
Comparative Study of Terminal Cortical Potentials Using Iridium and Ag/AgCl Electrodes


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
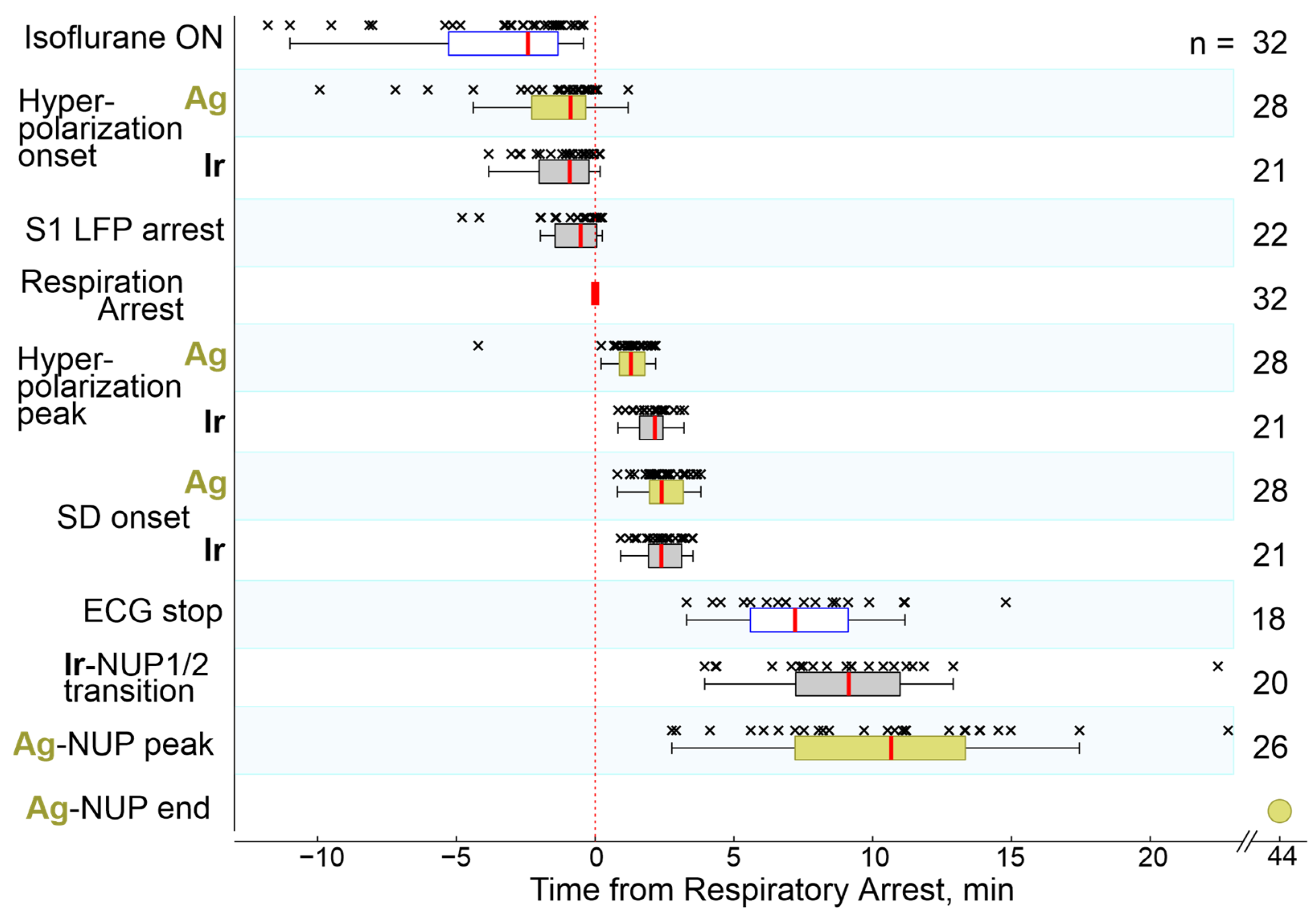
2.1. General Characteristics of the Cascade of Terminal Events
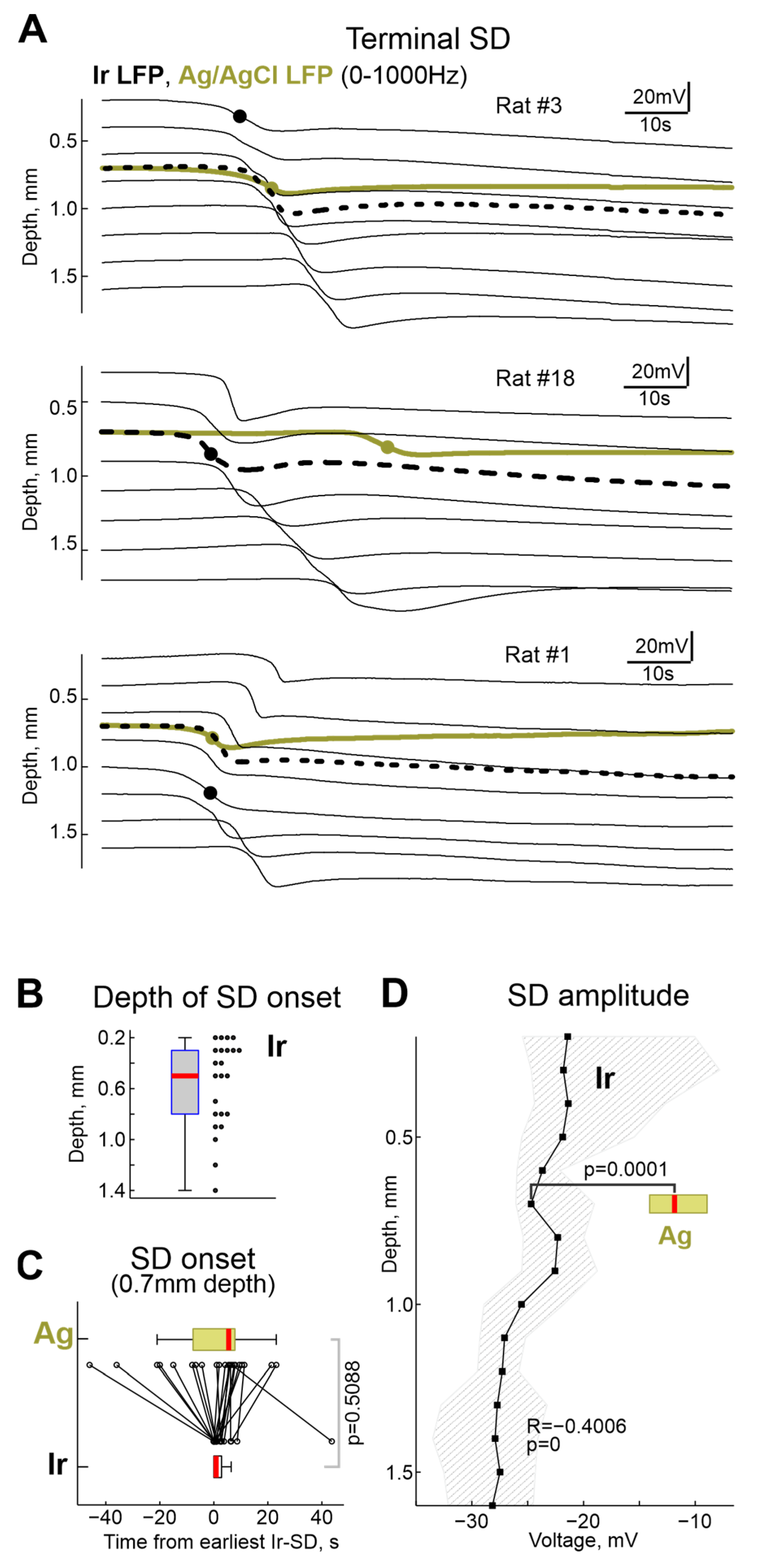
2.2. Terminal SD
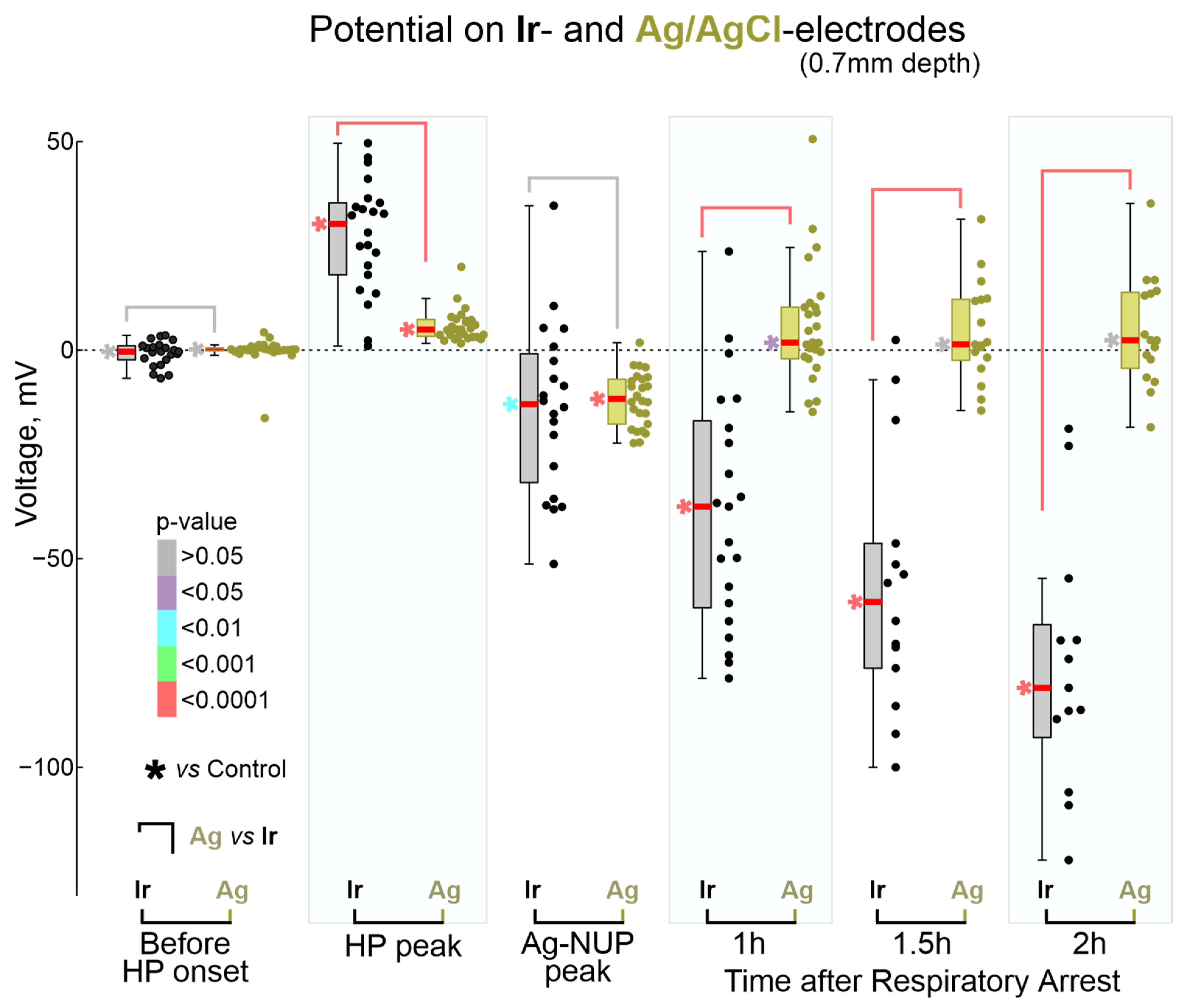
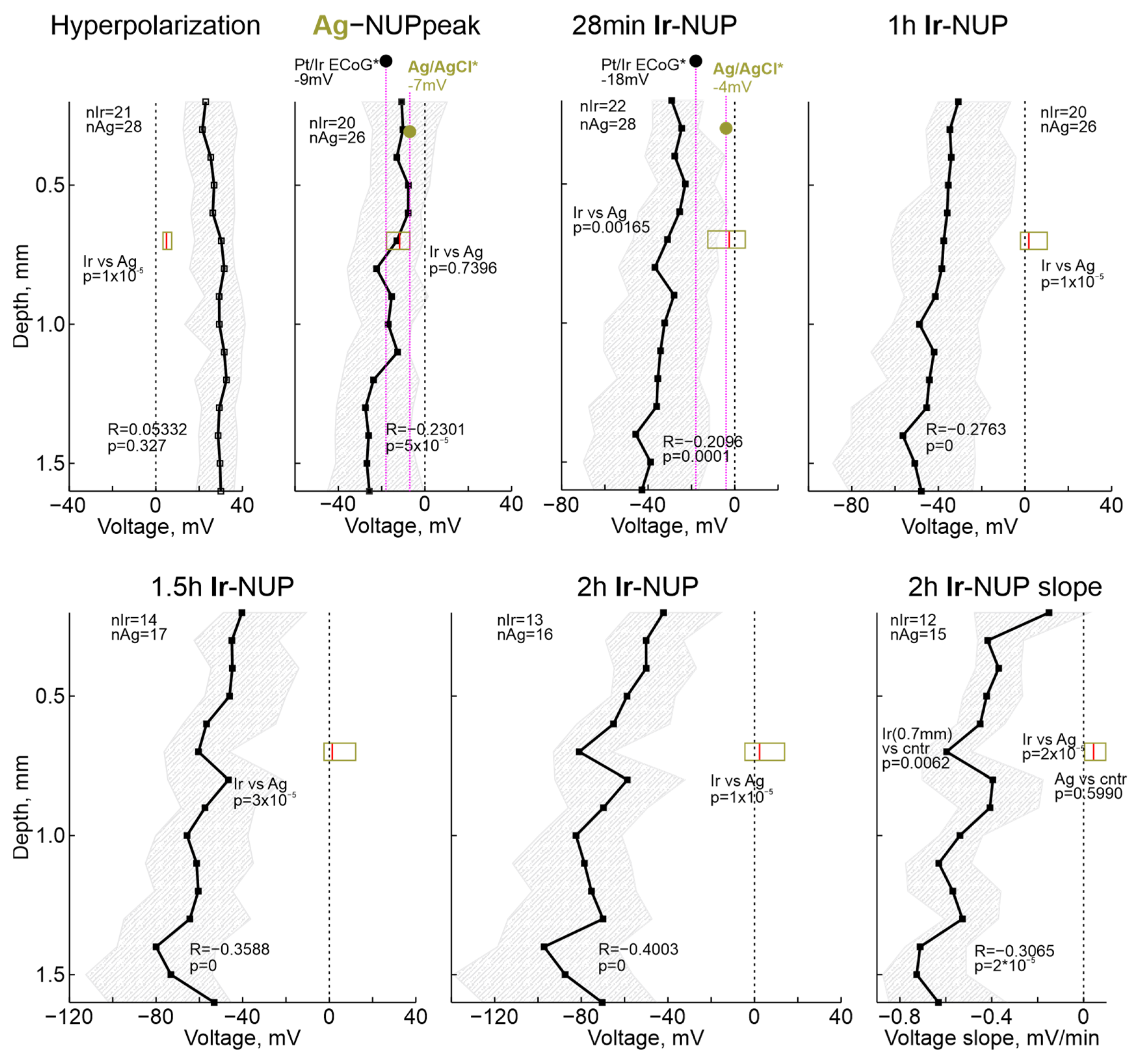
2.3. Terminal NUP
3. Discussion
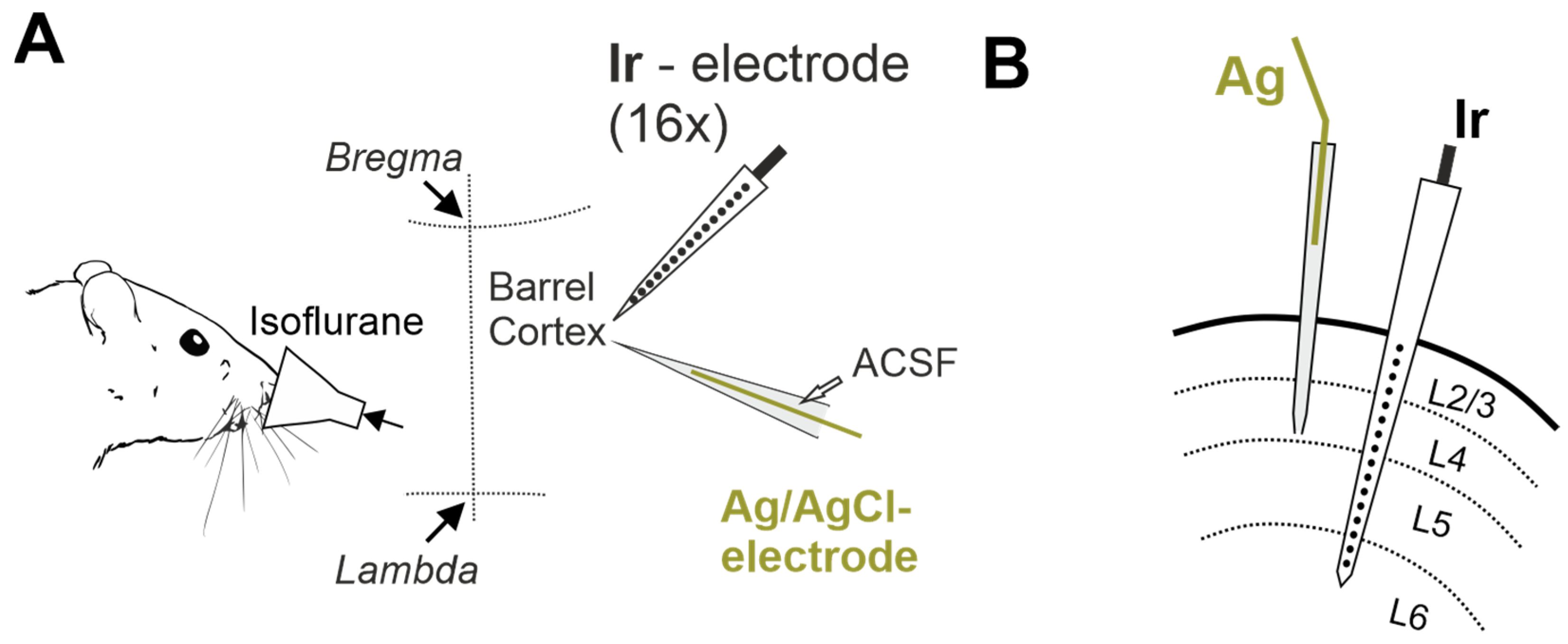
4. Materials and Methods
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Andrew, R.D.; Hartings, J.A.; Ayata, C.; Brennan, K.C.; wson-Scully, K.D.; Farkas, E.; Herreras, O.; Kirov, S.A.; Muller, M.; Ollen-Bittle, N.; et al. The Critical Role of Spreading Depolarizations in Early Brain Injury: Consensus and Contention. Neurocrit. Care 2022, 37, 83–101. [Google Scholar] [CrossRef]
- Ayata, C.; Lauritzen, M. Spreading Depression, Spreading Depolarizations, and the Cerebral Vasculature. Physiol. Rev. 2015, 95, 953–993. [Google Scholar] [CrossRef]
- Dreier, J.P. The role of spreading depression, spreading depolarization and spreading ischemia in neurological disease. Nat. Med. 2011, 17, 439–447. [Google Scholar] [CrossRef] [PubMed]
- Dreier, J.P.; Reiffurth, C. The stroke-migraine depolarization continuum. Neuron 2015, 86, 902–922. [Google Scholar] [CrossRef] [PubMed]
- Lemale, C.L.; Luckl, J.; Horst, V.; Reiffurth, C.; Major, S.; Hecht, N.; Woitzik, J.; Dreier, J.P. Migraine Aura, Transient Ischemic Attacks, Stroke, and Dying of the Brain Share the Same Key Pathophysiological Process in Neurons Driven by Gibbs-Donnan Forces, Namely Spreading Depolarization. Front. Cell. Neurosci. 2022, 16, 837650. [Google Scholar] [CrossRef] [PubMed]
- Giniatullin, R.; Khazipov, R.; van den Maagdenberg, A.; Jolkkonen, J. Common and distinct mechanisms of migraine and stroke. Front. Cell. Neurosci. 2023, 17, 1171836. [Google Scholar] [CrossRef] [PubMed]
- Meinert, F.; Lemale, C.L.G.; Major, S.; Helgers, S.O.A.; Domer, P.; Mencke, R.; Bergold, M.N.; Dreier, J.P.; Hecht, N.; Woitzik, J. Less-invasive subdural electrocorticography for investigation of spreading depolarizations in patients with subarachnoid hemorrhage. Front. Neurol. 2023, 13, 1091987. [Google Scholar] [CrossRef]
- Horst, V.; Kola, V.; Lemale, C.L.; Major, S.; Winkler, M.K.L.; Hecht, N.; Santos, E.; Platz, J.; Sakowitz, O.W.; Vatter, H.; et al. Spreading depolarization and angiographic spasm are separate mediators of delayed infarcts. Brain Commun. 2023, 5, fcad080. [Google Scholar] [CrossRef]
- Leao, A.A.P. Further Observations on the Spreading Depression of Activity in the Cerebral Cortex. J. Neurophysiol. 1947, 10, 409–414. [Google Scholar] [CrossRef]
- LEAO, A.A. The slow voltage variation of cortical spreading depression of activity. Electroencephalogr. Clin. Neurophysiol. 1951, 3, 315–321. [Google Scholar] [CrossRef]
- Strong, A.J.; Fabricius, M.; Boutelle, M.G.; Hibbins, S.J.; Hopwood, S.E.; Jones, R.; Parkin, M.C.; Lauritzen, M. Spreading and synchronous depressions of cortical activity in acutely injured human brain. Stroke 2002, 33, 2738–2743. [Google Scholar] [CrossRef]
- Somjen, G.G. Mechanisms of spreading depression and hypoxic spreading depression-like depolarization. Physiol. Rev. 2001, 81, 1065–1096. [Google Scholar] [CrossRef] [PubMed]
- Dreier, J.P.; Fabricius, M.; Ayata, C.; Sakowitz, O.W.; William, S.C.; Dohmen, C.; Graf, R.; Vajkoczy, P.; Helbok, R.; Suzuki, M.; et al. Recording, analysis, and interpretation of spreading depolarizations in neurointensive care: Review and recommendations of the COSBID research group. J. Cereb. Blood Flow. Metab. 2017, 37, 1595–1625. [Google Scholar] [CrossRef] [PubMed]
- Dreier, J.P.; Winkler, M.K.L.; Major, S.; Horst, V.; Lublinsky, S.; Kola, V.; Lemale, C.L.; Kang, E.J.; Maslarova, A.; Salur, I.; et al. Spreading depolarizations in ischaemia after subarachnoid haemorrhage, a diagnostic phase III study. Brain 2022, 145, 1264–1284. [Google Scholar] [CrossRef] [PubMed]
- Torteli, A.; Toth, R.; Berger, S.; Samardzic, S.; Bari, F.; Menyhart, A.; Farkas, E. Spreading depolarization causes reperfusion failure after cerebral ischemia. J. Cereb. Blood Flow. Metab. 2023, 43, 655–664. [Google Scholar] [CrossRef] [PubMed]
- Alsbrook, D.L.; Di Napoli, M.; Bhatia, K.; Desai, M.; Hinduja, A.; Rubinos, C.A.; Mansueto, G.; Singh, P.; Domeniconi, G.G.; Ikram, A.; et al. Pathophysiology of Early Brain Injury and Its Association with Delayed Cerebral Ischemia in Aneurysmal Subarachnoid Hemorrhage: A Review of Current Literature. J. Clin. Med. 2023, 12, 1015. [Google Scholar] [CrossRef]
- Dreier, J.P.; Isele, T.; Reiffurth, C.; Offenhauser, N.; Kirov, S.A.; Dahlem, M.A.; Herreras, O. Is spreading depolarization characterized by an abrupt, massive release of gibbs free energy from the human brain cortex? Neuroscientist 2013, 19, 25–42. [Google Scholar] [CrossRef]
- Luckl, J.; Lemale, C.L.; Kola, V.; Horst, V.; Khojasteh, U.; Oliveira-Ferreira, A.I.; Major, S.; Winkler, M.K.L.; Kang, E.J.; Schoknecht, K.; et al. The negative ultraslow potential, electrophysiological correlate of infarction in the human cortex. Brain 2018, 141, 1734–1752. [Google Scholar] [CrossRef]
- Dreier, J.P.; Major, S.; Lemale, C.L.; Kola, V.; Reiffurth, C.; Schoknecht, K.; Hecht, N.; Hartings, J.A.; Woitzik, J. Correlates of Spreading Depolarization, Spreading Depression, and Negative Ultraslow Potential in Epidural Versus Subdural Electrocorticography. Front. Neurosci. 2019, 13, 373. [Google Scholar] [CrossRef]
- Carlson, A.P.; Shuttleworth, C.W.; Major, S.; Lemale, C.L.; Dreier, J.P.; Hartings, J.A. Terminal spreading depolarizations causing electrocortical silencing prior to clinical brain death: Case report. J. Neurosurg. 2019, 131, 1773–1779. [Google Scholar] [CrossRef]
- Vinokurova, D.; Zakharov, A.; Chernova, K.; Burkhanova-Zakirova, G.; Horst, V.; Lemale, C.L.; Dreier, J.P.; Khazipov, R. Depth-profile of impairments in endothelin-1—Induced focal cortical ischemia. J. Cereb. Blood Flow. Metab. 2022, 42, 1944–1960. [Google Scholar] [CrossRef] [PubMed]
- Nasretdinov, A.; Evstifeev, A.; Vinokurova, D.; Burkhanova-Zakirova, G.; Chernova, K.; Churina, Z.; Khazipov, R. Full-Band EEG Recordings Using Hybrid AC/DC-Divider Filters. eNeuro 2021, 8, ENEURO.0246-21.2021. [Google Scholar] [CrossRef] [PubMed]
- Nasretdinov, A.; Lotfullina, N.; Vinokurova, D.; Lebedeva, J.; Burkhanova, G.; Chernova, K.; Zakharov, A.; Khazipov, R. Direct Current Coupled Recordings of Cortical Spreading Depression Using Silicone Probes. Front. Cell. Neurosci. 2017, 11, 408. [Google Scholar] [CrossRef]
- Hartikainen, K.; Rorarius, M.; Makela, K.; Yli-Hankala, A.; Jantti, V. Propofol and isoflurane induced EEG burst suppression patterns in rabbits. Acta Anaesthesiol. Scand. 1995, 39, 814–818. [Google Scholar] [CrossRef]
- Ferron, J.F.; Kroeger, D.; Chever, O.; Amzica, F. Cortical inhibition during burst suppression induced with isoflurane anesthesia. J. Neurosci. 2009, 29, 9850–9860. [Google Scholar] [CrossRef] [PubMed]
- Sitdikova, G.; Zakharov, A.; Janackova, S.; Gerasimova, E.; Lebedeva, J.; Inacio, A.R.; Zaynutdinova, D.; Minlebaev, M.; Holmes, G.L.; Khazipov, R. Isoflurane suppresses early cortical activity. Ann. Clin. Transl. Neurol. 2014, 1, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Salmi, M.; Del, G.F.; Minlebaev, M.; Zakharov, A.; Pauly, V.; Perron, P.; Pons-Bennaceur, A.; Corby-Pellegrino, S.; Aniksztejn, L.; Lenck-Santini, P.P.; et al. Impaired vocal communication, sleep-related discharges, and transient alteration of slow-wave sleep in developing mice lacking the GluN2A subunit of N-methyl-d-aspartate receptors. Epilepsia 2019, 60, 1424–1437. [Google Scholar] [CrossRef]
- Juzekaeva, E.; Nasretdinov, A.; Gainutdinov, A.; Sintsov, M.; Mukhtarov, M.; Khazipov, R. Preferential Initiation and Spread of Anoxic Depolarization in Layer 4 of Rat Barrel Cortex. Front. Cell. Neurosci. 2017, 11, 390. [Google Scholar] [CrossRef]
- Basarsky, T.A.; Duffy, S.N.; Andrew, R.D.; MacVicar, B.A. Imaging spreading depression and associated intracellular calcium waves in brain slices. J. Neurosci. 1998, 18, 7189–7199. [Google Scholar] [CrossRef]
- Joshi, I.; Andrew, R.D. Imaging anoxic depolarization during ischemia-like conditions in the mouse hemi-brain slice. J. Neurophysiol. 2001, 85, 414–424. [Google Scholar] [CrossRef]
- Kaufmann, D.; Theriot, J.J.; Zyuzin, J.; Service, C.A.; Chang, J.C.; Tang, Y.T.; Bogdanov, V.B.; Multon, S.; Schoenen, J.; Ju, Y.S.; et al. Heterogeneous incidence and propagation of spreading depolarizations. J. Cereb. Blood Flow. Metab. 2017, 37, 1748–1762. [Google Scholar] [CrossRef] [PubMed]
- Bogdanov, V.B.; Middleton, N.A.; Theriot, J.J.; Parker, P.D.; Abdullah, O.M.; Ju, Y.S.; Hartings, J.A.; Brennan, K.C. Susceptibility of Primary Sensory Cortex to Spreading Depolarizations. J. Neurosci. 2016, 36, 4733–4743. [Google Scholar] [CrossRef] [PubMed]
- Richter, F.; Lehmenkuhler, A. Spreading depression can be restricted to distinct depths of the rat cerebral cortex. Neurosci. Lett. 1993, 152, 65–68. [Google Scholar] [CrossRef] [PubMed]
- Dreier, J.P.; Major, S.; Foreman, B.; Winkler, M.K.L.; Kang, E.J.; Milakara, D.; Lemale, C.L.; DiNapoli, V.; Hinzman, J.M.; Woitzik, J.; et al. Terminal spreading depolarization and electrical silence in death of human cerebral cortex. Ann. Neurol. 2018, 83, 295–310. [Google Scholar] [CrossRef]
- Kroeger, D.; Amzica, F. Hypersensitivity of the anesthesia-induced comatose brain. J. Neurosci. 2007, 27, 10597–10607. [Google Scholar] [CrossRef]
- Dzhala, V.; Ben-Ari, Y.; Khazipov, R. Seizures accelerate anoxia-induced neuronal death in the neonatal rat hippocampus. Ann. Neurol. 2000, 48, 632–640. [Google Scholar] [CrossRef]
- Chen, S.; Mohajerani, M.H.; Xie, Y.; Murphy, T.H. Optogenetic analysis of neuronal excitability during global ischemia reveals selective deficits in sensory processing following reperfusion in mouse cortex. J. Neurosci. 2012, 32, 13510–13519. [Google Scholar] [CrossRef]
- Fowler, J.C. Adenosine antagonists delay hypoxia-induced depression of neuronal activity in hippocampal brain slice. Brain Res. 1989, 490, 378–384. [Google Scholar] [CrossRef]
- Katchman, A.N.; Hershkowitz, N. Adenosine antagonists prevent hypoxia-induced depression of excitatory but not inhibitory synaptic currents. Neurosci. Lett. 1993, 159, 123–126. [Google Scholar] [CrossRef]
- Khazipov, R.; Congar, P.; Ben-Ari, Y. Hippocampal CA1 lacunosum-moleculare interneurons: Comparison of effects of anoxia on excitatory and inhibitory postsynaptic currents. J. Neurophysiol. 1995, 74, 2138–2149. [Google Scholar] [CrossRef]
- Dzhala, V.; Desfreres, L.; Melyan, Z.; Ben-Ari, Y.; Khazipov, R. Epileptogenic action of caffeine during anoxia in the neonatal rat hippocampus. Ann. Neurol. 1999, 46, 95–102. [Google Scholar] [CrossRef]
- Khazipov, R.; Bregestovski, P.; Ben-Ari, Y. Hippocampal inhibitory interneurons are functionally disconnected from excitatory inputs by anoxia. J. Neurophysiol. 1993, 70, 2251–2259. [Google Scholar] [CrossRef] [PubMed]
- Franks, N.P.; Lieb, W.R. Volatile general anaesthetics activate a novel neuronal K+ current. Nature 1988, 333, 662–664. [Google Scholar] [CrossRef]
- Patel, A.J.; Honore, E.; Lesage, F.; Fink, M.; Romey, G.; Lazdunski, M. Inhalational anesthetics activate two-pore-domain background K+ channels. Nat. Neurosci. 1999, 2, 422–426. [Google Scholar] [CrossRef]
- Kuribayashi, J.; Sakuraba, S.; Kashiwagi, M.; Hatori, E.; Tsujita, M.; Hosokawa, Y.; Takeda, J.; Kuwana, S. Neural mechanisms of sevoflurane-induced respiratory depression in newborn rats. Anesthesiology 2008, 109, 233–242. [Google Scholar] [CrossRef] [PubMed]
- Ayata, C. Pearls and pitfalls in experimental models of spreading depression. Cephalalgia 2013, 33, 604–613. [Google Scholar] [CrossRef]
- Schramm, A.E.; Carton-Leclercq, A.; Diallo, S.; Navarro, V.; Chavez, M.; Mahon, S.; Charpier, S. Identifying neuronal correlates of dying and resuscitation in a model of reversible brain anoxia. Prog. Neurobiol. 2020, 185, 101733. [Google Scholar] [CrossRef] [PubMed]
- Lipton, P. Ischemic cell death in brain neurons. Physiol. Rev. 1999, 79, 1431–1568. [Google Scholar] [CrossRef]
- Zakharov, A.; Chernova, K.; Burkhanova, G.; Holmes, G.L.; Khazipov, R. Segregation of seizures and spreading depolarization across cortical layers. Epilepsia 2019, 60, 2386–2397. [Google Scholar] [CrossRef]
- Leao, A.A.P. Spreading depression of activity in the cerebral cortex. J. Neurophysiol. 1944, 7, 359–390. [Google Scholar] [CrossRef]
- Gainutdinov, A.; Juzekaeva, E.; Mukhtarov, M.; Khazipov, R. Anoxic spreading depolarization in the neonatal rat cortex in vitro. Front. Cell. Neurosci. 2023, 17, 1106268. [Google Scholar] [CrossRef]
- Juzekaeva, E.; Gainutdinov, A.; Mukhtarov, M.; Khazipov, R. Reappraisal of anoxic spreading depolarization as a terminal event during oxygen-glucose deprivation in brain slices in vitro. Sci. Rep. 2020, 10, 18970. [Google Scholar] [CrossRef]
- Murphy, T.H.; Li, P.; Betts, K.; Liu, R. Two-photon imaging of stroke onset in vivo reveals that NMDA-receptor independent ischemic depolarization is the major cause of rapid reversible damage to dendrites and spines. J. Neurosci. 2008, 28, 1756–1772. [Google Scholar] [CrossRef]
- Major, S.; Gajovic-Eichelmann, N.; Woitzik, J.; Dreier, J.P. Oxygen-Induced and pH-Induced Direct Current Artifacts on Invasive Platinum/Iridium Electrodes for Electrocorticography. Neurocrit. Care 2021, 35, 146–159. [Google Scholar] [CrossRef] [PubMed]
- Vinokurova, D.; Mingazov, B.; Zaharov, A.; Khazipov, R. Negative ultraslow potentials during kidney ischemia. Eur. J. Clin. Investig. 2022, 52, 1. [Google Scholar]
- Mutch, W.A.; Hansen, A.J. Extracellular pH changes during spreading depression and cerebral ischemia: Mechanisms of brain pH regulation. J. Cereb. Blood Flow. Metab. 1984, 4, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Cogan, S.F. Neural stimulation and recording electrodes. Annu. Rev. Biomed. Eng. 2008, 10, 275–309. [Google Scholar] [CrossRef]
- Elkin, B.S.; Shaik, M.A.; Morrison, B., 3rd. Fixed negative charge and the Donnan effect: A description of the driving forces associated with brain tissue swelling and oedema. Philos. Trans. A Math. Phys. Eng. Sci. 2010, 368, 585–603. [Google Scholar] [CrossRef] [PubMed]
- Stokum, J.A.; Gerzanich, V.; Simard, J.M. Molecular pathophysiology of cerebral edema. J. Cereb. Blood Flow. Metab. 2016, 36, 513–538. [Google Scholar] [CrossRef]
- Juzekaeva, E.; Gainutdinov, A.; Mukhtarov, M.; Khazipov, R. Dynamics of the Hypoxia-Induced Tissue Edema in the Rat Barrel Cortex in vitro. Front. Cell. Neurosci. 2018, 12, 502. [Google Scholar] [CrossRef]
- Charpak, S.; Audinat, E. Cardiac arrest in rodents: Maximal duration compatible with a recovery of neuronal activity. Proc. Natl. Acad. Sci. USA 1998, 95, 4748–4753. [Google Scholar] [CrossRef] [PubMed]
- Bratsch, S.G. Standard Electrode-Potentials and Temperature Coefficients in Water at 298.15-K. J. Phys. Chem. Ref. Data 1989, 18, 1–21. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mingazov, B.; Vinokurova, D.; Zakharov, A.; Khazipov, R. Comparative Study of Terminal Cortical Potentials Using Iridium and Ag/AgCl Electrodes. Int. J. Mol. Sci. 2023, 24, 10769. https://doi.org/10.3390/ijms241310769
Mingazov B, Vinokurova D, Zakharov A, Khazipov R. Comparative Study of Terminal Cortical Potentials Using Iridium and Ag/AgCl Electrodes. International Journal of Molecular Sciences. 2023; 24(13):10769. https://doi.org/10.3390/ijms241310769
Chicago/Turabian StyleMingazov, Bulat, Daria Vinokurova, Andrei Zakharov, and Roustem Khazipov. 2023. "Comparative Study of Terminal Cortical Potentials Using Iridium and Ag/AgCl Electrodes" International Journal of Molecular Sciences 24, no. 13: 10769. https://doi.org/10.3390/ijms241310769
APA StyleMingazov, B., Vinokurova, D., Zakharov, A., & Khazipov, R. (2023). Comparative Study of Terminal Cortical Potentials Using Iridium and Ag/AgCl Electrodes. International Journal of Molecular Sciences, 24(13), 10769. https://doi.org/10.3390/ijms241310769