
Cisplatin in Liver Cancer Therapy


Abstract
1. Introduction
2. Development of Cisplatin
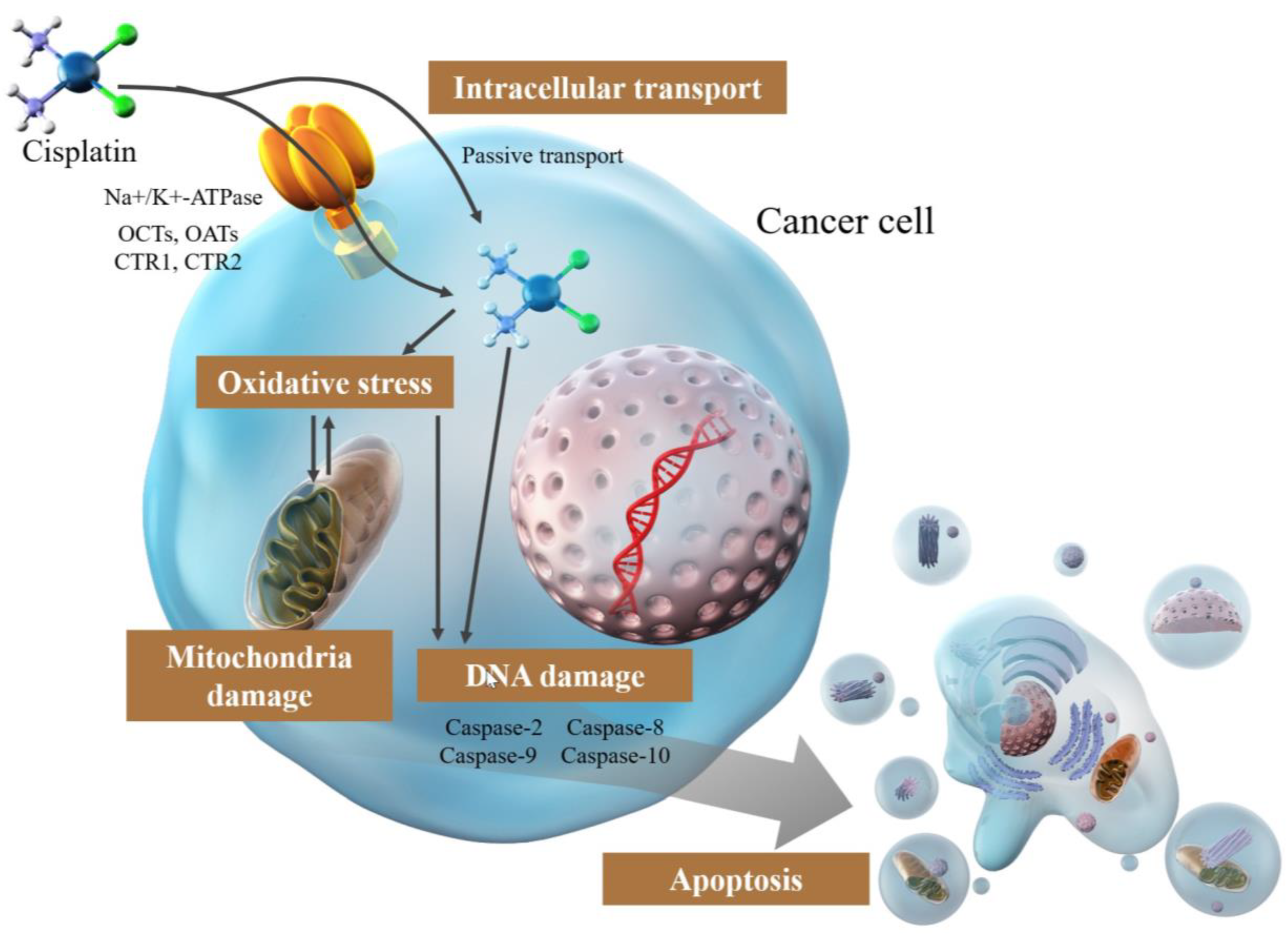
3. Mechanism of Cisplatin
3.1. Intracellular Transport of Cisplatin
3.2. Damage via DNA Cross-Linking by Cisplatin
3.3. Cisplatin-Induced DNA Replication and Transcription Arrest
3.4. Induction of Apoptosis by Cisplatin
3.5. Mitochondrial Damage by Cisplatin
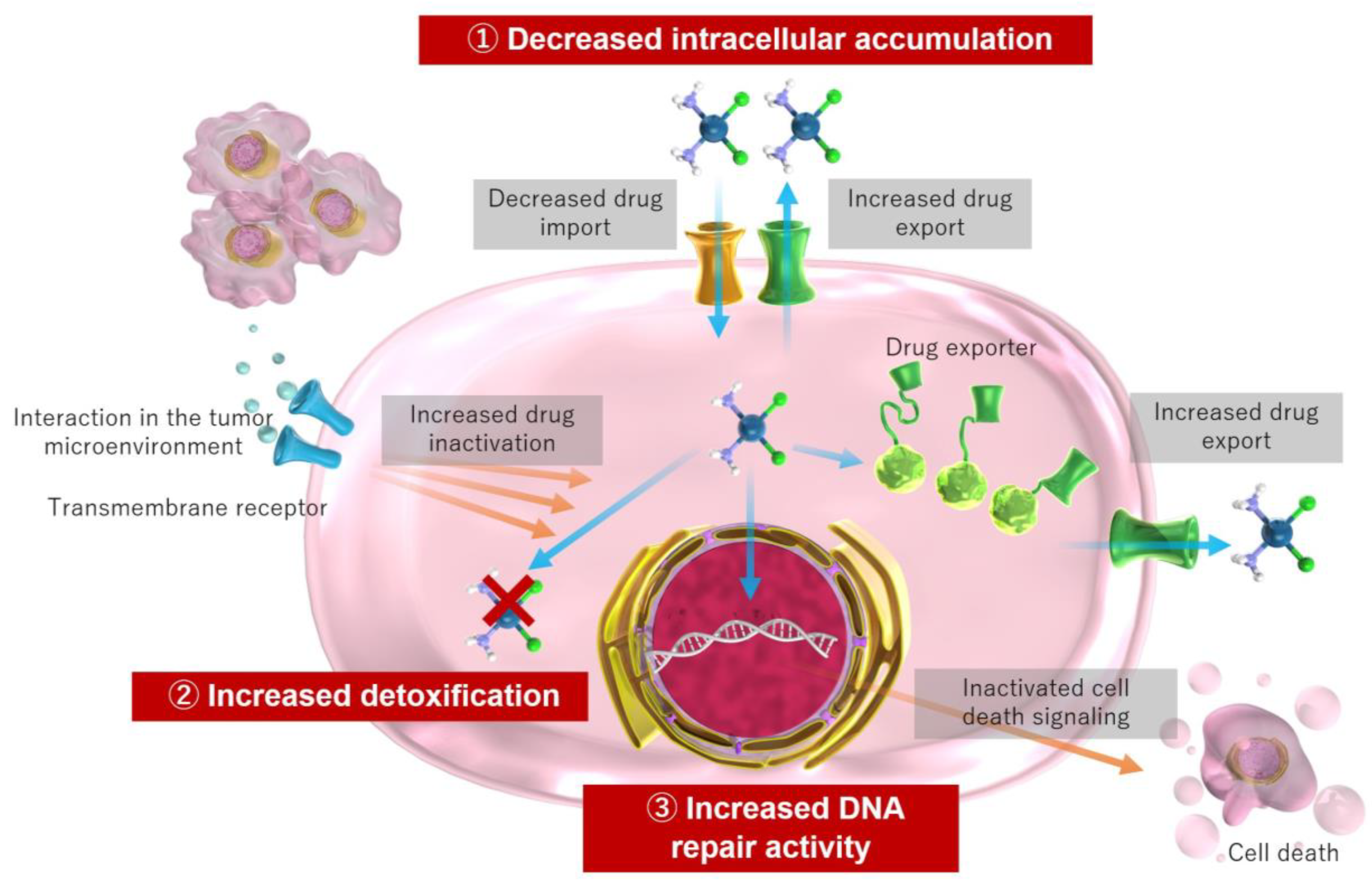
4. Mechanisms of Cisplatin Resistance
5. Adverse Events of Cisplatin

{kind=link}
{kind=link}
6. Cisplatin for HCC Treatment
6.1. Epidemiology and Treatment Algorithm for HCC
6.2. Molecular Mechanisms in TACE
6.3. Systemic Therapy and Cisplatin-Based TACE for HCC
| Drug | Target | References | |
|---|---|---|---|
| Tyrosine Kinase Inhibitors | Sorafenib | Almost 40 tyrosine kinases, such as c-RAF, B-RAF, VEGFR1-3, PDGFR-α/β, c-Kit, FLT-3, and RET | [9,156,164] |
| Regorafenib | c-RAF, wild-type and mutant (V600E) B-RAF, VEGFR1-3, FGFR1-2, PDGFR, KIT, RET, angiopoietin 1 receptor (TIE2), and p-38-α (greater potency to target VEGFR, KIT, TIE2, and RET compared to Sorafenib) | [9,164,165] | |
| Lenvatinib | VEGFR1–3, FGFR1-4, PDGFR-α, KIT, and RET | [9,15,164] | |
| Cabozantinib | VEGFR 1–3, KIT, RET, TIE2, FLT3, c-MET, and AXL | [9,158,164] | |
| VEGF Inhibitors | Ramucirumab | VEGFR-2 | [9,159,164] |
| Bevacizumab | VEGFR2 by binding VEGF-A | [9,164,165] | |
| Immune Checkpoint Inhibitor | Atezolizumab | PD-L1 | [9,164,165] |
7. Cisplatin and Treatment of Cholangiocarcinoma
8. Cisplatin for Pediatric Liver Tumors
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Omata, M.; Cheng, A.L.; Kokudo, N.; Kudo, M.; Lee, J.M.; Jia, J.; Tateishi, R.; Han, K.H.; Chawla, Y.K.; Shiina, S.; et al. Asia-Pacific clinical practice guidelines on the management of hepatocellular carcinoma: A 2017 update. Hepatol. Int. 2017, 11, 317–370. [Google Scholar] [CrossRef] [PubMed]
- European Association for the Study of the Liver. EASL Clinical Practice Guidelines: Management of hepatocellular carcinoma. J. Hepatol. 2018, 69, 182–236. [Google Scholar] [CrossRef] [PubMed]
- Marrero, J.A.; Kulik, L.M.; Sirlin, C.B.; Zhu, A.X.; Finn, R.S.; Abecassis, M.M.; Roberts, L.R.; Heimbach, J.K. Diagnosis, Staging, and Management of Hepatocellular Carcinoma: 2018 Practice Guidance by the American Association for the Study of Liver Diseases. Hepatology 2018, 68, 723–750. [Google Scholar] [CrossRef] [PubMed]
- DeVita, V.T., Jr.; Chu, E. A history of cancer chemotherapy. Cancer Res. 2008, 68, 8643–8653. [Google Scholar] [CrossRef]
- Su, S.; Chen, Y.; Zhang, P.; Ma, R.; Zhang, W.; Liu, J.; Li, T.; Niu, H.; Cao, Y.; Hu, B.; et al. The role of Platinum(IV)-based antitumor drugs and the anticancer immune response in medicinal inorganic chemistry. A systematic review from 2017 to 2022. Eur. J. Med. Chem. 2022, 243, 114680. [Google Scholar] [CrossRef]
- Ghosh, S. Cisplatin: The first metal based anticancer drug. Bioorg. Chem. 2019, 88, 102925. [Google Scholar] [CrossRef]
- Szefler, B.; Czelen, P. Will the Interactions of Some Platinum (II)-Based Drugs with B-Vitamins Reduce Their Therapeutic Effect in Cancer Patients? Comparison of Chemotherapeutic Agents such as Cisplatin, Carboplatin and Oxaliplatin-A Review. Int. J. Mol. Sci. 2023, 24, 1548. [Google Scholar] [CrossRef]
- Laface, C.; Laforgia, M.; Molinari, P.; Ugenti, I.; Gadaleta, C.D.; Porta, C.; Ranieri, G. Hepatic Arterial Infusion of Chemotherapy for Advanced Hepatobiliary Cancers: State of the Art. Cancers 2021, 13, 3091. [Google Scholar] [CrossRef]
- Laface, C.; Fedele, P.; Maselli, F.M.; Ambrogio, F.; Foti, C.; Molinari, P.; Ammendola, M.; Lioce, M.; Ranieri, G. Targeted Therapy for Hepatocellular Carcinoma: Old and New Opportunities. Cancers 2022, 14, 4028. [Google Scholar] [CrossRef]
- Laface, C.; Ranieri, G.; Maselli, F.M.; Ambrogio, F.; Foti, C.; Ammendola, M.; Laterza, M.; Cazzato, G.; Memeo, R.; Mastrandrea, G.; et al. Immunotherapy and the Combination with Targeted Therapies for Advanced Hepatocellular Carcinoma. Cancers 2023, 15, 654. [Google Scholar] [CrossRef]
- Moawad, A.W.; Morshid, A.; Khalaf, A.M.; Elmohr, M.M.; Hazle, J.D.; Fuentes, D.; Badawy, M.; Kaseb, A.O.; Hassan, M.; Mahvash, A.; et al. Multimodality annotated hepatocellular carcinoma data set including pre- and post-TACE with imaging segmentation. Sci. Data 2023, 10, 33. [Google Scholar] [CrossRef]
- Llovet, J.M.; Bruix, J. Systematic review of randomized trials for unresectable hepatocellular carcinoma: Chemoembolization improves survival. Hepatology 2003, 37, 429–442. [Google Scholar] [CrossRef]
- Lopez, P.M.; Villanueva, A.; Llovet, J.M. Systematic review: Evidence-based management of hepatocellular carcinoma—An updated analysis of randomized controlled trials. Aliment. Pharm. Ther. 2006, 23, 1535–1547. [Google Scholar] [CrossRef]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.L.; Forner, A.; et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef]
- Kudo, M.; Finn, R.S.; Qin, S.; Han, K.H.; Ikeda, K.; Piscaglia, F.; Baron, A.; Park, J.W.; Han, G.; Jassem, J.; et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: A randomised phase 3 non-inferiority trial. Lancet 2018, 391, 1163–1173. [Google Scholar] [CrossRef]
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.Y.; Kudo, M.; Breder, V.; Merle, P.; Kaseb, A.O.; et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N. Engl. J. Med. 2020, 382, 1894–1905. [Google Scholar] [CrossRef]
- National Institute of Diabetes and Digestive and Kidney Diseases. Platinum Coordination Complexes. In LiverTox: Clinical and Research Information on Drug-Induced Liver Injury; National Institute of Diabetes and Digestive and Kidney Diseases: Bethesda, MD, USA, 2012. [Google Scholar]
- Forgie, B.N.; Prakash, R.; Telleria, C.M. Revisiting the Anti-Cancer Toxicity of Clinically Approved Platinating Derivatives. Int. J. Mol. Sci. 2022, 23, 15410. [Google Scholar] [CrossRef]
- Dasari, S.; Tchounwou, P.B. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharm. 2014, 740, 364–378. [Google Scholar] [CrossRef]
- Rosenberg, B.; Vancamp, L.; Krigas, T. Inhibition of Cell Division in Escherichia Coli by Electrolysis Products from a Platinum Electrode. Nature 1965, 205, 698–699. [Google Scholar] [CrossRef]
- Rosenberg, B.; VanCamp, L.; Trosko, J.E.; Mansour, V.H. Platinum compounds: A new class of potent antitumour agents. Nature 1969, 222, 385–386. [Google Scholar] [CrossRef]
- Romani, A.M.P. Cisplatin in cancer treatment. Biochem. Pharm. 2022, 206, 115323. [Google Scholar] [CrossRef] [PubMed]
- Kelland, L. The resurgence of platinum-based cancer chemotherapy. Nat. Rev. Cancer 2007, 7, 573–584. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, A.I.; Christodoulou, J.; Parkinson, J.A.; Barnham, K.J.; Tucker, A.; Woodrow, J.; Sadler, P.J. Cisplatin binding sites on human albumin. J. Biol. Chem. 1998, 273, 14721–14730. [Google Scholar] [CrossRef] [PubMed]
- Gullo, J.J.; Litterst, C.L.; Maguire, P.J.; Sikic, B.I.; Hoth, D.F.; Woolley, P.V. Pharmacokinetics and protein binding of cis-dichlorodiammine platinum (II) administered as a one hour or as a twenty hour infusion. Cancer Chemother. Pharm. 1980, 5, 21–26. [Google Scholar] [CrossRef]
- Makovec, T. Cisplatin and beyond: Molecular mechanisms of action and drug resistance development in cancer chemotherapy. Radiol. Oncol. 2019, 53, 148–158. [Google Scholar] [CrossRef]
- Eljack, N.D.; Ma, H.Y.; Drucker, J.; Shen, C.; Hambley, T.W.; New, E.J.; Friedrich, T.; Clarke, R.J. Mechanisms of cell uptake and toxicity of the anticancer drug cisplatin. Metallomics 2014, 6, 2126–2133. [Google Scholar] [CrossRef]
- Lambert, I.H.; Sorensen, B.H. Facilitating the Cellular Accumulation of Pt-Based Chemotherapeutic Drugs. Int. J. Mol. Sci. 2018, 19, 2249. [Google Scholar] [CrossRef]
- Kishimoto, S.; Yasuda, M.; Suzuki, R.; Fukushima, S. Intracellular uptake of an antitumor-active azole-bridged dinuclear platinum(II) complex in cisplatin-resistant tumor cells. Biometals 2016, 29, 1075–1083. [Google Scholar] [CrossRef]
- Binks, S.P.; Dobrota, M. Kinetics and mechanism of uptake of platinum-based pharmaceuticals by the rat small intestine. Biochem. Pharm. 1990, 40, 1329–1336. [Google Scholar] [CrossRef]
- Yonezawa, A.; Masuda, S.; Yokoo, S.; Katsura, T.; Inui, K. Cisplatin and oxaliplatin, but not carboplatin and nedaplatin, are substrates for human organic cation transporters (SLC22A1-3 and multidrug and toxin extrusion family). J. Pharm. Exp. Ther. 2006, 319, 879–886. [Google Scholar] [CrossRef]
- Nieskens, T.T.G.; Peters, J.G.P.; Dabaghie, D.; Korte, D.; Jansen, K.; Van Asbeck, A.H.; Tavraz, N.N.; Friedrich, T.; Russel, F.G.M.; Masereeuw, R.; et al. Expression of Organic Anion Transporter 1 or 3 in Human Kidney Proximal Tubule Cells Reduces Cisplatin Sensitivity. Drug Metab. Dispos. 2018, 46, 592–599. [Google Scholar] [CrossRef]
- Wee, N.K.; Weinstein, D.C.; Fraser, S.T.; Assinder, S.J. The mammalian copper transporters CTR1 and CTR2 and their roles in development and disease. Int. J. Biochem. Cell Biol. 2013, 45, 960–963. [Google Scholar] [CrossRef]
- Ishida, S.; Lee, J.; Thiele, D.J.; Herskowitz, I. Uptake of the anticancer drug cisplatin mediated by the copper transporter Ctr1 in yeast and mammals. Proc. Natl. Acad. Sci. USA 2002, 99, 14298–14302. [Google Scholar] [CrossRef]
- Safaei, R. Role of copper transporters in the uptake and efflux of platinum containing drugs. Cancer Lett. 2006, 234, 34–39. [Google Scholar] [CrossRef]
- Kuo, M.T.; Fu, S.; Savaraj, N.; Chen, H.H. Role of the human high-affinity copper transporter in copper homeostasis regulation and cisplatin sensitivity in cancer chemotherapy. Cancer Res. 2012, 72, 4616–4621. [Google Scholar] [CrossRef]
- Katano, K.; Kondo, A.; Safaei, R.; Holzer, A.; Samimi, G.; Mishima, M.; Kuo, Y.M.; Rochdi, M.; Howell, S.B. Acquisition of resistance to cisplatin is accompanied by changes in the cellular pharmacology of copper. Cancer Res. 2002, 62, 6559–6565. [Google Scholar]
- Ivy, K.D.; Kaplan, J.H. A re-evaluation of the role of hCTR1, the human high-affinity copper transporter, in platinum-drug entry into human cells. Mol. Pharm. 2013, 83, 1237–1246. [Google Scholar] [CrossRef]
- Beretta, G.L.; Gatti, L.; Tinelli, S.; Corna, E.; Colangelo, D.; Zunino, F.; Perego, P. Cellular pharmacology of cisplatin in relation to the expression of human copper transporter CTR1 in different pairs of cisplatin-sensitive and-resistant cells. Biochem. Pharm. 2004, 68, 283–291. [Google Scholar] [CrossRef]
- Basu, A.; Krishnamurthy, S. Cellular responses to Cisplatin-induced DNA damage. J. Nucleic Acids 2010, 2010, 201367. [Google Scholar] [CrossRef]
- Lugones, Y.; Loren, P.; Salazar, L.A. Cisplatin Resistance: Genetic and Epigenetic Factors Involved. Biomolecules 2022, 12, 1365. [Google Scholar] [CrossRef]
- Plooy, A.C.; Fichtinger-Schepman, A.M.; Schutte, H.H.; van Dijk, M.; Lohman, P.H. The quantitative detection of various Pt-DNA-adducts in Chinese hamster ovary cells treated with cisplatin: Application of immunochemical techniques. Carcinogenesis 1985, 6, 561–566. [Google Scholar] [CrossRef] [PubMed]
- Eastman, A. Reevaluation of interaction of cis-dichloro(ethylenediamine)platinum(II) with DNA. Biochemistry 1986, 25, 3912–3915. [Google Scholar] [CrossRef] [PubMed]
- Deans, A.J.; West, S.C. DNA interstrand crosslink repair and cancer. Nat. Rev. Cancer 2011, 11, 467–480. [Google Scholar] [CrossRef] [PubMed]
- Florea, A.M.; Busselberg, D. Cisplatin as an anti-tumor drug: Cellular mechanisms of activity, drug resistance and induced side effects. Cancers 2011, 3, 1351–1371. [Google Scholar] [CrossRef]
- Ranasinghe, R.; Mathai, M.L.; Zulli, A. Cisplatin for cancer therapy and overcoming chemoresistance. Heliyon 2022, 8, e10608. [Google Scholar] [CrossRef]
- Vichi, P.; Coin, F.; Renaud, J.P.; Vermeulen, W.; Hoeijmakers, J.H.; Moras, D.; Egly, J.M. Cisplatin- and UV-damaged DNA lure the basal transcription factor TFIID/TBP. EMBO J. 1997, 16, 7444–7456. [Google Scholar] [CrossRef]
- Treiber, D.K.; Zhai, X.; Jantzen, H.M.; Essigmann, J.M. Cisplatin-DNA adducts are molecular decoys for the ribosomal RNA transcription factor hUBF (human upstream binding factor). Proc. Natl. Acad. Sci. USA 1994, 91, 5672–5676. [Google Scholar] [CrossRef]
- Zhai, X.; Beckmann, H.; Jantzen, H.M.; Essigmann, J.M. Cisplatin-DNA adducts inhibit ribosomal RNA synthesis by hijacking the transcription factor human upstream binding factor. Biochemistry 1998, 37, 16307–16315. [Google Scholar] [CrossRef]
- Mymryk, J.S.; Zaniewski, E.; Archer, T.K. Cisplatin inhibits chromatin remodeling, transcription factor binding, and transcription from the mouse mammary tumor virus promoter in vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 2076–2080. [Google Scholar] [CrossRef]
- Ober, M.; Lippard, S.J. A 1,2-d(GpG) cisplatin intrastrand cross-link influences the rotational and translational setting of DNA in nucleosomes. J. Am. Chem. Soc. 2008, 130, 2851–2861. [Google Scholar] [CrossRef]
- Todd, R.C.; Lippard, S.J. Consequences of cisplatin binding on nucleosome structure and dynamics. Chem. Biol. 2010, 17, 1334–1343. [Google Scholar] [CrossRef]
- Zhu, G.; Song, L.; Lippard, S.J. Visualizing inhibition of nucleosome mobility and transcription by cisplatin-DNA interstrand crosslinks in live mammalian cells. Cancer Res. 2013, 73, 4451–4460. [Google Scholar] [CrossRef]
- Achkar, I.W.; Abdulrahman, N.; Al-Sulaiti, H.; Joseph, J.M.; Uddin, S.; Mraiche, F. Cisplatin based therapy: The role of the mitogen activated protein kinase signaling pathway. J. Transl. Med. 2018, 16, 96. [Google Scholar] [CrossRef]
- Olivero, O.A.; Chang, P.K.; Lopez-Larraza, D.M.; Semino-Mora, M.C.; Poirier, M.C. Preferential formation and decreased removal of cisplatin-DNA adducts in Chinese hamster ovary cell mitochondrial DNA as compared to nuclear DNA. Mutat. Res. 1997, 391, 79–86. [Google Scholar] [CrossRef]
- Yang, Z.; Schumaker, L.M.; Egorin, M.J.; Zuhowski, E.G.; Guo, Z.; Cullen, K.J. Cisplatin preferentially binds mitochondrial DNA and voltage-dependent anion channel protein in the mitochondrial membrane of head and neck squamous cell carcinoma: Possible role in apoptosis. Clin. Cancer Res. 2006, 12, 5817–5825. [Google Scholar] [CrossRef]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim. Biophys. Acta 2016, 1863, 2977–2992. [Google Scholar] [CrossRef]
- Marullo, R.; Werner, E.; Degtyareva, N.; Moore, B.; Altavilla, G.; Ramalingam, S.S.; Doetsch, P.W. Cisplatin induces a mitochondrial-ROS response that contributes to cytotoxicity depending on mitochondrial redox status and bioenergetic functions. PLoS ONE 2013, 8, e81162. [Google Scholar] [CrossRef]
- Gupta, S.C.; Hevia, D.; Patchva, S.; Park, B.; Koh, W.; Aggarwal, B.B. Upsides and downsides of reactive oxygen species for cancer: The roles of reactive oxygen species in tumorigenesis, prevention, and therapy. Antioxid. Redox Signal. 2012, 16, 1295–1322. [Google Scholar] [CrossRef]
- Hampton, M.B.; Orrenius, S. Dual regulation of caspase activity by hydrogen peroxide: Implications for apoptosis. FEBS Lett. 1997, 414, 552–556. [Google Scholar] [CrossRef]
- Shrivastava, A.; Kuzontkoski, P.M.; Groopman, J.E.; Prasad, A. Cannabidiol induces programmed cell death in breast cancer cells by coordinating the cross-talk between apoptosis and autophagy. Mol. Cancer Ther. 2011, 10, 1161–1172. [Google Scholar] [CrossRef]
- Shen, D.W.; Pouliot, L.M.; Hall, M.D.; Gottesman, M.M. Cisplatin resistance: A cellular self-defense mechanism resulting from multiple epigenetic and genetic changes. Pharm. Rev. 2012, 64, 706–721. [Google Scholar] [CrossRef] [PubMed]
- Kalayda, G.V.; Wagner, C.H.; Jaehde, U. Relevance of copper transporter 1 for cisplatin resistance in human ovarian carcinoma cells. J. Inorg. Biochem. 2012, 116, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Kilari, D.; Guancial, E.; Kim, E.S. Role of copper transporters in platinum resistance. World J. Clin. Oncol. 2016, 7, 106–113. [Google Scholar] [CrossRef] [PubMed]
- Korita, P.V.; Wakai, T.; Shirai, Y.; Matsuda, Y.; Sakata, J.; Takamura, M.; Yano, M.; Sanpei, A.; Aoyagi, Y.; Hatakeyama, K.; et al. Multidrug resistance-associated protein 2 determines the efficacy of cisplatin in patients with hepatocellular carcinoma. Oncol. Rep. 2010, 23, 965–972. [Google Scholar]
- Pearson, S.A.; Cowan, J.A. Glutathione-coordinated metal complexes as substrates for cellular transporters. Metallomics 2021, 13, mfab015. [Google Scholar] [CrossRef]
- Chen, H.H.; Kuo, M.T. Role of glutathione in the regulation of Cisplatin resistance in cancer chemotherapy. Met. Based Drugs 2010, 2010, 430939. [Google Scholar] [CrossRef]
- Hinoshita, E.; Uchiumi, T.; Taguchi, K.; Kinukawa, N.; Tsuneyoshi, M.; Maehara, Y.; Sugimachi, K.; Kuwano, M. Increased expression of an ATP-binding cassette superfamily transporter, multidrug resistance protein 2, in human colorectal carcinomas. Clin. Cancer Res. 2000, 6, 2401–2407. [Google Scholar]
- Byun, S.S.; Kim, S.W.; Choi, H.; Lee, C.; Lee, E. Augmentation of cisplatin sensitivity in cisplatin-resistant human bladder cancer cells by modulating glutathione concentrations and glutathione-related enzyme activities. BJU Int. 2005, 95, 1086–1090. [Google Scholar] [CrossRef]
- Rocha, C.R.; Garcia, C.C.; Vieira, D.B.; Quinet, A.; de Andrade-Lima, L.C.; Munford, V.; Belizario, J.E.; Menck, C.F. Glutathione depletion sensitizes cisplatin- and temozolomide-resistant glioma cells in vitro and in vivo. Cell Death Dis. 2014, 5, e1505. [Google Scholar] [CrossRef]
- Konoshenko, M.; Lansukhay, Y.; Krasilnikov, S.; Laktionov, P. MicroRNAs as Predictors of Lung-Cancer Resistance and Sensitivity to Cisplatin. Int. J. Mol. Sci. 2022, 23, 7594. [Google Scholar] [CrossRef]
- Zou, Y.; Liu, Y.; Wu, X.; Shell, S.M. Functions of human replication protein A (RPA): From DNA replication to DNA damage and stress responses. J. Cell Physiol. 2006, 208, 267–273. [Google Scholar] [CrossRef]
- Nasrallah, N.A.; Wiese, B.M.; Sears, C.R. Xeroderma Pigmentosum Complementation Group C (XPC): Emerging Roles in Non-Dermatologic Malignancies. Front Oncol 2022, 12, 846965. [Google Scholar] [CrossRef]
- Fang, C.; Chen, Y.X.; Wu, N.Y.; Yin, J.Y.; Li, X.P.; Huang, H.S.; Zhang, W.; Zhou, H.H.; Liu, Z.Q. MiR-488 inhibits proliferation and cisplatin sensibility in non-small-cell lung cancer (NSCLC) cells by activating the eIF3a-mediated NER signaling pathway. Sci. Rep. 2017, 7, 40384. [Google Scholar] [CrossRef]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef]
- Sakai, W.; Swisher, E.M.; Karlan, B.Y.; Agarwal, M.K.; Higgins, J.; Friedman, C.; Villegas, E.; Jacquemont, C.; Farrugia, D.J.; Couch, F.J.; et al. Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature 2008, 451, 1116–1120. [Google Scholar] [CrossRef]
- Swisher, E.M.; Sakai, W.; Karlan, B.Y.; Wurz, K.; Urban, N.; Taniguchi, T. Secondary BRCA1 mutations in BRCA1-mutated ovarian carcinomas with platinum resistance. Cancer Res. 2008, 68, 2581–2586. [Google Scholar] [CrossRef]
- Zhao, J.; Fu, W.; Liao, H.; Dai, L.; Jiang, Z.; Pan, Y.; Huang, H.; Mo, Y.; Li, S.; Yang, G.; et al. The regulatory and predictive functions of miR-17 and miR-92 families on cisplatin resistance of non-small cell lung cancer. BMC Cancer 2015, 15, 731. [Google Scholar] [CrossRef]
- Nehme, A.; Baskaran, R.; Nebel, S.; Fink, D.; Howell, S.B.; Wang, J.Y.; Christen, R.D. Induction of JNK and c-Abl signalling by cisplatin and oxaliplatin in mismatch repair-proficient and -deficient cells. Br. J. Cancer 1999, 79, 1104–1110. [Google Scholar] [CrossRef]
- Martinez-Rivera, M.; Siddik, Z.H. Resistance and gain-of-resistance phenotypes in cancers harboring wild-type p53. Biochem. Pharm. 2012, 83, 1049–1062. [Google Scholar] [CrossRef]
- Tsang, R.Y.; Al-Fayea, T.; Au, H.J. Cisplatin overdose: Toxicities and management. Drug Saf. 2009, 32, 1109–1122. [Google Scholar] [CrossRef]
- Ali, R.; Aouida, M.; Alhaj Sulaiman, A.; Madhusudan, S.; Ramotar, D. Can Cisplatin Therapy Be Improved? Pathways That Can Be Targeted. Int. J. Mol. Sci. 2022, 23, 7241. [Google Scholar] [CrossRef] [PubMed]
- Crona, D.J.; Faso, A.; Nishijima, T.F.; McGraw, K.A.; Galsky, M.D.; Milowsky, M.I. A Systematic Review of Strategies to Prevent Cisplatin-Induced Nephrotoxicity. Oncologist 2017, 22, 609–619. [Google Scholar] [CrossRef] [PubMed]
- Volarevic, V.; Djokovic, B.; Jankovic, M.G.; Harrell, C.R.; Fellabaum, C.; Djonov, V.; Arsenijevic, N. Molecular mechanisms of cisplatin-induced nephrotoxicity: A balance on the knife edge between renoprotection and tumor toxicity. J. Biomed. Sci. 2019, 26, 25. [Google Scholar] [CrossRef] [PubMed]
- Fang, C.Y.; Lou, D.Y.; Zhou, L.Q.; Wang, J.C.; Yang, B.; He, Q.J.; Wang, J.J.; Weng, Q.J. Natural products: Potential treatments for cisplatin-induced nephrotoxicity. Acta Pharm. Sin. 2021, 42, 1951–1969. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.; Panichpisal, K.; Kurtzman, N.; Nugent, K. Cisplatin nephrotoxicity: A review. Am. J. Med. Sci. 2007, 334, 115–124. [Google Scholar] [CrossRef]
- Pabla, N.; Murphy, R.F.; Liu, K.; Dong, Z. The copper transporter Ctr1 contributes to cisplatin uptake by renal tubular cells during cisplatin nephrotoxicity. Am. J. Physiol. Ren. Physiol. 2009, 296, F505–F511. [Google Scholar] [CrossRef]
- Filipski, K.K.; Loos, W.J.; Verweij, J.; Sparreboom, A. Interaction of Cisplatin with the human organic cation transporter 2. Clin. Cancer Res. 2008, 14, 3875–3880. [Google Scholar] [CrossRef]
- Filipski, K.K.; Mathijssen, R.H.; Mikkelsen, T.S.; Schinkel, A.H.; Sparreboom, A. Contribution of organic cation transporter 2 (OCT2) to cisplatin-induced nephrotoxicity. Clin. Pharm. Ther. 2009, 86, 396–402. [Google Scholar] [CrossRef]
- Miller, R.P.; Tadagavadi, R.K.; Ramesh, G.; Reeves, W.B. Mechanisms of Cisplatin nephrotoxicity. Toxins 2010, 2, 2490–2518. [Google Scholar] [CrossRef]
- Deng, F.; Sharma, I.; Dai, Y.; Yang, M.; Kanwar, Y.S. Myo-inositol oxygenase expression profile modulates pathogenic ferroptosis in the renal proximal tubule. J. Clin. Investig. 2019, 129, 5033–5049. [Google Scholar] [CrossRef]
- Baliga, R.; Ueda, N.; Walker, P.D.; Shah, S.V. Oxidant mechanisms in toxic acute renal failure. Drug Metab. Rev. 1999, 31, 971–997. [Google Scholar] [CrossRef]
- Yang, C.; Kaushal, V.; Haun, R.S.; Seth, R.; Shah, S.V.; Kaushal, G.P. Transcriptional activation of caspase-6 and -7 genes by cisplatin-induced p53 and its functional significance in cisplatin nephrotoxicity. Cell Death Differ. 2008, 15, 530–544. [Google Scholar] [CrossRef]
- Tsuruya, K.; Ninomiya, T.; Tokumoto, M.; Hirakawa, M.; Masutani, K.; Taniguchi, M.; Fukuda, K.; Kanai, H.; Kishihara, K.; Hirakata, H.; et al. Direct involvement of the receptor-mediated apoptotic pathways in cisplatin-induced renal tubular cell death. Kidney Int. 2003, 63, 72–82. [Google Scholar] [CrossRef]
- Ramesh, G.; Reeves, W.B. TNF-alpha mediates chemokine and cytokine expression and renal injury in cisplatin nephrotoxicity. J. Clin. Investig. 2002, 110, 835–842. [Google Scholar] [CrossRef]
- Dong, Z.; Atherton, S.S. Tumor necrosis factor-alpha in cisplatin nephrotoxicity: A homebred foe? Kidney Int. 2007, 72, 5–7. [Google Scholar] [CrossRef]
- Zhang, B.; Ramesh, G.; Norbury, C.C.; Reeves, W.B. Cisplatin-induced nephrotoxicity is mediated by tumor necrosis factor-alpha produced by renal parenchymal cells. Kidney Int. 2007, 72, 37–44. [Google Scholar] [CrossRef]
- Liu, M.; Chien, C.C.; Burne-Taney, M.; Molls, R.R.; Racusen, L.C.; Colvin, R.B.; Rabb, H. A pathophysiologic role for T lymphocytes in murine acute cisplatin nephrotoxicity. J. Am. Soc. Nephrol. 2006, 17, 765–774. [Google Scholar] [CrossRef]
- Tiseo, M.; Martelli, O.; Mancuso, A.; Sormani, M.P.; Bruzzi, P.; Di Salvia, R.; De Marinis, F.; Ardizzoni, A. Short hydration regimen and nephrotoxicity of intermediate to high-dose cisplatin-based chemotherapy for outpatient treatment in lung cancer and mesothelioma. Tumori 2007, 93, 138–144. [Google Scholar] [CrossRef]
- Lavole, A.; Danel, S.; Baudrin, L.; Gounant, V.; Ruppert, A.M.; Epaud, C.; Belmont, L.; Rosencher, L.; Cadranel, J.; Milleron, B. Routine administration of a single dose of cisplatin >/= 75 mg/m2 after short hydration in an outpatient lung-cancer clinic. Bull. Cancer 2012, 99, E43–E48. [Google Scholar] [CrossRef]
- Ouchi, A.; Asano, M.; Aono, K.; Watanabe, T.; Kato, T. Comparison of short and continuous hydration regimen in chemotherapy containing intermediate- to high-dose Cisplatin. J. Oncol. 2014, 2014, 767652. [Google Scholar] [CrossRef]
- Ninomiya, K.; Hotta, K.; Hisamoto-Sato, A.; Ichihara, E.; Gotoda, H.; Morichika, D.; Tamura, T.; Kayatani, H.; Minami, D.; Kubo, T.; et al. Short-term low-volume hydration in cisplatin-based chemotherapy for patients with lung cancer: The second prospective feasibility study in the Okayama Lung Cancer Study Group Trial 1201. Int. J. Clin. Oncol. 2016, 21, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Hase, T.; Miyazaki, M.; Ichikawa, K.; Yogo, N.; Ozawa, N.; Hatta, T.; Ando, M.; Sato, M.; Kondo, M.; Yamada, K.; et al. Short hydration with 20 mEq of magnesium supplementation for lung cancer patients receiving cisplatin-based chemotherapy: A prospective study. Int. J. Clin. Oncol. 2020, 25, 1928–1935. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, Y.; Watanabe, K.; Tsukiyama, I.; Yabushita, H.; Matsuura, K.; Wakatsuki, A. Hydration with 15 mEq Magnesium Is Effective at Reducingthe Risk for Cisplatin-induced Nephrotoxicity in Patients Receiving Cisplatin (>/=50 mg/m2) Combination Chemotherapy. Anticancer Res. 2016, 36, 1873–1877. [Google Scholar] [PubMed]
- Miyoshi, T.; Hayashi, T.; Uoi, M.; Omura, F.; Tsumagari, K.; Maesaki, S.; Yokota, C.; Nakano, T.; Egawa, T. Preventive effect of 20 mEq and 8 mEq magnesium supplementation on cisplatin-induced nephrotoxicity: A propensity score-matched analysis. Support Care Cancer 2022, 30, 3345–3351. [Google Scholar] [CrossRef]
- Casanova, A.G.; Hernandez-Sanchez, M.T.; Lopez-Hernandez, F.J.; Martinez-Salgado, C.; Prieto, M.; Vicente-Vicente, L.; Morales, A.I. Systematic review and meta-analysis of the efficacy of clinically tested protectants of cisplatin nephrotoxicity. Eur. J. Clin. Pharm. 2020, 76, 23–33. [Google Scholar] [CrossRef]
- Yokoo, K.; Murakami, R.; Matsuzaki, T.; Yoshitome, K.; Hamada, A.; Saito, H. Enhanced renal accumulation of cisplatin via renal organic cation transporter deteriorates acute kidney injury in hypomagnesemic rats. Clin. Exp. Nephrol. 2009, 13, 578–584. [Google Scholar] [CrossRef]
- Solanki, M.H.; Chatterjee, P.K.; Xue, X.; Gupta, M.; Rosales, I.; Yeboah, M.M.; Kohn, N.; Metz, C.N. Magnesium protects against cisplatin-induced acute kidney injury without compromising cisplatin-mediated killing of an ovarian tumor xenograft in mice. Am. J. Physiol. Renal Physiol. 2015, 309, F35–F47. [Google Scholar] [CrossRef]
- Solanki, M.H.; Chatterjee, P.K.; Gupta, M.; Xue, X.; Plagov, A.; Metz, M.H.; Mintz, R.; Singhal, P.C.; Metz, C.N. Magnesium protects against cisplatin-induced acute kidney injury by regulating platinum accumulation. Am. J. Physiol. Renal Physiol. 2014, 307, F369–F384. [Google Scholar] [CrossRef]
- Ruggiero, A.; Rizzo, D.; Trombatore, G.; Maurizi, P.; Riccardi, R. The ability of mannitol to decrease cisplatin-induced nephrotoxicity in children: Real or not? Cancer Chemother. Pharm. 2016, 77, 19–26. [Google Scholar] [CrossRef]
- Sainamthip, P.; Saichaemchan, S.; Satirapoj, B.; Prasongsook, N. The Effect of Intravenous Mannitol Combined With Normal Saline in Preventing Cisplatin-Induced Nephrotoxicity: A Randomized, Double-Blind, Placebo-Controlled Trial. JCO Glob. Oncol. 2022, 8, e2100275. [Google Scholar] [CrossRef]
- McKibbin, T.; Cheng, L.L.; Kim, S.; Steuer, C.E.; Owonikoko, T.K.; Khuri, F.R.; Shin, D.M.; Saba, N.F. Mannitol to prevent cisplatin-induced nephrotoxicity in patients with squamous cell cancer of the head and neck (SCCHN) receiving concurrent therapy. Support Care Cancer 2016, 24, 1789–1793. [Google Scholar] [CrossRef]
- Morgan, K.P.; Buie, L.W.; Savage, S.W. The role of mannitol as a nephroprotectant in patients receiving cisplatin therapy. Ann. Pharmacother. 2012, 46, 276–281. [Google Scholar] [CrossRef]
- Ruggiero, A.; Ariano, A.; Triarico, S.; Capozza, M.A.; Romano, A.; Maurizi, P.; Mastrangelo, S.; Attina, G. Cisplatin-induced nephrotoxicity in children: What is the best protective strategy? J. Oncol. Pharm. Pract. 2021, 27, 180–186. [Google Scholar] [CrossRef]
- Callejo, A.; Sedo-Cabezon, L.; Juan, I.D.; Llorens, J. Cisplatin-Induced Ototoxicity: Effects, Mechanisms and Protection Strategies. Toxics 2015, 3, 268–293. [Google Scholar] [CrossRef]
- Rybak, L.P.; Mukherjea, D.; Ramkumar, V. Mechanisms of Cisplatin-Induced Ototoxicity and Prevention. Semin. Hear. 2019, 40, 197–204. [Google Scholar] [CrossRef]
- Zajaczkowska, R.; Kocot-Kepska, M.; Leppert, W.; Wrzosek, A.; Mika, J.; Wordliczek, J. Mechanisms of Chemotherapy-Induced Peripheral Neuropathy. Int. J. Mol. Sci. 2019, 20, 1451. [Google Scholar] [CrossRef]
- Brouwers, E.E.; Huitema, A.D.; Boogerd, W.; Beijnen, J.H.; Schellens, J.H. Persistent neuropathy after treatment with cisplatin and oxaliplatin. Acta Oncol. 2009, 48, 832–841. [Google Scholar] [CrossRef]
- Gregg, R.W.; Molepo, J.M.; Monpetit, V.J.; Mikael, N.Z.; Redmond, D.; Gadia, M.; Stewart, D.J. Cisplatin neurotoxicity: The relationship between dosage, time, and platinum concentration in neurologic tissues, and morphologic evidence of toxicity. J. Clin. Oncol. 1992, 10, 795–803. [Google Scholar] [CrossRef]
- Starobova, H.; Vetter, I. Pathophysiology of Chemotherapy-Induced Peripheral Neuropathy. Front. Mol. Neurosci. 2017, 10, 174. [Google Scholar] [CrossRef]
- Liu, M.; Liu, J.; Wang, L.; Wu, H.; Zhou, C.; Zhu, H.; Xu, N.; Xie, Y. Association of serum microRNA expression in hepatocellular carcinomas treated with transarterial chemoembolization and patient survival. PLoS ONE 2014, 9, e109347. [Google Scholar] [CrossRef]
- Kerckhove, N.; Collin, A.; Conde, S.; Chaleteix, C.; Pezet, D.; Balayssac, D. Long-Term Effects, Pathophysiological Mechanisms, and Risk Factors of Chemotherapy-Induced Peripheral Neuropathies: A Comprehensive Literature Review. Front. Pharm. 2017, 8, 86. [Google Scholar] [CrossRef] [PubMed]
- Santos, N.; Ferreira, R.S.; Santos, A.C.D. Overview of cisplatin-induced neurotoxicity and ototoxicity, and the protective agents. Food Chem. Toxicol. 2020, 136, 111079. [Google Scholar] [CrossRef] [PubMed]
- Olthoff, K.M.; Rosove, M.H.; Shackleton, C.R.; Imagawa, D.K.; Farmer, D.G.; Northcross, P.; Pakrasi, A.L.; Martin, P.; Goldstein, L.I.; Shaked, A.; et al. Adjuvant chemotherapy improves survival after liver transplantation for hepatocellular carcinoma. Ann. Surg. 1995, 221, 734–741, discussion 731–743. [Google Scholar] [CrossRef] [PubMed]
- Astolfi, L.; Ghiselli, S.; Guaran, V.; Chicca, M.; Simoni, E.; Olivetto, E.; Lelli, G.; Martini, A. Correlation of adverse effects of cisplatin administration in patients affected by solid tumours: A retrospective evaluation. Oncol. Rep. 2013, 29, 1285–1292. [Google Scholar] [CrossRef] [PubMed]
- Al-Malki, A.L.; Sayed, A.A. Thymoquinone attenuates cisplatin-induced hepatotoxicity via nuclear factor kappa-beta. BMC Complement Altern Med. 2014, 14, 282. [Google Scholar] [CrossRef]
- Hu, Y.; Sun, B.; Zhao, B.; Mei, D.; Gu, Q.; Tian, Z. Cisplatin-induced cardiotoxicity with midrange ejection fraction: A case report and review of the literature. Medicine 2018, 97, e13807. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Vogel, A.; Meyer, T.; Sapisochin, G.; Salem, R.; Saborowski, A. Hepatocellular carcinoma. Lancet 2022, 400, 1345–1362. [Google Scholar] [CrossRef]
- Petrick, J.L.; Florio, A.A.; Znaor, A.; Ruggieri, D.; Laversanne, M.; Alvarez, C.S.; Ferlay, J.; Valery, P.C.; Bray, F.; McGlynn, K.A. International trends in hepatocellular carcinoma incidence, 1978-2012. Int. J. Cancer 2020, 147, 317–330. [Google Scholar] [CrossRef]
- Younossi, Z.M. Non-alcoholic fatty liver disease—A global public health perspective. J. Hepatol. 2019, 70, 531–544. [Google Scholar] [CrossRef]
- Manjunatha, N.; Ganduri, V.; Rajasekaran, K.; Duraiyarasan, S.; Adefuye, M. Transarterial Chemoembolization and Unresectable Hepatocellular Carcinoma: A Narrative Review. Cureus 2022, 14, e28439. [Google Scholar] [CrossRef]
- Kasai, K.; Ushio, A.; Kasai, Y.; Sawara, K.; Miyamoto, Y.; Oikawa, K.; Takikawa, Y.; Suzuki, K. Therapeutic efficacy of transarterial chemo-embolization with a fine-powder formulation of cisplatin for hepatocellular carcinoma. World J. Gastroenterol. 2013, 19, 2242–2248. [Google Scholar] [CrossRef]
- Yodono, H.; Matsuo, K.; Shinohara, A. A retrospective comparative study of epirubicin-lipiodol emulsion and cisplatin-lipiodol suspension for use with transcatheter arterial chemoembolization for treatment of hepatocellular carcinoma. Anticancer Drugs 2011, 22, 277–282. [Google Scholar] [CrossRef]
- Maeda, N.; Osuga, K.; Higashihara, H.; Tomoda, K.; Mikami, K.; Nakazawa, T.; Nakamura, H.; Tomiyama, N. Transarterial chemoembolization with cisplatin as second-line treatment for hepatocellular carcinoma unresponsive to chemoembolization with epirubicin-Lipiodol emulsion. Cardiovasc. Interv. Radiol. 2012, 35, 82–89. [Google Scholar] [CrossRef]
- Kamada, K.; Nakanishi, T.; Kitamoto, M.; Aikata, H.; Kawakami, Y.; Ito, K.; Asahara, T.; Kajiyama, G. Long-term prognosis of patients undergoing transcatheter arterial chemoembolization for unresectable hepatocellular carcinoma: Comparison of cisplatin lipiodol suspension and doxorubicin hydrochloride emulsion. J. Vasc. Interv. Radiol. 2001, 12, 847–854. [Google Scholar] [CrossRef]
- De Baere, T.; Ronot, M.; Chung, J.W.; Golfieri, R.; Kloeckner, R.; Park, J.W.; Gebauer, B.; Kibriya, N.; Ananthakrishnan, G.; Miyayama, S. Initiative on Superselective Conventional Transarterial Chemoembolization Results (INSPIRE). Cardiovasc. Interv. Radiol. 2022, 45, 1430–1440. [Google Scholar] [CrossRef]
- Galle, P.R.; Tovoli, F.; Foerster, F.; Worns, M.A.; Cucchetti, A.; Bolondi, L. The treatment of intermediate stage tumours beyond TACE: From surgery to systemic therapy. J. Hepatol. 2017, 67, 173–183. [Google Scholar] [CrossRef]
- Kudo, M.; Ueshima, K.; Chan, S.; Minami, T.; Chishina, H.; Aoki, T.; Takita, M.; Hagiwara, S.; Minami, Y.; Ida, H.; et al. Lenvatinib as an Initial Treatment in Patients with Intermediate-Stage Hepatocellular Carcinoma Beyond Up-To-Seven Criteria and Child-Pugh A Liver Function: A Proof-Of-Concept Study. Cancers 2019, 11, 1084. [Google Scholar] [CrossRef]
- Reig, M.; Forner, A.; Rimola, J.; Ferrer-Fabrega, J.; Burrel, M.; Garcia-Criado, A.; Kelley, R.K.; Galle, P.R.; Mazzaferro, V.; Salem, R.; et al. BCLC strategy for prognosis prediction and treatment recommendation: The 2022 update. J. Hepatol. 2022, 76, 681–693. [Google Scholar] [CrossRef]
- Park, J.Y.; Ahn, S.H.; Yoon, Y.J.; Kim, J.K.; Lee, H.W.; Lee, D.Y.; Chon, C.Y.; Moon, Y.M.; Han, K.H. Repetitive short-course hepatic arterial infusion chemotherapy with high-dose 5-fluorouracil and cisplatin in patients with advanced hepatocellular carcinoma. Cancer 2007, 110, 129–137. [Google Scholar] [CrossRef]
- Kim, B.K.; Park, J.Y.; Choi, H.J.; Kim, D.Y.; Ahn, S.H.; Kim, J.K.; Lee, D.Y.; Lee, K.H.; Han, K.H. Long-term clinical outcomes of hepatic arterial infusion chemotherapy with cisplatin with or without 5-fluorouracil in locally advanced hepatocellular carcinoma. J. Cancer Res. Clin. Oncol. 2011, 137, 659–667. [Google Scholar] [CrossRef] [PubMed]
- Long, G.B.; Xiao, C.W.; Zhao, X.Y.; Zhang, J.; Li, X. Effects of hepatic arterial infusion chemotherapy in the treatment of hepatocellular carcinoma: A meta-analysis. Medicine 2020, 99, e20745. [Google Scholar] [CrossRef] [PubMed]
- Obi, S.; Sato, S.; Kawai, T. Current Status of Hepatic Arterial Infusion Chemotherapy. Liver Cancer 2015, 4, 188–199. [Google Scholar] [CrossRef] [PubMed]
- Shim, J.H.; Park, J.W.; Kim, J.H.; An, M.; Kong, S.Y.; Nam, B.H.; Choi, J.I.; Kim, H.B.; Lee, W.J.; Kim, C.M. Association between increment of serum VEGF level and prognosis after transcatheter arterial chemoembolization in hepatocellular carcinoma patients. Cancer Sci. 2008, 99, 2037–2044. [Google Scholar] [CrossRef]
- Pezzuto, A.; Carico, E. Role of HIF-1 in Cancer Progression: Novel Insights. A Review. Curr. Mol. Med. 2018, 18, 343–351. [Google Scholar] [CrossRef]
- Huang, Y.; Lin, D.; Taniguchi, C.M. Hypoxia inducible factor (HIF) in the tumor microenvironment: Friend or foe? Sci. China Life Sci. 2017, 60, 1114–1124. [Google Scholar] [CrossRef]
- Xiong, Z.P.; Yang, S.R.; Liang, Z.Y.; Xiao, E.H.; Yu, X.P.; Zhou, S.K.; Zhang, Z.S. Association between vascular endothelial growth factor and metastasis after transcatheter arterial chemoembolization in patients with hepatocellular carcinoma. Hepatobiliary Pancreat Dis. Int. 2004, 3, 386–390. [Google Scholar]
- Hsieh, M.Y.; Lin, Z.Y.; Chuang, W.L. Serial serum VEGF-A, angiopoietin-2, and endostatin measurements in cirrhotic patients with hepatocellular carcinoma treated by transcatheter arterial chemoembolization. Kaohsiung J. Med. Sci. 2011, 27, 314–322. [Google Scholar] [CrossRef]
- Liu, K.; Min, X.L.; Peng, J.; Yang, K.; Yang, L.; Zhang, X.M. The Changes of HIF-1alpha and VEGF Expression After TACE in Patients With Hepatocellular Carcinoma. J. Clin. Med. Res. 2016, 8, 297–302. [Google Scholar] [CrossRef]
- Wei, X.; Zhao, L.; Ren, R.; Ji, F.; Xue, S.; Zhang, J.; Liu, Z.; Ma, Z.; Wang, X.W.; Wong, L.; et al. MiR-125b Loss Activated HIF1alpha/pAKT Loop, Leading to Transarterial Chemoembolization Resistance in Hepatocellular Carcinoma. Hepatology 2021, 73, 1381–1398. [Google Scholar] [CrossRef]
- Ali, H.E.A.; Emam, A.A.; Zeeneldin, A.A.; Srour, R.; Tabashy, R.; El-Desouky, E.D.; Abd Elmageed, Z.Y.; Abdel-Wahab, A.A. Circulating miR-26a, miR-106b, miR-107 and miR-133b stratify hepatocellular carcinoma patients according to their response to transarterial chemoembolization. Clin. Biochem. 2019, 65, 45–52. [Google Scholar] [CrossRef]
- Suehiro, T.; Miyaaki, H.; Kanda, Y.; Shibata, H.; Honda, T.; Ozawa, E.; Miuma, S.; Taura, N.; Nakao, K. Serum exosomal microRNA-122 and microRNA-21 as predictive biomarkers in transarterial chemoembolization-treated hepatocellular carcinoma patients. Oncol. Lett. 2018, 16, 3267–3273. [Google Scholar] [CrossRef]
- Qin, L.; Zhan, Z.; Wei, C.; Li, X.; Zhang, T.; Li, J. Hsa-circRNA-G004213 promotes cisplatin sensitivity by regulating miR-513b-5p/PRPF39 in liver cancer. Mol. Med. Rep. 2021, 23, 1–12. [Google Scholar] [CrossRef]
- Shi, X.; Wang, B.; Feng, X.; Xu, Y.; Lu, K.; Sun, M. circRNAs and Exosomes: A Mysterious Frontier for Human Cancer. Mol. Ther. Nucleic. Acids 2020, 19, 384–392. [Google Scholar] [CrossRef]
- Cheng, A.L.; Kang, Y.K.; Chen, Z.; Tsao, C.J.; Qin, S.; Kim, J.S.; Luo, R.; Feng, J.; Ye, S.; Yang, T.S.; et al. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: A phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2009, 10, 25–34. [Google Scholar] [CrossRef]
- Bruix, J.; Qin, S.; Merle, P.; Granito, A.; Huang, Y.H.; Bodoky, G.; Pracht, M.; Yokosuka, O.; Rosmorduc, O.; Breder, V.; et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 389, 56–66. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K.; Meyer, T.; Cheng, A.L.; El-Khoueiry, A.B.; Rimassa, L.; Ryoo, B.Y.; Cicin, I.; Merle, P.; Chen, Y.; Park, J.W.; et al. Cabozantinib in Patients with Advanced and Progressing Hepatocellular Carcinoma. N. Engl. J. Med. 2018, 379, 54–63. [Google Scholar] [CrossRef]
- Zhu, A.X.; Kang, Y.K.; Yen, C.J.; Finn, R.S.; Galle, P.R.; Llovet, J.M.; Assenat, E.; Brandi, G.; Pracht, M.; Lim, H.Y.; et al. Ramucirumab after sorafenib in patients with advanced hepatocellular carcinoma and increased alpha-fetoprotein concentrations (REACH-2): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2019, 20, 282–296. [Google Scholar] [CrossRef]
- Kudo, M.; Ueshima, K.; Ikeda, M.; Torimura, T.; Tanabe, N.; Aikata, H.; Izumi, N.; Yamasaki, T.; Nojiri, S.; Hino, K.; et al. Final Results of TACTICS: A Randomized, Prospective Trial Comparing Transarterial Chemoembolization Plus Sorafenib to Transarterial Chemoembolization Alone in Patients with Unresectable Hepatocellular Carcinoma. Liver Cancer 2022, 11, 354–367. [Google Scholar] [CrossRef]
- Kuroda, H.; Oikawa, T.; Ninomiya, M.; Fujita, M.; Abe, K.; Okumoto, K.; Katsumi, T.; Sato, W.; Igarashi, G.; Iino, C.; et al. Objective Response by mRECIST to Initial Lenvatinib Therapy Is an Independent Factor Contributing to Deep Response in Hepatocellular Carcinoma Treated with Lenvatinib-Transcatheter Arterial Chemoembolization Sequential Therapy. Liver Cancer 2022, 11, 383–396. [Google Scholar] [CrossRef]
- Kudo, M. A New Treatment Option for Intermediate-Stage Hepatocellular Carcinoma with High Tumor Burden: Initial Lenvatinib Therapy with Subsequent Selective TACE. Liver Cancer 2019, 8, 299–311. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.; Bhardwaj, V. Therapeutic resistance in pancreatic ductal adenocarcinoma: Current challenges and future opportunities. World J. Gastroenterol. 2021, 27, 6527–6550. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Zhang, W.; Bi, X.; Yang, Z.; Tang, Y.; Jiang, L.; Bi, F.; Chen, M.; Cheng, S.; Chi, Y.; et al. Systemic Therapy for Hepatocellular Carcinoma: Chinese Consensus-Based Interdisciplinary Expert Statements. Liver Cancer 2022, 11, 192–208. [Google Scholar] [CrossRef] [PubMed]
- Granito, A.; Marinelli, S.; Forgione, A.; Renzulli, M.; Benevento, F.; Piscaglia, F.; Tovoli, F. Regorafenib Combined with Other Systemic Therapies: Exploring Promising Therapeutic Combinations in HCC. J. Hepatocell. Carcinoma 2021, 8, 477–492. [Google Scholar] [CrossRef]
- Kudo, M.; Ueshima, K.; Yokosuka, O.; Ogasawara, S.; Obi, S.; Izumi, N.; Aikata, H.; Nagano, H.; Hatano, E.; Sasaki, Y.; et al. Sorafenib plus low-dose cisplatin and fluorouracil hepatic arterial infusion chemotherapy versus sorafenib alone in patients with advanced hepatocellular carcinoma (SILIUS): A randomised, open label, phase 3 trial. Lancet Gastroenterol. Hepatol. 2018, 3, 424–432. [Google Scholar] [CrossRef]
- Hamaya, S.; Fujihara, S.; Iwama, H.; Fujita, K.; Shi, T.; Nakabayashi, R.; Mizuo, T.; Takuma, K.; Nakahara, M.; Oura, K.; et al. Characterization of Cisplatin Effects in Lenvatinib-resistant Hepatocellular Carcinoma Cells. Anticancer Res. 2022, 42, 1263–1275. [Google Scholar] [CrossRef]
- Oura, K.; Takuma, K.; Nakahara, M.; Tadokoro, T.; Fujita, K.; Mimura, S.; Tani, J.; Morishita, A.; Kobara, H.; Masaki, T. Multimodal treatment involving molecular targeted agents and on-demand transcatheter arterial chemoembolization for advanced hepatocellular carcinoma: A case report and review of the literature. Mol. Clin. Oncol. 2021, 15, 154. [Google Scholar] [CrossRef]
- Baterdene, O.; Miura, K.; Ueno, W.; Watanabe, S.; Tsukui, M.; Nomoto, H.; Goka, R.; Maeda, H.; Yamamoto, H.; Morimoto, N. A successful case of transarterial chemoembolization for hyperprogressive disease induced by immunotherapy in a patient with unresectable hepatocellular carcinoma. Clin. J. Gastroenterol. 2022, 15, 1101–1107. [Google Scholar] [CrossRef]
- Hasegawa, H.; Kawakubo, E.; Kitagawa, D.; Kishihara, F.; Funahashi, S.; Kitamura, M. Combined Modality Therapy for Giant Hepatocellular Carcinoma and Multiple Lung Metastases-A Case Study. Gan Kagaku Ryoho 2020, 47, 2251–2253. [Google Scholar]
- Montasser, A.; Beaufrere, A.; Cauchy, F.; Bouattour, M.; Soubrane, O.; Albuquerque, M.; Paradis, V. Transarterial chemoembolisation enhances programmed death-1 and programmed death-ligand 1 expression in hepatocellular carcinoma. Histopathology 2021, 79, 36–46. [Google Scholar] [CrossRef]
- Sun, L.; Xu, X.; Meng, F.; Liu, Q.; Wang, H.; Li, X.; Li, G.; Chen, F. Lenvatinib plus transarterial chemoembolization with or without immune checkpoint inhibitors for unresectable hepatocellular carcinoma: A review. Front. Oncol. 2022, 12, 980214. [Google Scholar] [CrossRef]
- Fournel, L.; Wu, Z.; Stadler, N.; Damotte, D.; Lococo, F.; Boulle, G.; Segal-Bendirdjian, E.; Bobbio, A.; Icard, P.; Tredaniel, J.; et al. Cisplatin increases PD-L1 expression and optimizes immune check-point blockade in non-small cell lung cancer. Cancer Lett. 2019, 464, 5–14. [Google Scholar] [CrossRef]
- Tran, L.; Allen, C.T.; Xiao, R.; Moore, E.; Davis, R.; Park, S.J.; Spielbauer, K.; Van Waes, C.; Schmitt, N.C. Cisplatin Alters Antitumor Immunity and Synergizes with PD-1/PD-L1 Inhibition in Head and Neck Squamous Cell Carcinoma. Cancer Immunol. Res. 2017, 5, 1141–1151. [Google Scholar] [CrossRef]
- Li, S.; Ji, J.; Zhang, Z.; Peng, Q.; Hao, L.; Guo, Y.; Zhou, W.; Cui, Q.; Shi, X. Cisplatin promotes the expression level of PD-L1 in the microenvironment of hepatocellular carcinoma through YAP1. Mol. Cell Biochem. 2020, 475, 79–91. [Google Scholar] [CrossRef]
- Zhang, Z.S.; Yang, R.H.; Yao, X.; Cheng, Y.Y.; Shi, H.X.; Yao, C.Y.; Gao, Z.X.; Qi, D.F.; Zhang, W.K.; Dou, Y.Y.; et al. HGF/c-MET pathway contributes to cisplatin-mediated PD-L1 expression in hepatocellular carcinoma. Cell Biol. Int. 2021, 45, 2521–2533. [Google Scholar] [CrossRef]
- Li, W.; Pei, Y.; Wang, Z.; Liu, J. Efficacy of transarterial chemoembolization monotherapy or combination conversion therapy in unresectable hepatocellular carcinoma: A systematic review and meta-analysis. Front. Oncol. 2022, 12, 930868. [Google Scholar] [CrossRef]
- Kaibori, M.; Matsushima, H.; Ishizaki, M.; Kosaka, H.; Matsui, K.; Nakatani, M.; Kariya, S.; Yamaguchi, T.; Yoshida, K.; Yoshii, K.; et al. The Impact of Sorafenib in Combination with Intermittent Hepatic Arterial Infusion Chemotherapy for Unresectable Hepatocellular Carcinoma with Major Vascular Invasion. Cancer Investig. 2022, 40, 81–89. [Google Scholar] [CrossRef]
- Kondo, M.; Morimoto, M.; Kobayashi, S.; Ohkawa, S.; Hidaka, H.; Nakazawa, T.; Aikata, H.; Hatanaka, T.; Takizawa, D.; Matsunaga, K.; et al. Randomized, phase II trial of sequential hepatic arterial infusion chemotherapy and sorafenib versus sorafenib alone as initial therapy for advanced hepatocellular carcinoma: SCOOP-2 trial. BMC Cancer 2019, 19, 954. [Google Scholar] [CrossRef]
- Choi, J.H.; Chung, W.J.; Bae, S.H.; Song, D.S.; Song, M.J.; Kim, Y.S.; Yim, H.J.; Jung, Y.K.; Suh, S.J.; Park, J.Y.; et al. Randomized, prospective, comparative study on the effects and safety of sorafenib vs. hepatic arterial infusion chemotherapy in patients with advanced hepatocellular carcinoma with portal vein tumor thrombosis. Cancer Chemother. Pharm. 2018, 82, 469–478. [Google Scholar] [CrossRef]
- Hatooka, M.; Kawaoka, T.; Aikata, H.; Inagaki, Y.; Morio, K.; Nakahara, T.; Murakami, E.; Tsuge, M.; Hiramatsu, A.; Imamura, M.; et al. Hepatic arterial infusion chemotherapy followed by sorafenib in patients with advanced hepatocellular carcinoma (HICS 55): An open label, non-comparative, phase II trial. BMC Cancer 2018, 18, 633. [Google Scholar] [CrossRef]
- Ishizaki, M.; Kaibori, M.; Matsui, K.; Ikeda, H.; Yoshida, K.; Okazaki, K.; Kariya, S.; Tanigawa, N.; Nakatake, R.; Matsushima, H.; et al. Phase I Study of Sorafenib in Combination with Intermittent Hepatic Arterial Infusion Chemotherapy for Unresectable Hepatocellular Carcinoma. Cancer Investig. 2017, 35, 271–276. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, M.; Shimizu, S.; Sato, T.; Morimoto, M.; Kojima, Y.; Inaba, Y.; Hagihara, A.; Kudo, M.; Nakamori, S.; Kaneko, S.; et al. Sorafenib plus hepatic arterial infusion chemotherapy with cisplatin versus sorafenib for advanced hepatocellular carcinoma: Randomized phase II trial. Ann. Oncol. 2016, 27, 2090–2096. [Google Scholar] [CrossRef] [PubMed]
- Dinh, V.Y.; Bhatia, S.; Narayanan, G.; Yrizarry, J.; Savaraj, N.; O’Brien, C.; Martin, P.; Feun, L. Pilot Study of Intrahepatic Artery Chemotherapy in Combination with Sorafenib in Hepatocellular Carcinoma. Anticancer Res. 2016, 36, 3555–3563. [Google Scholar] [PubMed]
- Hagihara, A.; Ikeda, M.; Ueno, H.; Morizane, C.; Kondo, S.; Nakachi, K.; Mitsunaga, S.; Shimizu, S.; Kojima, Y.; Suzuki, E.; et al. Phase I study of combination chemotherapy using sorafenib and transcatheter arterial infusion with cisplatin for advanced hepatocellular carcinoma. Cancer Sci. 2014, 105, 354–358. [Google Scholar] [CrossRef] [PubMed]
- Schmid, I.; Haberle, B.; Albert, M.H.; Corbacioglu, S.; Frohlich, B.; Graf, N.; Kammer, B.; Kontny, U.; Leuschner, I.; Scheel-Walter, H.G.; et al. Sorafenib and cisplatin/doxorubicin (PLADO) in pediatric hepatocellular carcinoma. Pediatr. Blood Cancer 2012, 58, 539–544. [Google Scholar] [CrossRef]
- Zhang, L.; Gong, J.F.; Pan, H.M.; Bai, Y.X.; Liu, T.S.; Cheng, Y.; Chen, Y.C.; Huang, J.Y.; Xu, T.T.; Ge, F.J.; et al. Atezolizumab therapy in Chinese patients with locally advanced or metastatic solid tumors: An open-label, phase Ⅰ study. Beijing Da Xue Xue Bao Yi Xue Ban 2022, 54, 971–980. [Google Scholar]
- Roth, G.S.; Neuzillet, C.; Sarabi, M.; Edeline, J.; Malka, D.; Lievre, A. Cholangiocarcinoma: What are the options in all comers and how has the advent of molecular profiling opened the way to personalised medicine ? Eur. J. Cancer 2023, 179, 1–14. [Google Scholar] [CrossRef]
- Vithayathil, M.; Khan, S.A. Current epidemiology of cholangiocarcinoma in Western countries. J. Hepatol. 2022, 77, 1690–1698. [Google Scholar] [CrossRef]
- Khan, S.A.; Thomas, H.C.; Davidson, B.R.; Taylor-Robinson, S.D. Cholangiocarcinoma. Lancet 2005, 366, 1303–1314. [Google Scholar] [CrossRef]
- Mosconi, S.; Beretta, G.D.; Labianca, R.; Zampino, M.G.; Gatta, G.; Heinemann, V. Cholangiocarcinoma. Crit. Rev. Oncol. Hematol. 2009, 69, 259–270. [Google Scholar] [CrossRef]
- Valle, J.W.; Wasan, H.; Johnson, P.; Jones, E.; Dixon, L.; Swindell, R.; Baka, S.; Maraveyas, A.; Corrie, P.; Falk, S.; et al. Gemcitabine alone or in combination with cisplatin in patients with advanced or metastatic cholangiocarcinomas or other biliary tract tumours: A multicentre randomised phase II study—The UK ABC-01 Study. Br. J. Cancer 2009, 101, 621–627. [Google Scholar] [CrossRef]
- Morizane, C.; Okusaka, T.; Mizusawa, J.; Takashima, A.; Ueno, M.; Ikeda, M.; Hamamoto, Y.; Ishii, H.; Boku, N.; Furuse, J. Randomized phase II study of gemcitabine plus S-1 versus S-1 in advanced biliary tract cancer: A Japan Clinical Oncology Group trial (JCOG 0805). Cancer Sci. 2013, 104, 1211–1216. [Google Scholar] [CrossRef]
- Okusaka, T.; Nakachi, K.; Fukutomi, A.; Mizuno, N.; Ohkawa, S.; Funakoshi, A.; Nagino, M.; Kondo, S.; Nagaoka, S.; Funai, J.; et al. Gemcitabine alone or in combination with cisplatin in patients with biliary tract cancer: A comparative multicentre study in Japan. Br. J. Cancer 2010, 103, 469–474. [Google Scholar] [CrossRef]
- Kanai, M.; Hatano, E.; Kobayashi, S.; Fujiwara, Y.; Marubashi, S.; Miyamoto, A.; Shiomi, H.; Kubo, S.; Ikuta, S.; Yanagimoto, H.; et al. A multi-institution phase II study of gemcitabine/cisplatin/S-1 (GCS) combination chemotherapy for patients with advanced biliary tract cancer (KHBO 1002). Cancer Chemother. Pharm. 2015, 75, 293–300. [Google Scholar] [CrossRef]
- Sakai, D.M.; Zornow, K.A.; Campoy, L.; Cable, C.; Appel, L.D.; Putnam, H.J.; Martin-Flores, M. Intravenous rocuronium 0.3 mg/kg improves the conditions for tracheal intubation in cats: A randomized, placebo-controlled trial. J. Feline Med. Surg. 2018, 20, 1124–1129. [Google Scholar] [CrossRef]
- Banales, J.M.; Marin, J.J.G.; Lamarca, A.; Rodrigues, P.M.; Khan, S.A.; Roberts, L.R.; Cardinale, V.; Carpino, G.; Andersen, J.B.; Braconi, C.; et al. Cholangiocarcinoma 2020: The next horizon in mechanisms and management. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 557–588. [Google Scholar] [CrossRef]
- Valle, J.W.; Wasan, H.; Lopes, A.; Backen, A.C.; Palmer, D.H.; Morris, K.; Duggan, M.; Cunningham, D.; Anthoney, D.A.; Corrie, P.; et al. Cediranib or placebo in combination with cisplatin and gemcitabine chemotherapy for patients with advanced biliary tract cancer (ABC-03): A randomised phase 2 trial. Lancet Oncol. 2015, 16, 967–978. [Google Scholar] [CrossRef]
- Oh, D.Y.; Lee, K.H.; Lee, D.W.; Yoon, J.; Kim, T.Y.; Bang, J.H.; Nam, A.R.; Oh, K.S.; Kim, J.M.; Lee, Y.; et al. Gemcitabine and cisplatin plus durvalumab with or without tremelimumab in chemotherapy-naive patients with advanced biliary tract cancer: An open-label, single-centre, phase 2 study. Lancet Gastroenterol. Hepatol. 2022, 7, 522–532. [Google Scholar] [CrossRef]
- Zheng, H.C. The molecular mechanisms of chemoresistance in cancers. Oncotarget 2017, 8, 59950–59964. [Google Scholar] [CrossRef]
- Kiss, R.C.; Xia, F.; Acklin, S. Targeting DNA Damage Response and Repair to Enhance Therapeutic Index in Cisplatin-Based Cancer Treatment. Int. J. Mol. Sci. 2021, 22, 8199. [Google Scholar] [CrossRef]
- Kawaguchi, H.; Terai, Y.; Tanabe, A.; Sasaki, H.; Takai, M.; Fujiwara, S.; Ashihara, K.; Tanaka, Y.; Tanaka, T.; Tsunetoh, S.; et al. Gemcitabine as a molecular targeting agent that blocks the Akt cascade in platinum-resistant ovarian cancer. J. Ovarian Res. 2014, 7, 38. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Nie, Y.H.; Cai, M.B.; Li, Z.M.; Zhu, H.B.; Tan, Y.R. Gemcitabine Combined with Cisplatin Has a Better Effect in the Treatment of Recurrent/Metastatic Advanced Nasopharyngeal Carcinoma. Drug Des. Devel Ther. 2022, 16, 1191–1198. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, L.; Hu, G.Q.; Zhang, N.; Zhu, X.D.; Yang, K.Y.; Jin, F.; Shi, M.; Chen, Y.P.; Hu, W.H.; et al. Gemcitabine and Cisplatin Induction Chemotherapy in Nasopharyngeal Carcinoma. N. Engl. J. Med. 2019, 381, 1124–1135. [Google Scholar] [CrossRef] [PubMed]
- Ding, D.Y.; Gan, X.J.; Zhang, J.N.; Hou, G.J.; Tao, Q.F.; Sun, D.P.; Li, W.; Yang, Y.; Ding, W.B.; Yu, J.; et al. Serum thrombospondin-1 serves as a novel biomarker and agonist of gemcitabine-based chemotherapy in intrahepatic cholangiocarcinoma. Br. J. Cancer 2023, 128, 907–917. [Google Scholar] [CrossRef]
- Prasopporn, S.; Suppramote, O.; Ponvilawan, B.; Jamyuang, C.; Chanthercrob, J.; Chaiboonchoe, A.; More-Krong, P.; Kongsri, K.; Suntiparpluacha, M.; Chanwat, R.; et al. Combining the SMAC mimetic LCL161 with Gemcitabine plus Cisplatin therapy inhibits and prevents the emergence of multidrug resistance in cholangiocarcinoma. Front. Oncol. 2022, 12, 1021632. [Google Scholar] [CrossRef]
- Huang, C.S.; Zhu, Y.Q.; Xu, Q.C.; Chen, S.; Huang, Y.; Zhao, G.; Ni, X.; Liu, B.; Zhao, W.; Yin, X.Y. YTHDF2 promotes intrahepatic cholangiocarcinoma progression and desensitises cisplatin treatment by increasing CDKN1B mRNA degradation. Clin. Transl. Med. 2022, 12, e848. [Google Scholar] [CrossRef]
- Schmid, I.; von Schweinitz, D. Pediatric hepatocellular carcinoma: Challenges and solutions. J. Hepatocell. Carcinoma 2017, 4, 15–21. [Google Scholar] [CrossRef]
- Hager, J.; Sergi, C.M. Hepatoblastoma. In Liver Cancer; Sergi, C.M., Ed.; Exon Publications: Brisbane, Australia, 2021. [Google Scholar]
- Turcotte, L.M.; Georgieff, M.K.; Ross, J.A.; Feusner, J.H.; Tomlinson, G.E.; Malogolowkin, M.H.; Krailo, M.D.; Miller, N.; Fonstad, R.; Spector, L.G. Neonatal medical exposures and characteristics of low birth weight hepatoblastoma cases: A report from the Children’s Oncology Group. Pediatr. Blood Cancer 2014, 61, 2018–2023. [Google Scholar] [CrossRef]
- Digiacomo, G.; Serra, R.P.; Turrini, E.; Tiri, A.; Cavazzoni, A.; Alfieri, R.; Bertolini, P. State of the art and perspectives in pediatric hepatocellular carcinoma. Biochem. Pharm. 2023, 207, 115373. [Google Scholar] [CrossRef]
- Khanna, R.; Verma, S.K. Pediatric hepatocellular carcinoma. World J. Gastroenterol. 2018, 24, 3980–3999. [Google Scholar] [CrossRef]
- Chan, K.L.; Fan, S.T.; Tam, P.K.; Chiang, A.K.; Chan, G.C.; Ha, S.Y. Paediatric hepatoblastoma and hepatocellular carcinoma: Retrospective study. Hong Kong Med. J. 2002, 8, 13–17. [Google Scholar]
- Pritchard, J.; Brown, J.; Shafford, E.; Perilongo, G.; Brock, P.; Dicks-Mireaux, C.; Keeling, J.; Phillips, A.; Vos, A.; Plaschkes, J. Cisplatin, doxorubicin, and delayed surgery for childhood hepatoblastoma: A successful approach--results of the first prospective study of the International Society of Pediatric Oncology. J. Clin. Oncol. 2000, 18, 3819–3828. [Google Scholar] [CrossRef]
- Czauderna, P.; Mackinlay, G.; Perilongo, G.; Brown, J.; Shafford, E.; Aronson, D.; Pritchard, J.; Chapchap, P.; Keeling, J.; Plaschkes, J.; et al. Hepatocellular carcinoma in children: Results of the first prospective study of the International Society of Pediatric Oncology group. J. Clin. Oncol. 2002, 20, 2798–2804. [Google Scholar] [CrossRef]
- Czauderna, P. Adult type vs. Childhood hepatocellular carcinoma--are they the same or different lesions? Biology, natural history, prognosis, and treatment. Med. Pediatr. Oncol. 2002, 39, 519–523. [Google Scholar] [CrossRef]
- Perilongo, G.; Shafford, E.; Maibach, R.; Aronson, D.; Brugieres, L.; Brock, P.; Childs, M.; Czauderna, P.; MacKinlay, G.; Otte, J.B.; et al. Risk-adapted treatment for childhood hepatoblastoma. final report of the second study of the International Society of Paediatric Oncology--SIOPEL 2. Eur. J. Cancer 2004, 40, 411–421. [Google Scholar] [CrossRef]
- Czauderna, P. Hepatoblastoma throughout SIOPEL trials—Clinical lessons learnt. Front. Biosci. (Elite Ed) 2012, 4, 470–479. [Google Scholar] [CrossRef]
- Murawski, M.; Weeda, V.B.; Maibach, R.; Morland, B.; Roebuck, D.J.; Zimmerman, A.; Casanova, M.; Perilongo, G.; Laithier, V.; Kebudi, R.; et al. Hepatocellular Carcinoma in Children: Does Modified Platinum- and Doxorubicin-Based Chemotherapy Increase Tumor Resectability and Change Outcome? Lessons Learned From the SIOPEL 2 and 3 Studies. J. Clin. Oncol. 2016, 34, 1050–1056. [Google Scholar] [CrossRef]
- Czauderna, P.; Haeberle, B.; Hiyama, E.; Rangaswami, A.; Krailo, M.; Maibach, R.; Rinaldi, E.; Feng, Y.; Aronson, D.; Malogolowkin, M.; et al. The Children’s Hepatic tumors International Collaboration (CHIC): Novel global rare tumor database yields new prognostic factors in hepatoblastoma and becomes a research model. Eur. J. Cancer 2016, 52, 92–101. [Google Scholar] [CrossRef]
- Meyers, R.L.; Maibach, R.; Hiyama, E.; Haberle, B.; Krailo, M.; Rangaswami, A.; Aronson, D.C.; Malogolowkin, M.H.; Perilongo, G.; von Schweinitz, D.; et al. Risk-stratified staging in paediatric hepatoblastoma: A unified analysis from the Children’s Hepatic tumors International Collaboration. Lancet Oncol. 2017, 18, 122–131. [Google Scholar] [CrossRef]
- Knight, K.R.; Kraemer, D.F.; Neuwelt, E.A. Ototoxicity in children receiving platinum chemotherapy: Underestimating a commonly occurring toxicity that may influence academic and social development. J. Clin. Oncol. 2005, 23, 8588–8596. [Google Scholar] [CrossRef]
- Bass, J.K.; Knight, K.R.; Yock, T.I.; Chang, K.W.; Cipkala, D.; Grewal, S.S. Evaluation and Management of Hearing Loss in Survivors of Childhood and Adolescent Cancers: A Report From the Children’s Oncology Group. Pediatr. Blood Cancer 2016, 63, 1152–1162. [Google Scholar] [CrossRef] [PubMed]
- Romano, A.; Capozza, M.A.; Mastrangelo, S.; Maurizi, P.; Triarico, S.; Rolesi, R.; Attina, G.; Fetoni, A.R.; Ruggiero, A. Assessment and Management of Platinum-Related Ototoxicity in Children Treated for Cancer. Cancers 2020, 12, 1266. [Google Scholar] [CrossRef] [PubMed]
- Grewal, S.; Merchant, T.; Reymond, R.; McInerney, M.; Hodge, C.; Shearer, P. Auditory late effects of childhood cancer therapy: A report from the Children’s Oncology Group. Pediatrics 2010, 125, e938–e950. [Google Scholar] [CrossRef] [PubMed]
- Clemens, E.; van den Heuvel-Eibrink, M.M.; Mulder, R.L.; Kremer, L.C.M.; Hudson, M.M.; Skinner, R.; Constine, L.S.; Bass, J.K.; Kuehni, C.E.; Langer, T.; et al. Recommendations for ototoxicity surveillance for childhood, adolescent, and young adult cancer survivors: A report from the International Late Effects of Childhood Cancer Guideline Harmonization Group in collaboration with the PanCare Consortium. Lancet Oncol. 2019, 20, e29–e41. [Google Scholar] [CrossRef]
- Brock, P.R.; Maibach, R.; Neuwelt, E.A. Sodium Thiosulfate and Cisplatin-Induced Hearing Loss. N. Engl. J. Med. 2018, 379, 1181. [Google Scholar] [CrossRef]
- Freyer, D.R.; Brock, P.R.; Chang, K.W.; Dupuis, L.L.; Epelman, S.; Knight, K.; Mills, D.; Phillips, R.; Potter, E.; Risby, D.; et al. Prevention of cisplatin-induced ototoxicity in children and adolescents with cancer: A clinical practice guideline. Lancet Child Adolesc. Health 2020, 4, 141–150. [Google Scholar] [CrossRef]
- Neuwelt, E.A.; Gilmer-Knight, K.; Lacy, C.; Nicholson, H.S.; Kraemer, D.F.; Doolittle, N.D.; Hornig, G.W.; Muldoon, L.L. Toxicity profile of delayed high dose sodium thiosulfate in children treated with carboplatin in conjunction with blood-brain-barrier disruption. Pediatr. Blood Cancer 2006, 47, 174–182. [Google Scholar] [CrossRef]
- Fouladi, M.; Chintagumpala, M.; Ashley, D.; Kellie, S.; Gururangan, S.; Hassall, T.; Gronewold, L.; Stewart, C.F.; Wallace, D.; Broniscer, A.; et al. Amifostine protects against cisplatin-induced ototoxicity in children with average-risk medulloblastoma. J. Clin. Oncol. 2008, 26, 3749–3755. [Google Scholar] [CrossRef]
- Campbell, K.C.; Meech, R.P.; Klemens, J.J.; Gerberi, M.T.; Dyrstad, S.S.; Larsen, D.L.; Mitchell, D.L.; El-Azizi, M.; Verhulst, S.J.; Hughes, L.F. Prevention of noise- and drug-induced hearing loss with D-methionine. Hear. Res. 2007, 226, 92–103. [Google Scholar] [CrossRef]


| Molecular targeted agents | ||
| Sorafenib | HCC with portal vein invasion | [178] |
| Advanced HCC | [179] | |
| HCC with portal vein tumor thrombosis | [180] | |
| Advanced HCC | [181] | |
| Advanced HCC | [166] | |
| Unresectable HCC | [182] | |
| Advanced HCC | [183] | |
| Unresectable HCC | [184] | |
| Advanced HCC | [185] | |
| Pediatric HCC | [186] | |
| Immune checkpoint inhibitors | ||
| Atezolizumab | Advanced HCC | [187] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hamaya, S.; Oura, K.; Morishita, A.; Masaki, T. Cisplatin in Liver Cancer Therapy. Int. J. Mol. Sci. 2023, 24, 10858. https://doi.org/10.3390/ijms241310858
Hamaya S, Oura K, Morishita A, Masaki T. Cisplatin in Liver Cancer Therapy. International Journal of Molecular Sciences. 2023; 24(13):10858. https://doi.org/10.3390/ijms241310858
Chicago/Turabian StyleHamaya, Sae, Kyoko Oura, Asahiro Morishita, and Tsutomu Masaki. 2023. "Cisplatin in Liver Cancer Therapy" International Journal of Molecular Sciences 24, no. 13: 10858. https://doi.org/10.3390/ijms241310858
APA StyleHamaya, S., Oura, K., Morishita, A., & Masaki, T. (2023). Cisplatin in Liver Cancer Therapy. International Journal of Molecular Sciences, 24(13), 10858. https://doi.org/10.3390/ijms241310858