International Journal of Molecular Sciences, 24(13), 11105. https://doi.org/10.3390/ijms241311105