
Talk to Me—Interplay between Mitochondria and Microbiota in Aging



Abstract
1. Introduction
1.1. Common Evolutionary History of Mitochondria and Bacteria
1.2. Mitochondria in the Aging Organism
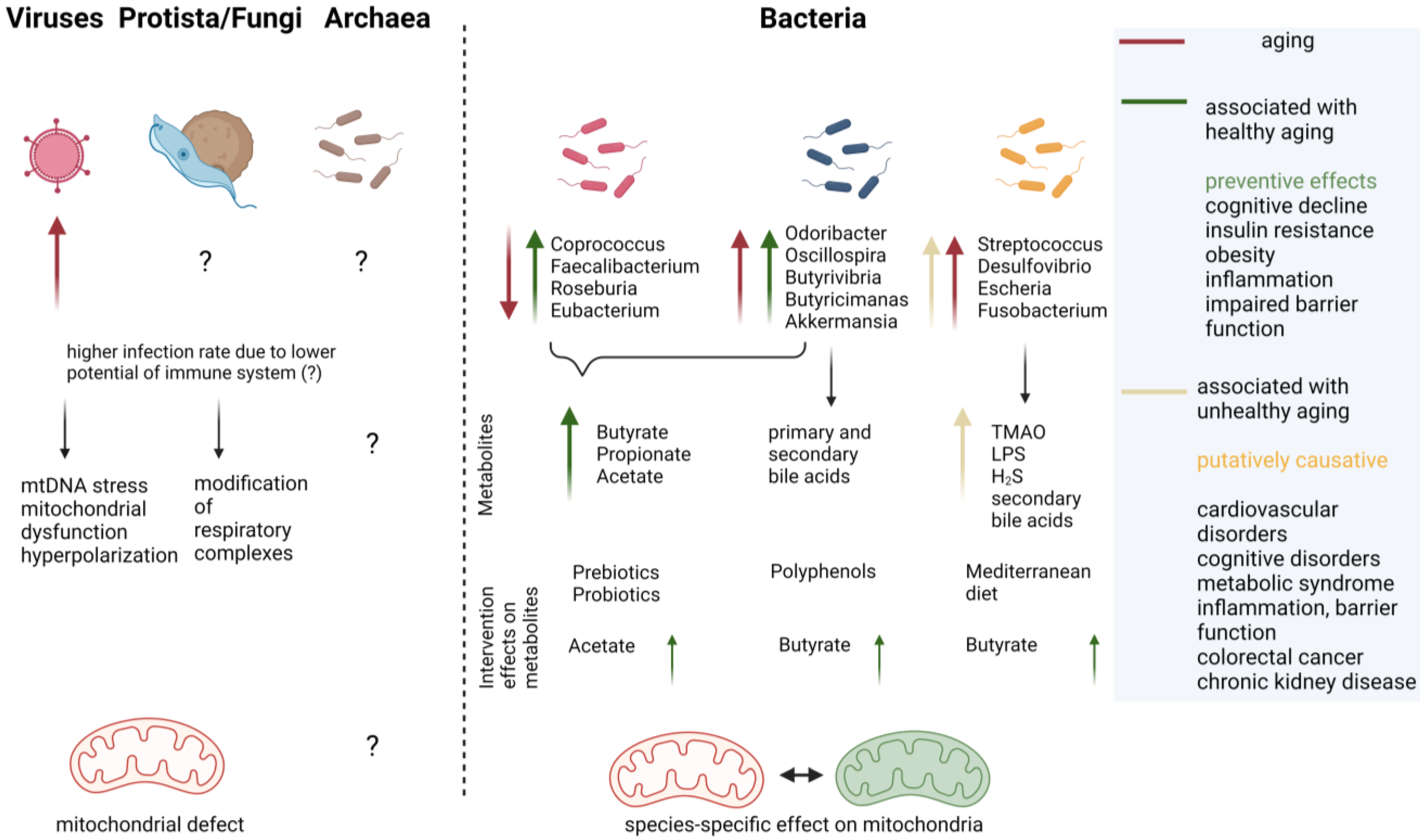
1.3. Microbiota and Aging of the Host
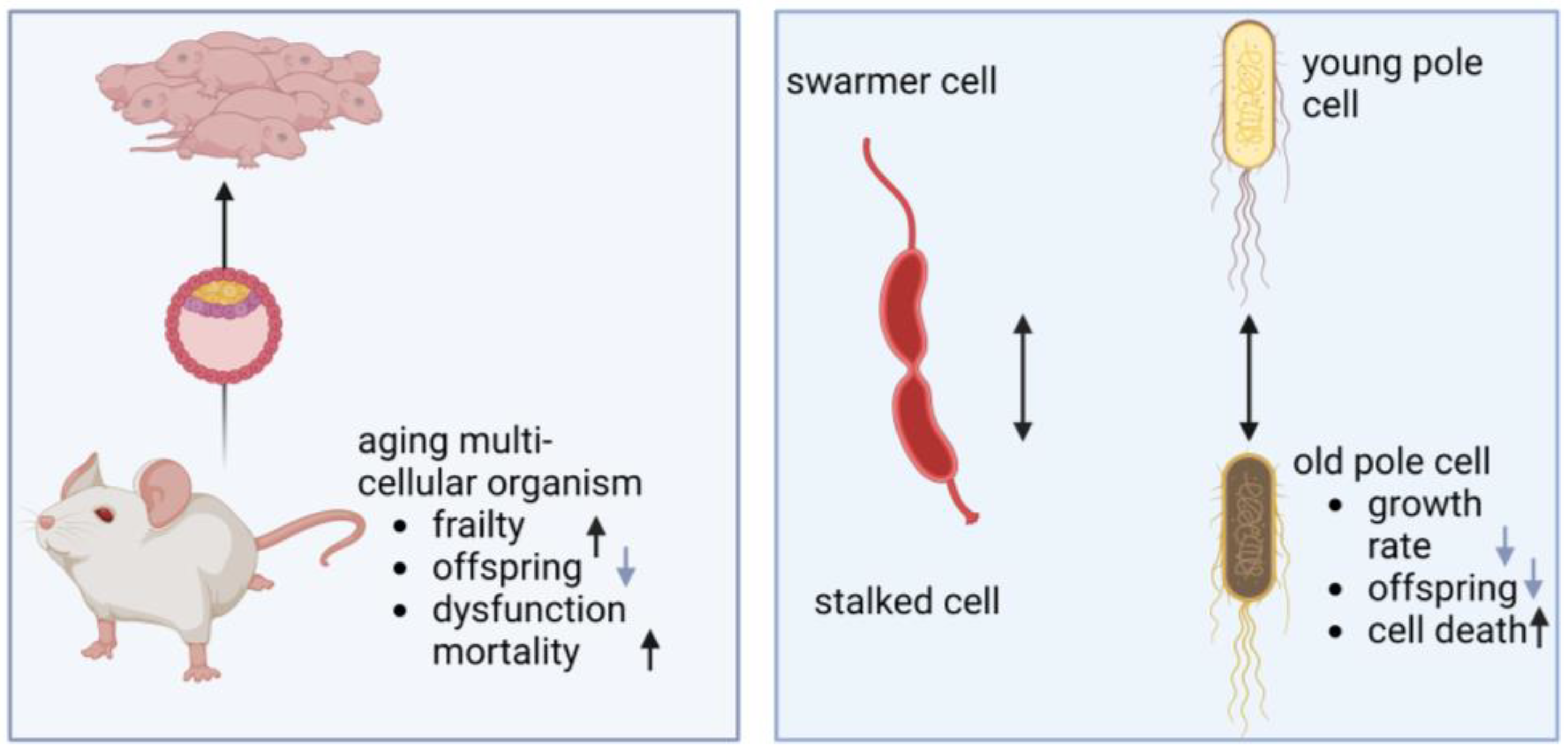
2. Aging of Bacteria and Mitochondria

{kind=link}
{kind=link}
{kind=link}
| Hallmark | Investigation Tools | Organism | Observation | Ref |
|---|---|---|---|---|
| Genomic instability | fYFP-labeled MutL mismatch repair proteins | E. coli | Replication errors occur at a constant rate across ages. | [88] |
| Growth arrest, DNA repair mutants | E. coli | Oxidative damage to DNA does not limit survival during stasis. | [89] | |
| Loss of Proteostasis | Coomassie Blue-stained PAA gels | E. coli | Aggregates accumulate concomitantly with cell death. Dead cells contain increased amounts of aggregates. Aggregation occurs before cell death. | [90] |
| Fluorescently tagged chaperone (IbpA) | E. coli | Aggregates migrate to the “old pole” cell. | [91,92,93] | |
| Fluorescently tagged chaperone (IbpA) | E. coli | Ejection of minicells with aggregates promotes stress resistancy of cells. | [94] | |
| Growth arrest | E. coli | Accelerated protein oxidation | [89] |
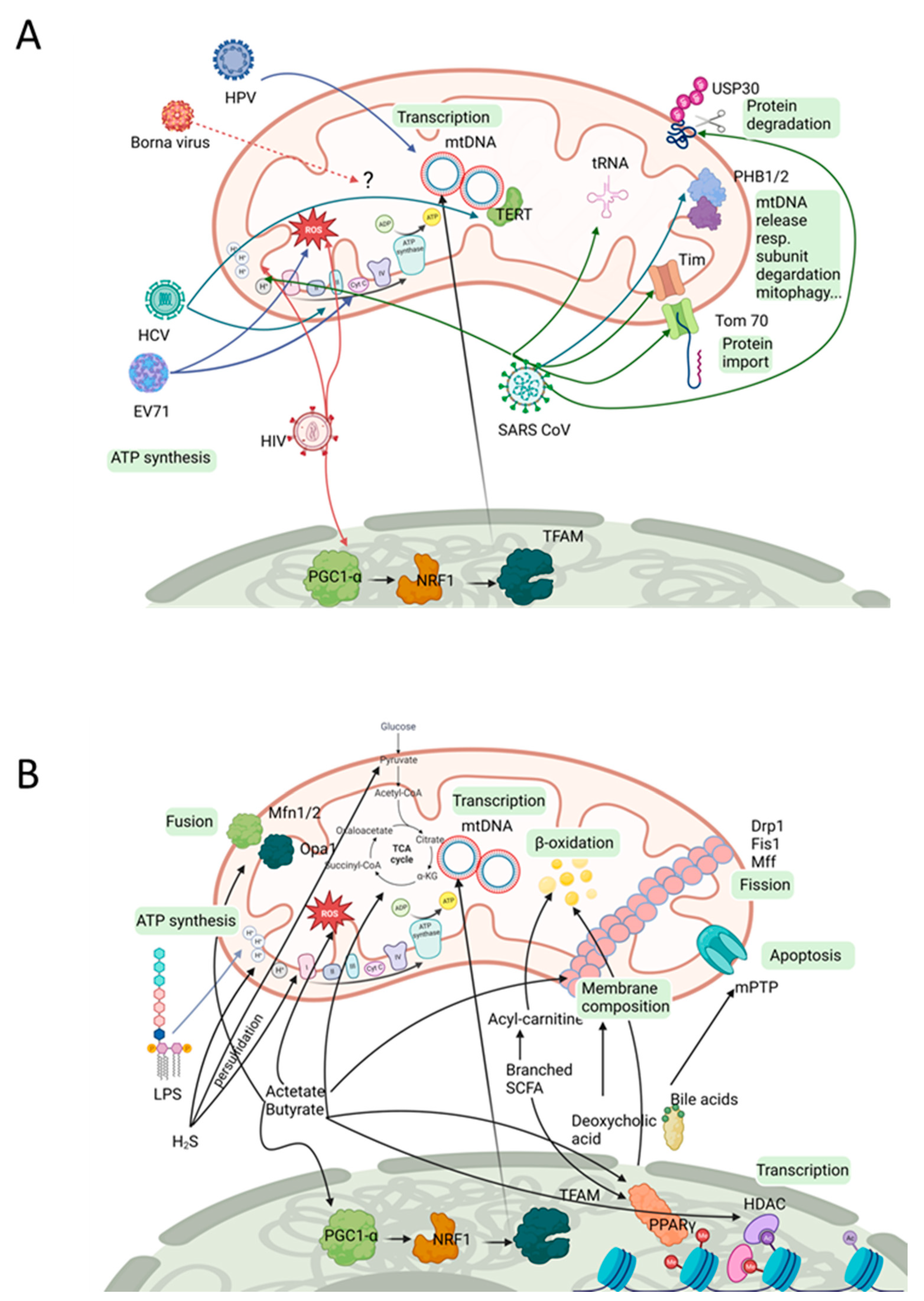
3. Viral Impact on Mitochondria
4. Eukaryotic Microbial Organisms with Mitochondrial Effectors
5. Bacterial Compounds Affecting Mitochondria
6. Deducing Therapeutic Interventions from Current Knowledge
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Anderson, S.; Bankier, A.T.; Barrell, B.G.; De Bruijn, M.H.L.; Coulson, A.R.; Drouin, J.; Eperon, I.C.; Nierlich, D.P.; Roe, B.A.; Sanger, F.; et al. Sequence and Organization of the Human Mitochondrial Genome. Nature 1981, 290, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Phan, H.T.L.; Lee, H.; Kim, K. Trends and Prospects in Mitochondrial Genome Editing. Exp. Mol. Med. 2023, 55, 871–878. [Google Scholar] [CrossRef] [PubMed]
- Quirós, P.M.; Mottis, A.; Auwerx, J. Mitonuclear Communication in Homeostasis and Stress. Nat. Rev. Mol. Cell Biol. 2016, 17, 213–226. [Google Scholar] [CrossRef] [PubMed]
- Westermann, B. Mitochondrial Fusion and Fission in Cell Life and Death. Nat. Rev. Mol. Cell Biol. 2010, 11, 872–884. [Google Scholar] [CrossRef]
- Friedman, J.R.; Nunnari, J. Mitochondrial Form and Function. Nature 2014, 505, 335–343. [Google Scholar] [CrossRef]
- Chan, D.C. Fusion and Fission: Interlinked Processes Critical for Mitochondrial Health. Annu. Rev. Genet. 2012, 46, 265–287. [Google Scholar] [CrossRef]
- Yapa, N.M.B.; Lisnyak, V.; Reljic, B.; Ryan, M.T. Mitochondrial Dynamics in Health and Disease. FEBS Lett. 2021, 595, 1184–1204. [Google Scholar] [CrossRef]
- Eisner, V.; Picard, M.; Hajnóczky, G. Mitochondrial Dynamics in Adaptive and Maladaptive Cellular Stress Responses. Nat. Cell Biol. 2018, 20, 755–765. [Google Scholar] [CrossRef]
- Pfeiffer, A.; Jaeckel, M.; Lewerenz, J.; Noack, R.; Pouya, A.; Schacht, T.; Hoffmann, C.; Winter, J.; Schweiger, S.; Schäfer, M.K.E.; et al. Mitochondrial Function and Energy Metabolism in Neuronal HT22 Cells Resistant to Oxidative Stress. Br. J. Pharmacol. 2014, 171, 2147–2158. [Google Scholar] [CrossRef]
- Favaro, G.; Romanello, V.; Varanita, T.; Desbats, M.A.; Morbidoni, V.; Tezze, C.; Albiero, M.; Canato, M.; Gherardi, G.; De Stefani, D.; et al. DRP1-Mediated Mitochondrial Shape Controls Calcium Homeostasis and Muscle Mass. Nat. Commun. 2019, 10, 2576. [Google Scholar] [CrossRef]
- Giacomello, M.; Pyakurel, A.; Glytsou, C.; Scorrano, L. The Cell Biology of Mitochondrial Membrane Dynamics. Nat. Rev. Mol. Cell Biol. 2020, 21, 204–224. [Google Scholar] [CrossRef]
- Gregory, A.C.; Zablocki, O.; Zayed, A.A.; Howell, A.; Bolduc, B.; Sullivan, M.B. The Gut Virome Database Reveals Age-Dependent Patterns of Virome Diversity in the Human Gut. Cell Host Microbe 2020, 28, 724–740. [Google Scholar] [CrossRef]
- Panel, M.; Ghaleh, B.; Morin, D. Mitochondria and Aging: A Role for the Mitochondrial Transition Pore? Aging Cell 2018, 17, e12793. [Google Scholar] [CrossRef]
- Janikiewicz, J.; Szymański, J.; Malinska, D.; Patalas-Krawczyk, P.; Michalska, B.; Duszyński, J.; Giorgi, C.; Bonora, M.; Dobrzyn, A.; Wieckowski, M.R. Mitochondria-Associated Membranes in Aging and Senescence: Structure, Function, and Dynamics. Cell Death Dis. 2018, 9, 332. [Google Scholar] [CrossRef]
- Giorgi, C.; Marchi, S.; Simoes, I.C.M.; Ren, Z.; Morciano, G.; Perrone, M.; Patalas-Krawczyk, P.; Borchard, S.; Jędrak, P.; Pierzynowska, K.; et al. Mitochondria and Reactive Oxygen Species in Aging and Age-Related Diseases. Int. Rev. Cell Mol. Biol. 2018, 340, 209–344. [Google Scholar]
- Kauppila, T.E.S.; Kauppila, J.H.K.; Larsson, N.G. Mammalian Mitochondria and Aging: An Update. Cell Metab. 2017, 25, 57–71. [Google Scholar] [CrossRef]
- Miwa, S.; Kashyap, S.; Chini, E.; von Zglinicki, T. Mitochondrial Dysfunction in Cell Senescence and Aging. J. Clin. Investig. 2022, 132, e158447. [Google Scholar] [CrossRef]
- Carreras-Sureda, A.; Kroemer, G.; Cardenas, J.C.; Hetz, C. Balancing Energy and Protein Homeostasis at ER-Mitochondria Contact Sites. Sci. Signal. 2022, 15, eabm7524. [Google Scholar] [CrossRef]
- Müller, M.; Ahumada-Castro, U.; Sanhueza, M.; Gonzalez-Billault, C.; Court, F.A.; Cárdenas, C. Mitochondria and Calcium Regulation as Basis of Neurodegeneration Associated with Aging. Front. Neurosci. 2018, 12, 470. [Google Scholar] [CrossRef]
- Lima, T.; Li, T.Y.; Mottis, A.; Auwerx, J. Pleiotropic Effects of Mitochondria in Aging. Nat. Aging 2022, 2, 199–213. [Google Scholar] [CrossRef]
- Torres, A.K.; Jara, C.; Llanquinao, J.; Lira, M.; Cortés-Díaz, D.; Tapia-Rojas, C. Mitochondrial Bioenergetics, Redox Balance, and Calcium Homeostasis Dysfunction with Defective Ultrastructure and Quality Control in the Hippocampus of Aged Female C57BL/6J Mice. Int. J. Mol. Sci. 2023, 24, 5476. [Google Scholar] [CrossRef] [PubMed]
- Grimm, A.; Friedland, K.; Eckert, A. Mitochondrial Dysfunction: The Missing Link between Aging and Sporadic Alzheimer’s Disease. Biogerontology 2016, 17, 281–296. [Google Scholar] [CrossRef] [PubMed]
- Cunnane, S.C.; Trushina, E.; Morland, C.; Prigione, A.; Casadesus, G.; Andrews, Z.B.; Beal, M.F.; Bergersen, L.H.; Brinton, R.D.; de la Monte, S.; et al. Brain Energy Rescue: An Emerging Therapeutic Concept for Neurodegenerative Disorders of Ageing. Nat. Rev. Drug Discov. 2020, 19, 609–633. [Google Scholar] [CrossRef] [PubMed]
- Theurey, P.; Pizzo, P. The Aging Mitochondria. Genes 2018, 9, 22. [Google Scholar] [CrossRef] [PubMed]
- Gemma, C.; Vila, J.; Bachstetter, A.; Bickford, P.C. Oxidative Stress and the Aging Brain: From Theory to Prevention. In Brain Aging; CRC Press: Boca Raton, FL, USA, 2019; pp. 353–374. [Google Scholar]
- Harman, D. Aging: A Theory Based on Free Radical and Radiation Chemistry. J. Gerontol. 1956, 11, 298–300. [Google Scholar] [CrossRef]
- Trushina, E.; Trushin, S.; Hasan, M.F. Mitochondrial Complex I as a Therapeutic Target for Alzheimer’s Disease. Acta Pharm. Sin. B 2022, 12, 483–495. [Google Scholar] [CrossRef]
- Murphy, M.P.; Hartley, R.C. Mitochondria as a Therapeutic Target for Common Pathologies. Nat. Rev. Drug Discov. 2018, 17, 865–886. [Google Scholar] [CrossRef]
- Jörg, M.; Plehn, J.E.; Friedland, K.; Müller, W.E. Mitochondrial Dysfunction as a Causative Factor in Alzheimer’s Disease-Spectrum Disorders: Lymphocytes as a Window to the Brain. Curr. Alzheimer Res. 2021, 18, 733–752. [Google Scholar] [CrossRef]
- Wen, J.J.; Garg, N. Oxidative Modification of Mitochondrial Respiratory Complexes in Response to the Stress of Trypanosoma Cruzi Infection. Free Radic. Biol. Med. 2004, 37, 2072–2081. [Google Scholar] [CrossRef]
- Overmyer, K.A.; Evans, C.R.; Qi, N.R.; Minogue, C.E.; Carson, J.J.; Chermside-Scabbo, C.J.; Koch, L.G.; Britton, S.L.; Pagliarini, D.J.; Coon, J.J.; et al. Maximal Oxidative Capacity during Exercise Is Associated with Skeletal Muscle Fuel Selection and Dynamic Changes in Mitochondrial Protein Acetylation. Cell Metab. 2015, 21, 468–478. [Google Scholar] [CrossRef]
- Short, K.R.; Bigelow, M.L.; Kahl, J.; Singh, R.; Coenen-Schimke, J.; Raghavakaimal, S.; Nair, K.S. Decline in Skeletal Muscle Mitochondrial Function with Aging in Humans. Proc. Natl. Acad. Sci. USA 2005, 102, 5618–5623. [Google Scholar] [CrossRef]
- Lane, R.K.; Hilsabeck, T.; Rea, S.L. The Role of Mitochondrial Dysfunction in Age-Related Diseases. Biochim. Biophys. Acta Bioenerg. 2015, 1847, 1387–1400. [Google Scholar] [CrossRef]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The Hallmarks of Aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef]
- López-Lluch, G.; Irusta, P.M.; Navas, P.; de Cabo, R. Mitochondrial Biogenesis and Healthy Aging. Exp. Gerontol. 2008, 43, 813–819. [Google Scholar] [CrossRef]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. Hallmarks of Aging: An Expanding Universe. Cell 2023, 186, 243–278. [Google Scholar] [CrossRef]
- Beckervordersandforth, R.; Ebert, B.; Schäffner, I.; Moss, J.; Fiebig, C.; Shin, J.; Moore, D.L.; Ghosh, L.; Trinchero, M.F.; Stockburger, C.; et al. Role of Mitochondrial Metabolism in the Control of Early Lineage Progression and Aging Phenotypes in Adult Hippocampal Neurogenesis. Neuron 2017, 93, 560–573.e6. [Google Scholar] [CrossRef]
- Picca, A.; Pesce, V.; Fracasso, F.; Joseph, A.M.; Leeuwenburgh, C.; Lezza, A.M.S. Aging and Calorie Restriction Oppositely Affect Mitochondrial Biogenesis through TFAM Binding at Both Origins of Mitochondrial DNA Replication in Rat Liver. PLoS ONE 2013, 8, e74644. [Google Scholar] [CrossRef]
- Yan, C.; Duanmu, X.; Zeng, L.; Liu, B.; Song, Z. Mitochondrial DNA: Distribution, Mutations, and Elimination. Cells 2019, 8, 379. [Google Scholar] [CrossRef]
- Smith, A.L.M.; Whitehall, J.C.; Greaves, L.C. Mitochondrial DNA Mutations in Ageing and Cancer. Mol. Oncol. 2022, 16, 3276–3294. [Google Scholar] [CrossRef]
- Wei, W.; Keogh, M.J.; Wilson, I.; Coxhead, J.; Ryan, S.; Rollinson, S.; Griffin, H.; Kurzawa-Akanbi, M.; Santibanez-Koref, M.; Talbot, K.; et al. Mitochondrial DNA Point Mutations and Relative Copy Number in 1363 Disease and Control Human Brains. Acta Neuropathol. Commun. 2017, 5, 13. [Google Scholar] [CrossRef]
- Sun, J.; Brown, T.T.; Samuels, D.C.; Hulgan, T.; D’Souza, G.; Jamieson, B.D.; Erlandson, K.M.; Martinson, J.; Palella, F.J.; Margolick, J.B.; et al. The Role of Mitochondrial DNA Variation in Age-Related Decline in Gait Speed among Older Men Living with Human Immunodeficiency Virus. Clin. Infect. Dis. 2018, 67, 778–784. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Slone, J.; Fei, L.; Huang, T. Mitochondrial Dna Variants and Common Diseases: A Mathematical Model for the Diversity of Age-Related Mtdna Mutations. Cells 2019, 8, 608. [Google Scholar] [CrossRef] [PubMed]
- Rossignol, R.; Faustin, B.; Rocher, C.; Malgat, M.; Mazat, J.P.; Letellier, T. Mitochondrial Threshold Effects. Biochem. J. 2003, 370, 751–762. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Smith, H.J.; Yao, P.; Mair, W.B. Causal Roles of Mitochondrial Dynamics in Longevity and Healthy Aging. EMBO Rep. 2019, 20, e48395. [Google Scholar] [CrossRef]
- Chaudhari, S.N.; Kipreos, E.T. Increased Mitochondrial Fusion Allows the Survival of Older Animals in Diverse C. Elegans Longevity Pathways. Nat. Commun. 2017, 8, 182. [Google Scholar] [CrossRef]
- Byrne, J.J.; Soh, M.S.; Chandhok, G.; Vijayaraghavan, T.; Teoh, J.S.; Crawford, S.; Cobham, A.E.; Yapa, N.M.B.; Mirth, C.K.; Neumann, B. Disruption of Mitochondrial Dynamics Affects Behaviour and Lifespan in Caenorhabditis Elegans. Cell. Mol. Life Sci. 2019, 76, 1967–1985. [Google Scholar] [CrossRef]
- Sun, N.; Youle, R.J.; Finkel, T. The Mitochondrial Basis of Aging. Mol. Cell 2016, 61, 654–666. [Google Scholar] [CrossRef]
- Doblado, L.; Lueck, C.; Rey, C.; Samhan-arias, A.K.; Prieto, I.; Stacchiotti, A.; Monsalve, M. Mitophagy in Human Diseases. Int. J. Mol. Sci. 2021, 22, 3903. [Google Scholar] [CrossRef]
- Palikaras, K.; Lionaki, E.; Tavernarakis, N. Mechanisms of Mitophagy in Cellular Homeostasis, Physiology and Pathology. Nat. Cell Biol. 2018, 20, 1013–1022. [Google Scholar] [CrossRef]
- Sun, N.; Yun, J.; Liu, J.; Malide, D.; Liu, C.; Rovira, I.I.; Holmström, K.M.; Fergusson, M.M.; Yoo, Y.H.; Combs, C.A.; et al. Measuring In Vivo Mitophagy. Mol. Cell 2015, 60, 685–696. [Google Scholar] [CrossRef]
- Dutta, D.; Calvani, R.; Bernabei, R.; Leeuwenburgh, C.; Marzetti, E. Contribution of Impaired Mitochondrial Autophagy to Cardiac Aging: Mechanisms and Therapeutic Opportunities. Circ. Res. 2012, 110, 1125–1138. [Google Scholar] [CrossRef]
- Xu, C.; Cao, Y.; Liu, R.; Liu, L.; Zhang, W.; Fang, X.; Jia, S.; Ye, J.; Liu, Y.; Weng, L.; et al. Mitophagy-Regulated Mitochondrial Health Strongly Protects the Heart against Cardiac Dysfunction after Acute Myocardial Infarction. J. Cell. Mol. Med. 2022, 26, 1315–1326. [Google Scholar] [CrossRef]
- Liang, W.J.; Gustafsson, Å.B. The Aging Heart: Mitophagy at the Center of Rejuvenation. Front. Cardiovasc. Med. 2020, 7, 18. [Google Scholar] [CrossRef]
- Madreiter-Sokolowski, C.T.; Thomas, C.; Ristow, M. Interrelation between ROS and Ca2+ in Aging and Age-Related Diseases. Redox Biol. 2020, 36, 101678. [Google Scholar] [CrossRef]
- Mather, M.; Rottenberg, H. Aging Enhances the Activation of the Permeability Transition Pore in Mitochondria. Biochem. Biophys. Res. Commun. 2000, 273, 603–608. [Google Scholar] [CrossRef]
- Mills, E.L.; Kelly, B.; O’Neill, L.A.J. Mitochondria Are the Powerhouses of Immunity. Nat. Immunol. 2017, 18, 488–498. [Google Scholar] [CrossRef]
- Vringer, E.; Tait, S.W.G. Mitochondria and Cell Death-Associated Inflammation. Cell Death Differ. 2023, 30, 304–312. [Google Scholar] [CrossRef]
- Andrieux, P.; Chevillard, C.; Cunha-Neto, E.; Nunes, J.P.S. Mitochondria as a Cellular Hub in Infection and Inflammation. Int. J. Mol. Sci. 2021, 22, 11338. [Google Scholar] [CrossRef]
- Li, X.; Straub, J.; Medeiros, T.C.; Mehra, C.; Den Brave, F.; Peker, E.; Atanassov, I.; Stillger, K.; Michaelis, J.B.; Burbridge, E.; et al. Mitochondria Shed Their Outer Membrane in Response to Infection-Induced Stress. Science 2022, 375, eabi4343. [Google Scholar] [CrossRef]
- Swanson, K.V.; Deng, M.; Ting, J.P.Y. The NLRP3 Inflammasome: Molecular Activation and Regulation to Therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.; Huang, Y.; Chen, M.; Yang, Y.; Li, X.; Zhang, W. Mitochondrial DNA in NLRP3 Inflammasome Activation. Int. Immunopharmacol. 2022, 108, 108719. [Google Scholar] [CrossRef] [PubMed]
- Iyer, S.S.; He, Q.; Janczy, J.R.; Elliott, E.I.; Zhong, Z.; Olivier, A.K.; Sadler, J.J.; Knepper-Adrian, V.; Han, R.; Qiao, L.; et al. Mitochondrial Cardiolipin Is Required for Nlrp3 Inflammasome Activation. Immunity 2013, 39, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Shkoporov, A.N.; Hill, C. Bacteriophages of the Human Gut: The “Known Unknown” of the Microbiome. Cell Host Microbe 2019, 25, 195–209. [Google Scholar] [CrossRef]
- Jeffery, I.B.; Das, A.; O’Herlihy, E.; Coughlan, S.; Cisek, K.; Moore, M.; Bradley, F.; Carty, T.; Pradhan, M.; Dwibedi, C.; et al. Differences in Fecal Microbiomes and Metabolomes of People with vs without Irritable Bowel Syndrome and Bile Acid Malabsorption. Gastroenterology 2020, 158, 1016–1028.e8. [Google Scholar] [CrossRef]
- Shanahan, F.; Ghosh, T.S.; O’Toole, P.W. The Healthy Microbiome—What Is the Definition of a Healthy Gut Microbiome? Gastroenterology 2021, 160, 483–494. [Google Scholar] [CrossRef]
- Lopez-Otín, C.; Kroemer, G. Decelerating Ageing and Biological Clocks by Autophagy. Nat. Rev. Mol. Cell Biol. 2019, 20, 385–386. [Google Scholar] [CrossRef]
- Ghosh, T.S.; Arnoux, J.; O’Toole, P.W. Metagenomic Analysis Reveals Distinct Patterns of Gut Lactobacillus Prevalence, Abundance, and Geographical Variation in Health and Disease. Gut Microbes 2020, 12, 1822729. [Google Scholar] [CrossRef]
- Ghosh, T.S.; Shanahan, F.; O’Toole, P.W. The Gut Microbiome as a Modulator of Healthy Ageing. Nat. Rev. Gastroenterol. Hepatol. 2022, 19, 565–584. [Google Scholar] [CrossRef]
- Wu, L.; Zeng, T.; Deligios, M.; Milanesi, L.; Langille, M.G.I.; Zinellu, A.; Rubino, S.; Carru, C.; Kelvin, D.J. Age-Related Variation of Bacterial and Fungal Communities in Different Body Habitats across the Young, Elderly, and Centenarians in Sardinia. mSphere 2020, 5, e00558-19. [Google Scholar] [CrossRef]
- Parker, A.; James, S.A.; Purse, C.; Brion, A.; Goldson, A.; Telatin, A.; Baker, D.; Carding, S.R. Absence of Bacteria Permits Fungal Gut-to-Brain Translocation and Invasion in Germfree Mice but Ageing Alone Does Not Drive Pathobiont Expansion in Conventionally Raised Mice. Front. Aging Neurosci. 2022, 14, 828429. [Google Scholar] [CrossRef]
- Biagi, E.; Nylund, L.; Candela, M.; Ostan, R.; Bucci, L.; Pini, E.; Nikkïla, J.; Monti, D.; Satokari, R.; Franceschi, C.; et al. Through Ageing, and beyond: Gut Microbiota and Inflammatory Status in Seniors and Centenarians. PLoS ONE 2010, 5, e10667. [Google Scholar] [CrossRef]
- Mayneris-Perxachs, J.; Arnoriaga-Rodríguez, M.; Garre-Olmo, J.; Puig, J.; Ramos, R.; Trelis, M.; Burokas, A.; Coll, C.; Zapata-Tona, C.; Pedraza, S.; et al. Presence of Blastocystis in Gut Microbiota Is Associated with Cognitive Traits and Decreased Executive Function. ISME J. 2022, 16, 2181–2197. [Google Scholar] [CrossRef]
- Tito, R.Y.; Chaffron, S.; Caenepeel, C.; Lima-Mendez, G.; Wang, J.; Vieira-Silva, S.; Falony, G.; Hildebrand, F.; Darzi, Y.; Rymenans, L.; et al. Population-Level Analysis of Blastocystis Subtype Prevalence and Variation in the Human Gut Microbiota. Gut 2019, 68, 1180–1189. [Google Scholar] [CrossRef]
- Johansen, J.; Atarashi, K.; Arai, Y.; Hirose, N.; Sørensen, S.J.; Vatanen, T.; Knip, M.; Honda, K.; Xavier, R.J.; Rasmussen, S.; et al. Centenarians Have a Diverse Gut Virome with the Potential to Modulate Metabolism and Promote Healthy Lifespan. Nat. Microbiol. 2023, 8, 1064–1078. [Google Scholar] [CrossRef]
- Paul, B.D.; Snyder, S.H.; Kashfi, K. Effects of Hydrogen Sulfide on Mitochondrial Function and Cellular Bioenergetics. Redox Biol. 2021, 38, 101772. [Google Scholar] [CrossRef]
- Alharshawi, K.; Cox, B.; Ariza, M.E. Examination of Control Asymptomatic Cohorts Reveals Heightened Anti-EBV and HHV-6 A/B DUTPase Antibodies in the Aging Populations. J. Med. Virol. 2022, 94, 3464–3468. [Google Scholar] [CrossRef]
- Autio, A.; Kettunen, J.; Nevalainen, T.; Kimura, B.; Hurme, M. Herpesviruses and Their Genetic Diversity in the Blood Virome of Healthy Individuals: Effect of Aging. Immun. Ageing 2022, 19, 15. [Google Scholar] [CrossRef]
- Ackermann, M.; Stearns, S.C.; Jenal, U. Senescence in a Bacterium with Asymmetric Division. Science 2003, 300, 1920. [Google Scholar] [CrossRef]
- Stewart, E.J.; Madden, R.; Paul, G.; Taddei, F. Aging and Death in an Organism That Reproduces by Morphologically Symmetric Division. PLoS Biol. 2005, 3, 0295–0300. [Google Scholar] [CrossRef]
- Bergman, J.M.; Wrande, M.; Hughes, D. Acetate Availability and Utilization Supports the Growth of Mutant Sub-Populations on Aging Bacterial Colonies. PLoS ONE 2014, 9, e109255. [Google Scholar] [CrossRef] [PubMed]
- Abram, F.; Arcari, T.; Guerreiro, D.; O’Byrne, C.P. Evolutionary Trade-Offs between Growth and Survival: The Delicate Balance between Reproductive Success and Longevity in Bacteria. Adv. Microb. Physiol. 2021, 79, 133–162. [Google Scholar] [PubMed]
- Steiner, U.K. Senescence in Bacteria and Its Underlying Mechanisms. Front. Cell Dev. Biol. 2021, 9, 668915. [Google Scholar] [CrossRef] [PubMed]
- Wozniak, K.J.; Simmons, L.A. Bacterial DNA Excision Repair Pathways. Nat. Rev. Microbiol. 2022, 20, 465–477. [Google Scholar] [CrossRef]
- Govers, S.K.; Mortier, J.; Adam, A.; Aertsen, A. Protein Aggregates Encode Epigenetic Memory of Stressful Encounters in Individual Escherichia coli Cells. PLoS Biol. 2018, 16, e2003853. [Google Scholar] [CrossRef]
- Roberts, R.O.; Aakre, J.A.; Kremers, W.K.; Vassilaki, M.; Knopman, D.S.; Mielke, M.M.; Alhurani, R.; Geda, Y.E.; Machulda, M.M.; Coloma, P.; et al. Prevalence and Outcomes of Amyloid Positivity Among Persons Without Dementia in a Longitudinal, Population-Based Setting. JAMA Neurol. 2018, 75, 970–979. [Google Scholar] [CrossRef]
- Robert, L.; Ollion, J.; Robert, J.; Song, X.; Matic, I.; Elez, M. Mutation Dynamics and Fitness Effects Followed in Single Cells. Science 2018, 359, 1283–1286. [Google Scholar] [CrossRef]
- Dukan, S.; Nyström, T. Oxidative Stress Defense and Deterioration of Growth-Arrested Escherichia coli Cells. J. Biol. Chem. 1999, 274, 26027–26032. [Google Scholar] [CrossRef]
- Maisonneuve, E.; Fraysse, L.; Moinier, D.; Dukan, S. Existence of Abnormal Protein Aggregates in Healthy Escherichia coli Cells. J. Bacteriol. 2008, 190, 887–893. [Google Scholar] [CrossRef]
- Lindner, A.B.; Madden, R.; Demarez, A.; Stewart, E.J.; Taddei, F. Asymmetric Segregation of Protein Aggregates Is Associated with Cellular Aging and Rejuvenation. Proc. Natl. Acad. Sci. USA 2008, 105, 3076–3081. [Google Scholar] [CrossRef]
- Winkler, J.; Seybert, A.; König, L.; Pruggnaller, S.; Haselmann, U.; Sourjik, V.; Weiss, M.; Frangakis, A.S.; Mogk, A.; Bukau, B. Quantitative and Spatio-Temporal Features of Protein Aggregation in Escherichia coli and Consequences on Protein Quality Control and Cellular Ageing. EMBO J. 2010, 29, 910–923. [Google Scholar] [CrossRef]
- Proenca, A.M.; Rang, C.U.; Qiu, A.; Shi, C.; Chao, L. Cell Aging Preserves Cellular Immortality in the Presence of Lethal Levels of Damage. PLoS Biol. 2019, 17, e3000266. [Google Scholar] [CrossRef]
- Rang, C.U.; Proenca, A.; Buetz, C.; Shi, C.; Chao, L. Minicells as a Damage Disposal Mechanism in Escherichia coli. mSphere 2018, 3, e00428-18. [Google Scholar] [CrossRef]
- Sánchez-Romero, M.A.; Casadesús, J. The Bacterial Epigenome. Nat. Rev. Microbiol. 2020, 18, 7–20. [Google Scholar] [CrossRef]
- Veening, J.W.; Stewart, E.J.; Berngruber, T.W.; Taddei, F.; Kuipers, O.P.; Hamoen, L.W. Bet-Hedging and Epigenetic Inheritance in Bacterial Cell Development. Proc. Natl. Acad. Sci. USA 2008, 105, 4393–4398. [Google Scholar] [CrossRef]
- Galperin, M.Y. What Bacteria Want. Environ. Microbiol. 2018, 20, 4221–4229. [Google Scholar] [CrossRef]
- Biselli, E.; Schink, S.J.; Gerland, U. Slower Growth of Escherichia coli Leads to Longer Survival in Carbon Starvation Due to a Decrease in the Maintenance Rate. Mol. Syst. Biol. 2020, 16, e9478. [Google Scholar] [CrossRef]
- Dakic, T.; Jevdjovic, T.; Vujovic, P.; Mladenovic, A. The Less We Eat, the Longer We Live: Can Caloric Restriction Help Us Become Centenarians? Int. J. Mol. Sci. 2022, 23, 6546. [Google Scholar] [CrossRef]
- Liu, X.; Liu, Z.; Wu, Z.; Ren, J.; Fan, Y.; Sun, L.; Cao, G.; Niu, Y.; Zhang, B.; Ji, Q.; et al. Resurrection of Endogenous Retroviruses during Aging Reinforces Senescence. Cell 2023, 186, 287–304.e26. [Google Scholar] [CrossRef]
- Vorobjev, I.A.; Zorov, D.B. Diazepam Inhibits Cell Respiration and Induces Fragmentation of Mitochondrial Reticulum. FEBS Lett. 1983, 163, 311–314. [Google Scholar] [CrossRef]
- Zorov, D.B.; Vorobjev, I.A.; Plotnikov, E.Y.; Silachev, D.N.; Zorova, L.D.; Pevzner, I.B.; Babenko, V.A.; Zorov, S.D.; Jankauskas, S.S.; Popkov, V.A. Specific Issues of Mitochondrial Fragmentation (Fission). Biochem. Suppl. Ser. A Membr. Cell Biol. 2015, 9, 278–284. [Google Scholar] [CrossRef]
- Zorov, D.B.; Bannikova, S.Y.; Belousov, V.V.; Vyssokikh, M.Y.; Zorova, L.D.; Isaev, N.K.; Krasnikov, B.F.; Plotnikov, E.Y. Reactive Oxygen and Nitrogen Species: Friends or Foes? Biochemestry 2005, 70, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Popkov, V.A.; Plotnikov, E.Y.; Lyamzaev, K.G.; Silachev, D.N.; Zorova, L.D.; Pevzner, I.B.; Jankauskas, S.S.; Zorov, S.D.; Babenko, V.A.; Zorov, D.B. Mitodiversity. Biochemistry 2015, 80, 532–541. [Google Scholar] [CrossRef] [PubMed]
- Eppenberger-Eberhardt, M.; Riesinger, I.; Messerli, M.; Schwarb, P.; Muller, M.; Eppenberger, H.M.; Wallimann, T. Adult Rat Cardiomyocytes Cultured in Creatine-Deficient Medium Display Large Mitochondria with Paracrystalline Inclusions, Enriched for Creatine Kinase. J. Cell Biol. 1991, 113, 289–302. [Google Scholar] [CrossRef] [PubMed]
- Katajisto, P.; Döhla, J.; Chaffer, C.L.; Pentinmikko, N.; Marjanovic, N.; Iqbal, S.; Zoncu, R.; Chen, W.; Weinberg, R.A.; Sabatini, D.M. Stem Cells. Asymmetric Apportioning of Aged Mitochondria between Daughter Cells Is Required for Stemness. Science 2015, 348, 340–343. [Google Scholar] [CrossRef]
- Liu, X.; Weaver, D.; Shirihai, O.; Hajnóczky, G. Mitochondrial “Kiss-and-Run”: Interplay between Mitochondrial Motility and Fusion-Fission Dynamics. EMBO J. 2009, 28, 3074–3089. [Google Scholar] [CrossRef]
- Shpilka, T.; Haynes, C.M. The Mitochondrial UPR: Mechanisms, Physiological Functions and Implications in Ageing. Nat. Rev. Mol. Cell Biol. 2018, 19, 109–120. [Google Scholar] [CrossRef]
- Patron, M.; Tarasenko, D.; Nolte, H.; Kroczek, L.; Ghosh, M.; Ohba, Y.; Lasarzewski, Y.; Ahmadi, Z.A.; Cabrera-Orefice, A.; Eyiama, A.; et al. Regulation of Mitochondrial Proteostasis by the Proton Gradient. EMBO J. 2022, 41, e110476. [Google Scholar] [CrossRef]
- Cole, L.W. The Evolution of Per-Cell Organelle Number. Front. Cell Dev. Biol. 2016, 4, 85. [Google Scholar] [CrossRef]
- Claessen, D.; Errington, J. Cell Wall Deficiency as a Coping Strategy for Stress. Trends Microbiol. 2019, 27, 1025–1033. [Google Scholar] [CrossRef]
- Shitut, S.; Shen, M.-J.; Claushuis, B.; Derks, R.J.E.; Giera, M.; Rozen, D.; Claessen, D.; Kros, A. Generating Heterokaryotic Cells via Bacterial Cell-Cell Fusion. Microbiol. Spectr. 2022, 10, e0169322. [Google Scholar] [CrossRef]
- Vincent, A.E.; Turnbull, D.M.; Eisner, V.; Hajnóczky, G.; Picard, M. Mitochondrial Nanotunnels. Trends Cell Biol. 2017, 27, 787–799. [Google Scholar] [CrossRef]
- Vincent, A.E.; White, K.; Davey, T.; Philips, J.; Ogden, R.T.; Lawess, C.; Warren, C.; Hall, M.G.; Ng, Y.S.; Falkous, G.; et al. Quantitative 3D Mapping of the Human Skeletal Muscle Mitochondrial Network. Cell Rep. 2019, 26, 996–1009.e4. [Google Scholar] [CrossRef]
- Pereira, F.C.; Berry, D. Microbial Nutrient Niches in the Gut. Environ. Microbiol. 2017, 19, 1366–1378. [Google Scholar] [CrossRef]
- Lian, S.; Liu, J.; Wu, Y.; Xia, P.; Zhu, G. Bacterial and Viral Co-Infection in the Intestine: Competition Scenario and Their Effect on Host Immunity. Int. J. Mol. Sci. 2022, 23, 2311. [Google Scholar] [CrossRef]
- Stanley Schneierson, S.; Shore, B. Antibacterial Activity of Herpes Simplex Virus Grown in Tissue Culture. Nature 1963, 199, 721–722. [Google Scholar] [CrossRef]
- Neu, U.; Mainou, B.A. Virus Interactions with Bacteria: Partners in the Infectious Dance. PLoS Pathog. 2020, 16, e1008234. [Google Scholar] [CrossRef]
- Wang, Y.; Santerre, M.; Tempera, I.; Martin, K.; Mukerjee, R.; Sawaya, B.E. HIV-1 Vpr Disrupts Mitochondria Axonal Transport and Accelerates Neuronal Aging. Neuropharmacology 2017, 117, 364–375. [Google Scholar] [CrossRef]
- Piciocchi, M.; Cardin, R.; Cillo, U.; Vitale, A.; Cappon, A.; Mescoli, C.; Guido, M.; Rugge, M.; Burra, P.; Floreani, A.; et al. Differential Timing of Oxidative DNA Damage and Telomere Shortening in Hepatitis C and B Virus-Related Liver Carcinogenesis. Transl. Res. 2016, 168, 122–133. [Google Scholar] [CrossRef]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; O’Meara, M.J.; Rezelj, V.V.; Guo, J.Z.; Swaney, D.L.; et al. A SARS-CoV-2 Protein Interaction Map Reveals Targets for Drug Repurposing. Nature 2020, 583, 459–468. [Google Scholar] [CrossRef]
- Kato, H.; Lu, Q.; Rapaport, D.; Kozjak-Pavlovic, V. Tom70 Is Essential for PINK1 Import into Mitochondria. PLoS ONE 2013, 8, e58435. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Wang, L.; Dong, C.; Che, Y.; Jiang, L.; Liu, L.; Zhao, H.; Liao, Y.; Sheng, Y.; Dong, S.; et al. The Interaction of the SARS Coronavirus Non-Structural Protein 10 with the Cellular Oxido-Reductase System Causes an Extensive Cytopathic Effect. J. Clin. Virol. 2005, 34, 133–139. [Google Scholar] [CrossRef] [PubMed]
- West, A.P.; Khoury-Hanold, W.; Staron, M.; Tal, M.C.; Pineda, C.M.; Lang, S.M.; Bestwick, M.; Duguay, B.A.; Raimundo, N.; MacDuff, D.A.; et al. Mitochondrial DNA Stress Primes the Antiviral Innate Immune Response. Nature 2015, 520, 553–557. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.M.; Wang, W.; Song, Z.G.; Hu, Y.; Tao, Z.W.; Tian, J.H.; Pei, Y.Y.; et al. A New Coronavirus Associated with Human Respiratory Disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.-S.; Qi, H.-Y.; Boularan, C.; Huang, N.-N.; Abu-Asab, M.; Shelhamer, J.H.; Kehrl, J.H. SARS-Coronavirus Open Reading Frame-9b Suppresses Innate Immunity by Targeting Mitochondria and the MAVS/TRAF3/TRAF6 Signalosome. J. Immunol. 2014, 193, 3080–3089. [Google Scholar] [CrossRef]
- Chen, C.Y.; Ping, Y.H.; Lee, H.C.; Chen, K.H.; Lee, Y.M.; Chan, Y.J.; Lien, T.C.; Jap, T.S.; Lin, C.H.; Kao, L.S.; et al. Open Reading Frame 8a of the Human Severe Acute Respiratory Syndrome Coronavirus Not Only Promotes Viral Replication but Also Induces Apoptosis. J. Infect. Dis. 2007, 196, 405–415. [Google Scholar] [CrossRef]
- Cornillez-Ty, C.T.; Liao, L.; Yates, J.R.; Kuhn, P.; Buchmeier, M.J. Severe Acute Respiratory Syndrome Coronavirus Nonstructural Protein 2 Interacts with a Host Protein Complex Involved in Mitochondrial Biogenesis and Intracellular Signaling. J. Virol. 2009, 83, 10314–10318. [Google Scholar] [CrossRef]
- Pasquier, C.; Robichon, A. Computational Search of Hybrid Human/SARS-CoV-2 DsRNA Reveals Unique Viral Sequences That Diverge from Those of Other Coronavirus Strains. Heliyon 2021, 7, e07284. [Google Scholar] [CrossRef]
- Perry, S.W.; Norman, J.P.; Litzburg, A.; Zhang, D.; Dewhurst, S.; Gelbard, H.A. HIV-1 Transactivator of Transcription Protein Induces Mitochondrial Hyperpolarization and Synaptic Stress Leading to Apoptosis. J. Immunol. 2005, 174, 4333–4344. [Google Scholar] [CrossRef]
- Gorwood, J.; Ejlalmanesh, T.; Bourgeois, C.; Mantecon, M.; Rose, C.; Atlan, M.; Desjardins, D.; Le Grand, R.; Fève, B.; Lambotte, O.; et al. SIV Infection and the HIV Proteins Tat and Nef Induce Senescence in Adipose Tissue and Human Adipose Stem Cells, Resulting in Adipocyte Dysfunction. Cells 2020, 9, 854. [Google Scholar] [CrossRef]
- Chen, N.C.; Partridge, A.T.; Tuzer, F.; Cohen, J.; Nacarelli, T.; Navas-Martín, S.; Sell, C.; Torres, C.; Martín-García, J. Induction of a Senescence-Like Phenotype in Cultured Human Fetal Microglia During HIV-1 Infection. J. Gerontol. A Biol. Sci. Med. Sci. 2018, 73, 1187–1196. [Google Scholar] [CrossRef]
- Teodorof-Diedrich, C.; Spector, S.A. Human Immunodeficiency Virus Type 1 and Methamphetamine-Mediated Mitochondrial Damage and Neuronal Degeneration in Human Neurons. J. Virol. 2020, 94, e00924-20. [Google Scholar] [CrossRef]
- Teodorof-Diedrich, C.; Spector, S.A. Human Immunodeficiency Virus Type 1 Gp120 and Tat Induce Mitochondrial Fragmentation and Incomplete Mitophagy in Human Neurons. J. Virol. 2018, 92, e00993-18. [Google Scholar] [CrossRef]
- Cheng, M.L.; Weng, S.F.; Kuo, C.H.; Ho, H.Y. Enterovirus 71 Induces Mitochondrial Reactive Oxygen Species Generation That Is Required for Efficient Replication. PLoS ONE 2014, 9, e113234. [Google Scholar] [CrossRef]
- Poenisch, M.; Burger, N.; Staeheli, P.; Bauer, G.; Schneider, U. Protein X of Borna Disease Virus Inhibits Apoptosis and Promotes Viral Persistence in the Central Nervous Systems of Newborn-Infected Rats. J. Virol. 2009, 83, 4297–4307. [Google Scholar] [CrossRef]
- Perrin-Cocon, L.; Kundlacz, C.; Jacquemin, C.; Hanoulle, X.; Aublin-Gex, A.; Figl, M.; Manteca, J.; André, P.; Vidalain, P.O.; Lotteau, V.; et al. Domain 2 of Hepatitis C Virus Protein NS5A Activates Glucokinase and Induces Lipogenesis in Hepatocytes. Int. J. Mol. Sci. 2022, 23, 919. [Google Scholar] [CrossRef]
- Makiuchi, T.; Nozaki, T. Highly Divergent Mitochondrion-Related Organelles in Anaerobic Parasitic Protozoa. Biochimie 2014, 100, 3–17. [Google Scholar] [CrossRef]
- Müller, M.; Mentel, M.; van Hellemond, J.J.; Henze, K.; Woehle, C.; Gould, S.B.; Yu, R.-Y.; van der Giezen, M.; Tielens, A.G.M.; Martin, W.F. Biochemistry and Evolution of Anaerobic Energy Metabolism in Eukaryotes. Microbiol. Mol. Biol. Rev. 2012, 76, 444–495. [Google Scholar] [CrossRef]
- Wen, J.J.; Vyatkina, G.; Garg, N. Oxidative Damage during Chagasic Cardiomyopathy Development: Role of Mitochondrial Oxidant Release and Inefficient Antioxidant Defense. Free Radic. Biol. Med. 2004, 37, 1821–1833. [Google Scholar] [CrossRef]
- Verma, S.; Shakya, V.P.S.; Idnurm, A. Exploring and Exploiting the Connection between Mitochondria and the Virulence of Human Pathogenic Fungi. Virulence 2018, 9, 426–446. [Google Scholar] [CrossRef]
- Koh, A.; De Vadder, F.; Kovatcheva-Datchary, P.; Bäckhed, F. From Dietary Fiber to Host Physiology: Short-Chain Fatty Acids as Key Bacterial Metabolites. Cell 2016, 165, 1332–1345. [Google Scholar] [CrossRef] [PubMed]
- Smith, P.M.; Howitt, M.R.; Panikov, N.; Michaud, M.; Gallini, C.A.; Bohlooly, Y.M.; Glickman, J.N.; Garrett, W.S. The Microbial Metabolites, Short-Chain Fatty Acids, Regulate Colonic T Reg Cell Homeostasis. Science 2013, 341, 569–573. [Google Scholar] [CrossRef] [PubMed]
- Russell, W.R.; Hoyles, L.; Flint, H.J.; Dumas, M.E. Colonic Bacterial Metabolites and Human Health. Curr. Opin. Microbiol. 2013, 16, 246–254. [Google Scholar] [CrossRef] [PubMed]
- Flint, H.J. Gut Microbial Metabolites in Health and Disease. Gut Microbes 2016, 7, 187–188. [Google Scholar] [CrossRef]
- Tomasova, L.; Grman, M.; Ondrias, K.; Ufnal, M. The Impact of Gut Microbiota Metabolites on Cellular Bioenergetics and Cardiometabolic Health. Nutr. Metab. 2021, 18, 72. [Google Scholar] [CrossRef]
- Louis, P.; Flint, H.J. Formation of Propionate and Butyrate by the Human Colonic Microbiota. Environ. Microbiol. 2017, 19, 29–41. [Google Scholar] [CrossRef]
- Macfarlane, S.; Macfarlane, G.T. Regulation of Short-Chain Fatty Acid Production. Proc. Nutr. Soc. 2003, 62, 67–72. [Google Scholar] [CrossRef]
- Hu, S.; Kuwabara, R.; de Haan, B.J.; Smink, A.M.; de Vos, P. Acetate and Butyrate Improve β-Cell Metabolism and Mitochondrial Respiration under Oxidative Stress. Int. J. Mol. Sci. 2020, 21, 1542. [Google Scholar] [CrossRef]
- Macia, L.; Tan, J.; Vieira, A.T.; Leach, K.; Stanley, D.; Luong, S.; Maruya, M.; Ian McKenzie, C.; Hijikata, A.; Wong, C.; et al. Metabolite-Sensing Receptors GPR43 and GPR109A Facilitate Dietary Fibre-Induced Gut Homeostasis through Regulation of the Inflammasome. Nat. Commun. 2015, 6, 6734. [Google Scholar] [CrossRef]
- Bolognini, D.; Tobin, A.B.; Milligan, G.; Moss, C.E. The Pharmacology and Function of Receptors for Short-Chain Fatty Acids. Mol. Pharmacol. 2016, 89, 388–398. [Google Scholar] [CrossRef]
- Canfora, E.E.; Jocken, J.W.; Blaak, E.E. Short-Chain Fatty Acids in Control of Body Weight and Insulin Sensitivity. Nat. Rev. Endocrinol. 2015, 11, 577–591. [Google Scholar] [CrossRef]
- Cummings, J.H.; Pomare, E.W.; Branch, H.W.J.; Naylor, C.P.E.; MacFarlane, G.T. Short Chain Fatty Acids in Human Large Intestine, Portal, Hepatic and Venous Blood. Gut 1987, 28, 1221–1227. [Google Scholar] [CrossRef]
- Den Besten, G.; Van Eunen, K.; Groen, A.K.; Venema, K.; Reijngoud, D.J.; Bakker, B.M. The Role of Short-Chain Fatty Acids in the Interplay between Diet, Gut Microbiota, and Host Energy Metabolism. J. Lipid Res. 2013, 54, 2325–2340. [Google Scholar] [CrossRef]
- Barcenilla, A.; Pryde, S.E.; Martin, J.C.; Duncan, S.H.; Stewart, C.S.; Henderson, C.; Flint, H.J. Phylogenetic Relationships of Butyrate-Producing Bacteria from the Human Gut. Appl. Environ. Microbiol. 2000, 66, 1654–1661. [Google Scholar] [CrossRef]
- Rivière, A.; Selak, M.; Lantin, D.; Leroy, F.; De Vuyst, L. Bifidobacteria and Butyrate-Producing Colon Bacteria: Importance and Strategies for Their Stimulation in the Human Gut. Front. Microbiol. 2016, 7, 979. [Google Scholar] [CrossRef]
- Kelly, C.J.; Zheng, L.; Campbell, E.L.; Saeedi, B.; Scholz, C.C.; Bayless, A.J.; Wilson, K.E.; Glover, L.E.; Kominsky, D.J.; Magnuson, A.; et al. Crosstalk between Microbiota-Derived Short-Chain Fatty Acids and Intestinal Epithelial HIF Augments Tissue Barrier Function. Cell Host Microbe 2015, 17, 662–671. [Google Scholar] [CrossRef]
- Fachi, J.L.; de Felipe, J.S.; Pral, L.P.; da Silva, B.K.; Corrêa, R.O.; de Andrade, M.C.P.; da Fonseca, D.M.; Basso, P.J.; Câmara, N.O.S.; de Sales e Souza, É.L.; et al. Butyrate Protects Mice from Clostridium Difficile-Induced Colitis through an HIF-1-Dependent Mechanism. Cell Rep. 2019, 27, 750–761.e7. [Google Scholar] [CrossRef]
- Martin-Gallausiaux, C.; Béguet-Crespel, F.; Marinelli, L.; Jamet, A.; Ledue, F.; Blottière, H.M.; Lapaque, N. Butyrate Produced by Gut Commensal Bacteria Activates TGF-Beta1 Expression through the Transcription Factor SP1 in Human Intestinal Epithelial Cells. Sci. Rep. 2018, 8, 9742. [Google Scholar] [CrossRef]
- Huang, W.; Man, Y.; Gao, C.; Zhou, L.; Gu, J.; Xu, H.; Wan, Q.; Long, Y.; Chai, L.; Xu, Y.; et al. Short-Chain Fatty Acids Ameliorate Diabetic Nephropathy via GPR43-Mediated Inhibition of Oxidative Stress and NF- κ B Signaling. Oxid. Med. Cell. Longev. 2020, 2020, 4074832. [Google Scholar] [CrossRef]
- Wang, F.; Wu, H.; Fan, M.; Yu, R.; Zhang, Y.; Liu, J.; Zhou, X.; Cai, Y.; Huang, S.; Hu, Z.; et al. Sodium Butyrate Inhibits Migration and Induces AMPK-MTOR Pathway-Dependent Autophagy and ROS-Mediated Apoptosis via the MiR-139-5p/Bmi-1 Axis in Human Bladder Cancer Cells. FASEB J. 2020, 34, 4266–4282. [Google Scholar] [CrossRef]
- Li, X.; Wang, C.; Zhu, J.; Lin, Q.; Yu, M.; Wen, J.; Feng, J.; Hu, C. Sodium Butyrate Ameliorates Oxidative Stress-Induced Intestinal Epithelium Barrier Injury and Mitochondrial Damage through AMPK-Mitophagy Pathway. Oxid. Med. Cell. Longev. 2022, 2022, 3745135. [Google Scholar] [CrossRef] [PubMed]
- Donohoe, D.R.; Garge, N.; Zhang, X.; Sun, W.; O’Connell, T.M.; Bunger, M.K.; Bultman, S.J. The Microbiome and Butyrate Regulate Energy Metabolism and Autophagy in the Mammalian Colon. Cell Metab. 2011, 13, 517–526. [Google Scholar] [CrossRef] [PubMed]
- Louis, P.; Hold, G.L.; Flint, H.J. The Gut Microbiota, Bacterial Metabolites and Colorectal Cancer. Nat. Rev. Microbiol. 2014, 12, 661–672. [Google Scholar] [CrossRef] [PubMed]
- Byndloss, M.X.; Olsan, E.E.; Rivera-Chávez, F.; Tiffany, C.R.; Cevallos, S.A.; Lokken, K.L.; Torres, T.P.; Byndloss, A.J.; Faber, F.; Gao, Y.; et al. Microbiota-Activated PPAR-γ Signaling Inhibits Dysbiotic Enterobacteriaceae Expansion. Science 2017, 357, 570–575. [Google Scholar] [CrossRef]
- Den Besten, G.; Bleeker, A.; Gerding, A.; Van Eunen, K.; Havinga, R.; Van Dijk, T.H.; Oosterveer, M.H.; Jonker, J.W.; Groen, A.K.; Reijngoud, D.J.; et al. Short-Chain Fatty Acids Protect against High-Fat Diet-Induced Obesity via a Pparg-Dependent Switch from Lipogenesis to Fat Oxidation. Diabetes 2015, 64, 2398–2408. [Google Scholar] [CrossRef]
- Zhang, X.; Gérard, P. Diet-Gut Microbiota Interactions on Cardiovascular Disease. Comput. Struct. Biotechnol. J. 2022, 20, 1528–1540. [Google Scholar] [CrossRef]
- Chiang, J.Y.L.; Ferrell, J.M. Bile Acid Receptors FXR and TGR5 Signaling in Fatty Liver Diseases and Therapy. Am. J. Physiol. Gastrointest. Liver Physiol. 2020, 318, G554–G573. [Google Scholar] [CrossRef]
- Sousa, T.; Castro, R.E.; Pinto, S.N.; Coutinho, A.; Lucas, S.D.; Moreira, R.; Rodrigues, C.M.P.; Prieto, M.; Fernandes, F. Deoxycholic Acid Modulates Cell Death Signaling through Changes in Mitochondrial Membrane Properties. J. Lipid Res. 2015, 56, 2158–2171. [Google Scholar] [CrossRef]
- Jia, W.; Xie, G.; Jia, W. Bile Acid–Microbiota Crosstalk in Gastrointestinal Inflammation and Carcinogenesis. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 111–128. [Google Scholar] [CrossRef]
- Gao, K.; Mu, C.L.; Farzi, A.; Zhu, W.Y. Tryptophan Metabolism: A Link between the Gut Microbiota and Brain. Adv. Nutr. 2020, 11, 709–723. [Google Scholar] [CrossRef]
- Jiao, N.; Wang, L.; Wang, Y.; Xu, D.; Zhang, X.; Yin, J. Cysteine Exerts an Essential Role in Maintaining Intestinal Integrity and Function Independent of Glutathione. Mol. Nutr. Food Res. 2022, 66, e2100728. [Google Scholar] [CrossRef]
- Murphy, B.; Bhattacharya, R.; Mukherjee, P. Hydrogen Sulfide Signaling in Mitochondria and Disease. FASEB J. 2019, 33, 13098–13125. [Google Scholar] [CrossRef]
- Saad, M.J.A.; Santos, A.; Prada, P.O. Linking Gut Microbiota and Inflammation to Obesity and Insulin Resistance. Physiology 2016, 31, 283–293. [Google Scholar] [CrossRef]
- Go, M.; Kou, J.; Lim, J.E.; Yang, J.; Fukuchi, K. ichiro Microglial Response to LPS Increases in Wild-Type Mice during Aging but Diminishes in an Alzheimer’s Mouse Model: Implication of TLR4 Signaling in Disease Progression. Biochem. Biophys. Res. Commun. 2016, 479, 331–337. [Google Scholar] [CrossRef]
- Park, B.S.; Song, D.H.; Kim, H.M.; Choi, B.S.; Lee, H.; Lee, J.O. The Structural Basis of Lipopolysaccharide Recognition by the TLR4-MD-2 Complex. Nature 2009, 458, 1191–1195. [Google Scholar] [CrossRef]
- Tripathi, A.; Paliwal, P.; Krishnamurthy, S. Piracetam Attenuates LPS-Induced Neuroinflammation and Cognitive Impairment in Rats. Cell. Mol. Neurobiol. 2017, 37, 1373–1386. [Google Scholar] [CrossRef]
- Constantino-Jonapa, L.A.; Espinoza-Palacios, Y.; Escalona-Montaño, A.R.; Hernández-Ruiz, P.; Amezcua-Guerra, L.M.; Amedei, A.; Aguirre-García, M.M. Contribution of Trimethylamine N-Oxide (TMAO) to Chronic Inflammatory and Degenerative Diseases. Biomedicines 2023, 11, 431. [Google Scholar] [CrossRef]
- Ghosh, T.S.; Shanahan, F.; O’Toole, P.W. Toward an Improved Definition of a Healthy Microbiome for Healthy Aging. Nat. Aging 2022, 2, 1054–1069. [Google Scholar] [CrossRef]
- Fung, T.C.; Olson, C.A.; Hsiao, E.Y. Interactions between the Microbiota, Immune and Nervous Systems in Health and Disease. Nat. Neurosci. 2017, 20, 145–155. [Google Scholar] [CrossRef]
- Rao, M.; Gershon, M.D. The Bowel and beyond: The Enteric Nervous System in Neurological Disorders. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 517–528. [Google Scholar] [CrossRef]
- Rutsch, A.; Kantsjö, J.B.; Ronchi, F. The Gut-Brain Axis: How Microbiota and Host Inflammasome Influence Brain Physiology and Pathology. Front. Immunol. 2020, 11, 604179. [Google Scholar] [CrossRef] [PubMed]
- Mitrea, L.; Nemeş, S.A.; Szabo, K.; Teleky, B.E.; Vodnar, D.C. Guts Imbalance Imbalances the Brain: A Review of Gut Microbiota Association With Neurological and Psychiatric Disorders. Front. Med. 2022, 9, 813204. [Google Scholar] [CrossRef] [PubMed]
- Morais, L.H.; Schreiber, H.L.; Mazmanian, S.K. The Gut Microbiota–Brain Axis in Behaviour and Brain Disorders. Nat. Rev. Microbiol. 2021, 19, 241–255. [Google Scholar] [CrossRef] [PubMed]
- Borbolis, F.; Mytilinaiou, E.; Palikaras, K. The Crosstalk between Microbiome and Mitochondrial Homeostasis in Neurodegeneration. Cells 2023, 12, 429. [Google Scholar] [CrossRef] [PubMed]
- Imdad, S.; Lim, W.; Kim, J.H.; Kang, C. Intertwined Relationship of Mitochondrial Metabolism, Gut Microbiome and Exercise Potential. Int. J. Mol. Sci. 2022, 23, 2679. [Google Scholar] [CrossRef]
- Ramos, C.; Gibson, G.R.; Walton, G.E.; Magistro, D.; Kinnear, W.; Hunter, K. Systematic Review of the Effects of Exercise and Physical Activity on the Gut Microbiome of Older Adults. Nutrients 2022, 14, 674. [Google Scholar] [CrossRef]
- Clarke, S.F.; Murphy, E.F.; O’Sullivan, O.; Lucey, A.J.; Humphreys, M.; Hogan, A.; Hayes, P.; O’Reilly, M.; Jeffery, I.B.; Wood-Martin, R.; et al. Exercise and Associated Dietary Extremes Impact on Gut Microbial Diversity. Gut 2014, 63, 1913–1920. [Google Scholar] [CrossRef]
- Katsirma, Z.; Dimidi, E.; Rodriguez-Mateos, A.; Whelan, K. Fruits and Their Impact on the Gut Microbiota, Gut Motility and Constipation. Food Funct. 2021, 12, 8850–8866. [Google Scholar] [CrossRef]
- Lee, S.; Keirsey, K.I.; Kirkland, R.; Grunewald, Z.I.; Fischer, J.G.; de La Serre, C.B. Blueberry Supplementation Influences the Gut Microbiota, Inflammation, and Insulin Resistance in High-Fat-Diet-Fed Rats. J. Nutr. 2018, 148, 209–219. [Google Scholar] [CrossRef]
- Morissette, A.; Kropp, C.; Songpadith, J.P.; Moreira, R.J.; Costa, J.; Mariné-Casadó, R.; Pilon, G.; Varin, T.V.; Dudonné, S.; Boutekrabt, L.; et al. Blueberry Proanthocyanidins and Anthocyanins Improve Metabolic Health through a Gut Microbiota-Dependent Mechanism in Diet-Induced Obese Mice. Am. J. Physiol. Endocrinol. Metab. 2020, 318, E965–E980. [Google Scholar] [CrossRef]
- Wankhade, U.D.; Zhong, Y.; Lazarenko, O.P.; Chintapalli, S.V.; Piccolo, B.D.; Chen, J.R.; Shankar, K. Sex-Specific Changes in Gut Microbiome Composition Following Blueberry Consumption in C57Bl/6J Mice. Nutrients 2019, 11, 313. [Google Scholar] [CrossRef]
- Vendrame, S.; Guglielmetti, S.; Riso, P.; Arioli, S.; Klimis-Zacas, D.; Porrini, M. Six-Week Consumption of a Wild Blueberry Powder Drink Increases Bifidobacteria in the Human Gut. J. Agric. Food Chem. 2011, 59, 12815–12820. [Google Scholar] [CrossRef]
- Zhang, T.; Li, Q.; Cheng, L.; Buch, H.; Zhang, F. Akkermansia Muciniphila Is a Promising Probiotic. Microb. Biotechnol. 2019, 12, 1109–1125. [Google Scholar] [CrossRef]
- Depommier, C.; Everard, A.; Druart, C.; Plovier, H.; Van Hul, M.; Vieira-Silva, S.; Falony, G.; Raes, J.; Maiter, D.; Delzenne, N.M.; et al. Supplementation with Akkermansia Muciniphila in Overweight and Obese Human Volunteers: A Proof-of-Concept Exploratory Study. Nat. Med. 2019, 25, 1096–1103. [Google Scholar] [CrossRef]
- Kriebs, A. Microbiota Supplements to Improve Metabolic Health. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 514. [Google Scholar] [CrossRef]
- Ghosh, T.S.; Rampelli, S.; Jeffery, I.B.; Santoro, A.; Neto, M.; Capri, M.; Giampieri, E.; Jennings, A.; Candela, M.; Turroni, S.; et al. Mediterranean Diet Intervention Alters the Gut Microbiome in Older People Reducing Frailty and Improving Health Status: The NU-AGE 1-Year Dietary Intervention across Five European Countries. Gut 2020, 69, 1218–1228. [Google Scholar] [CrossRef]
- Cani, P.D.; Van Hul, M. Mediterranean Diet, Gut Microbiota and Health: When Age and Calories Do Not Add Up! Gut 2020, 69, 1167–1168. [Google Scholar] [CrossRef]
- Zeng, X.; Li, X.; Li, X.; Wei, C.; Shi, C.; Hu, K.; Kong, D.; Luo, Q.; Xu, Y.; Shan, W.; et al. Fecal Microbiota Transplantation from Young Mice Rejuvenates Aged Hematopoietic Stem Cells by Suppressing Inflammation. Blood 2023, 141, 1691–1707. [Google Scholar] [CrossRef]
- Villani, R.; Sangineto, M.; Pontrelli, P.; Bellanti, F.; Bukke, V.N.; Moola, A.; Gesualdo, L.; Vendemiale, G.; Grandaliano, G.; Stallone, G.; et al. Eradication of HCV by Direct Antiviral Agents Restores Mitochondrial Function and Energy Homeostasis in Peripheral Blood Mononuclear Cells. FASEB J. 2022, 36, e22650. [Google Scholar] [CrossRef]
- Valeri, F.; Dos Santos Guilherme, M.; He, F.; Stoye, N.M.; Schwiertz, A.; Endres, K. Impact of the Age of Cecal Material Transfer Donors on Alzheimer’s Disease Pathology in 5xfad Mice. Microorganisms 2021, 9, 2548. [Google Scholar] [CrossRef]
- D’Amato, A.; Di Cesare Mannelli, L.; Lucarini, E.; Man, A.L.; Le Gall, G.; Branca, J.J.V.; Ghelardini, C.; Amedei, A.; Bertelli, E.; Regoli, M.; et al. Faecal Microbiota Transplant from Aged Donor Mice Affects Spatial Learning and Memory via Modulating Hippocampal Synaptic Plasticity-and Neurotransmission-Related Proteins in Young Recipients. Microbiome 2020, 8, 140. [Google Scholar] [CrossRef] [PubMed]



| Virus | Viral Component | Mitochondrial Component | Observation/Theoretical Impact | Ref. |
|---|---|---|---|---|
| Herpes virus | Whole virus | mtDNA | Virus infection induces mtDNA stress (increase in larger nucleoids, decrease in total nucleoid number). | [124] |
| SARS-CoV | 5′/3′ untranslated regions contain putative mitochondrial residency sites | Localization in host mitochondria. | [125] | |
| Viral nonstructured protein Orf9b | Tom70 | Localization in host mitochondria. | [121] | |
| Nsp5 (C145A) | tRNA methyltransferase 1 (TRMT1) | Enzyme for dimethylation of guanosine (m2,2G) on mitochondrial tRNAs. | [121] | |
| ORF9b | Localization in host mitochondria. | [126] | ||
| ORF8a | Localization in host mitochondria, increases in mitochondrial transmembrane potential. | [127] | ||
| Nonstructural protein 2 (Nsp2) | Prohibitin 1 (PHB1) and PHB2 (also other potential host interaction partners identified for Nsp1 and 2) | Mitochondrial biogenesis. | [128] | |
| ORF3a | Mitochondrial ubiquitin-specific peptidase 30 (USP30) | [129] | ||
| Nsp10 | NADH 4L subunit and cytochrome oxidase II | NADH cytochrome activity altered, inner mitochondrial membrane depolarized. | [123] | |
| Global analysis of SARS-CoV-2 host-interacting proteins: 26 of the 29 SARS-CoV-2 proteins) Nsp4, Nsp8, Orf9c, structural protein M | 332 high-confidence SARS-CoV-2-human protein–protein interactions; with mitochondrial relevance: Tim complex, mitochondrial ribosome, electron transport | Mitochondrial function. | [121] | |
| HIV-1 | Tat (transactivator of transcription protein) | ? | Hyperpolarization of mitochondria in cortical neurons. | [130] |
| HIV-1 viral protein R (Vpr) | ? (probably microtubule structure of the host cell) | Disturbed axonal transport. Reduced peroxisome proliferator-activated receptor-gamma coactivator 1 alpha (PGC-1α) expression via increase in the methylation of the PGC-1α promoter. | [119] | |
| Tat and negative-regulating factor (Nef) | ? | Increased ROS production and mitochondrial mass destabilization of the mitochondrial membrane potential. | [131] | |
| Tat | Mitochondrial KATP channels discussed | Mitochondrial hyperpolarization. | [130] | |
| Infection | ? | Increased mitochondrial ROS levels. Decreased ATP-linked respiration. | [132] | |
| HIV gp120 and Tat | Inhibition of mitophagic flux. Increased mitochondrial fragmentation. Increased sequestosome 1 translocation to damaged mitochondria. | [133,134] | ||
| Enterovirus 71 (EV71) | Infection | ? | Decrease in mitochondrial electrochemical potential. ΔΨ(m) increase in oligomycin-insensitive oxygen consumption. Induced ROS production. | [135] |
| Borna disease virus (BDV) | Accessory protein X | ? | Colocalizes with mitochondria. | [136] |
| Hepatitis C virus (HCV) | D2 domain of NS5A (NS5A-D2) | (might be due to glucokinase activation of the host cell) | Reduced spare respiration capacity. | [137] |
| Infection | ? | TERT localization in mitochondria. | [120] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Endres, K.; Friedland, K. Talk to Me—Interplay between Mitochondria and Microbiota in Aging. Int. J. Mol. Sci. 2023, 24, 10818. https://doi.org/10.3390/ijms241310818
Endres K, Friedland K. Talk to Me—Interplay between Mitochondria and Microbiota in Aging. International Journal of Molecular Sciences. 2023; 24(13):10818. https://doi.org/10.3390/ijms241310818
Chicago/Turabian StyleEndres, Kristina, and Kristina Friedland. 2023. "Talk to Me—Interplay between Mitochondria and Microbiota in Aging" International Journal of Molecular Sciences 24, no. 13: 10818. https://doi.org/10.3390/ijms241310818
APA StyleEndres, K., & Friedland, K. (2023). Talk to Me—Interplay between Mitochondria and Microbiota in Aging. International Journal of Molecular Sciences, 24(13), 10818. https://doi.org/10.3390/ijms241310818