
Pain Biomarkers in Fibromyalgia Syndrome: Current Understanding and Future Directions



Abstract
1. Introduction
2. Pathophysiology of Pain in FM Syndrome
2.1. Central Sensitization
2.2. Neuroinflammation
2.3. Neuroendocrine and Autonomic Nervous System Dysfunction
3. Biomarkers of Pain in Fibromyalgia Syndrome
3.1. Glutamate
3.2. Substance P
3.3. Nerve Growth Factor
3.4. Brain-Derived Neurotrophic Factor
3.5. Mu Opioid Receptor
3.6. Mast Cells and Cytokine Production
3.7. Pentraxin-3
3.8. Neuropeptide Y
4. Potential Future Directions
4.1. Vitamin D
4.2. Gut Microbiome
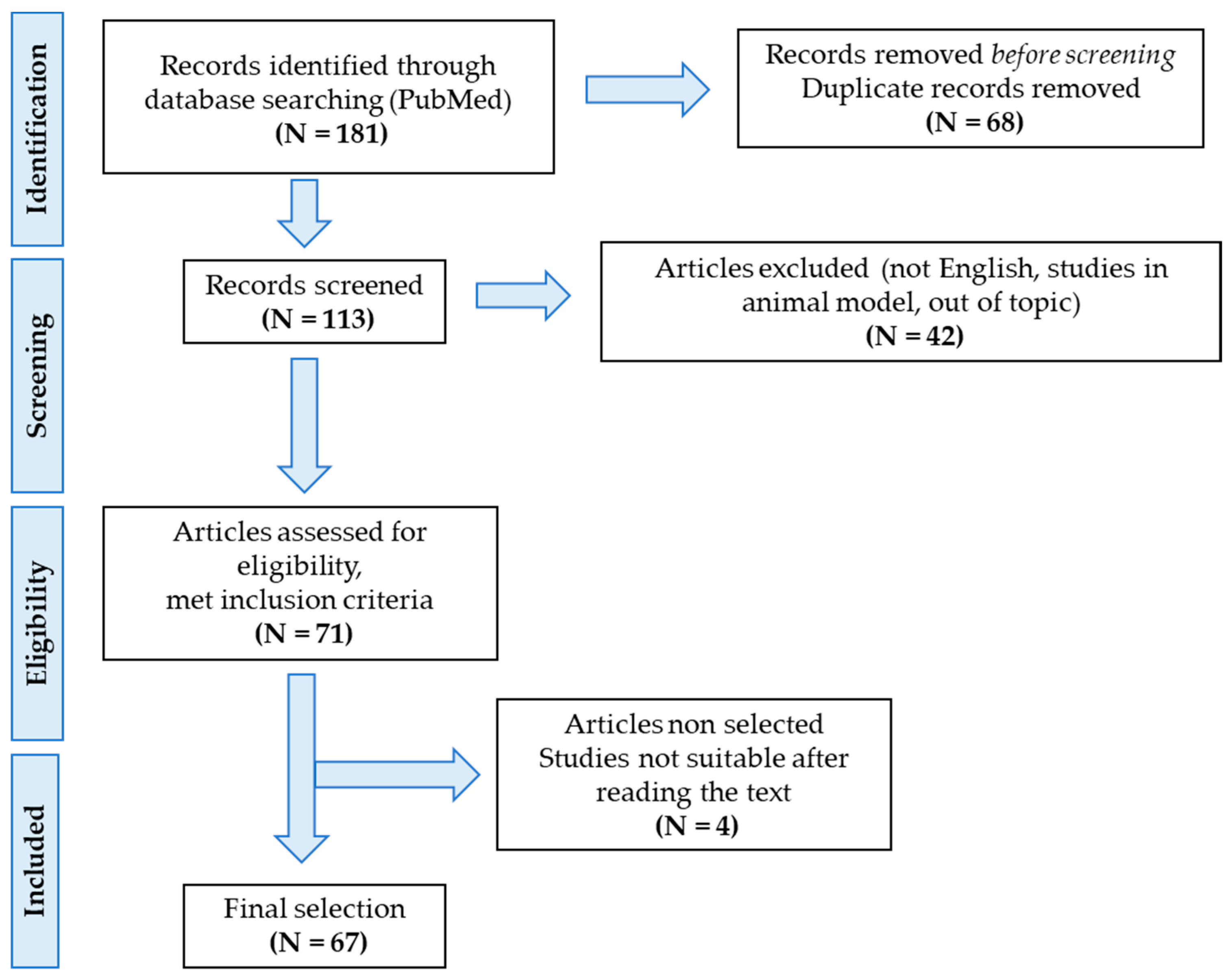
5. Materials and Methods
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ACTH | Adrenocorticotropic hormone |
| ACR | American College of Rheumatology |
| AMPA-R | Amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors |
| ANS | Autonomic nervous system |
| ATP | Adenosine triphosphate |
| BBB | Blood–brain barrier |
| BDNF | Brain-derived neurotrophic factor |
| CGRP | Calcitonin gene-related peptide |
| COMT | Catechol-O-methyl transferase |
| CNS | Central nervous system |
| CPM | Conditioned pain modulation |
| CPP | Chronic primary pain |
| CRH | Corticotropin releasing hormone |
| CS | Central sensitization |
| CSF | Cerebrospinal fluid |
| CSS | Central sensitization syndromes |
| CXCL | Chemokine ligand |
| DH | Dorsal horn |
| DRG | Dorsal root ganglia |
| FM | Fibromyalgia |
| fMRI | Functional magnetic resonance imaging |
| Glu | Glutamate |
| HC | Healthy control |
| 1H-MRS | Proton magnetic resonance spectroscopy |
| HPA | Hypothalamic–pituitary–adrenal |
| HRV | Heart rate variability |
| IASP | International Association for the Study of Pain |
| IL | Interleukin |
| KCC2 | Potassium-chloride cotransporter 2 |
| LTP | Long-term potentiation |
| MCs | Mast cells |
| MCP | Monocyte chemotactic protein |
| MOR | Mu opioid receptor |
| NGF | Nerve growth factor |
| NK | Neurokinin |
| NMDA-R | N-methyl-D-aspartate receptor |
| NPY | Neuropeptide Y |
| PAG | Periaqueductal grey |
| PET | Positron emission tomography |
| PFC | Prefrontal cortex |
| PNS | Peripheral nervous system |
| PTX-3 | Pentraxin-3 |
| rs-fMRI | Resting-state functional magnetic resonance imaging |
| SFN | Small fiber neuropathy |
| SNP | Single-nucleotide polymorphism |
| SP | Substance P |
| SSS | Symptom Severity Scale |
| TLR | Toll-like receptor |
| TNF | Tumor necrosis factor |
| Trk | Tyrosine-kinase |
| VBM | Voxel-based morphometry |
| VDR | Vitamin D receptor |
| WP | Widespread pain |
| WPI | Widespread Pain Index |
References
- Queiroz, L.P. Worldwide Epidemiology of Fibromyalgia. Curr. Pain Headache Rep. 2013, 17, 356. [Google Scholar] [CrossRef] [PubMed]
- Di Franco, M.; Iannuccelli, C.; Bazzichi, L.; Atzeni, F.; Consensi, A.; Salaffi, F.; Pietropaolo, M.; Alessandri, C.; Basili, S.; Olivieri, M.; et al. Misdiagnosis in fibromyalgia: A multicentre study. Ann. Rheum. Dis. 2012, 29, S104–S108. [Google Scholar]
- Häuser, W.; Sarzi-Puttini, P.; Fitzcharles, M.A. Fibromyalgia syndrome: Under-, over- and misdiagnosis. Clin. Exp. Rheumatol. 2019, 37, 90–97. [Google Scholar]
- Giorgi, V.; Sirotti, S.; Romano, M.E.; Marotto, D.; Ablin, J.N.; Salaffi, F.; Sarzi-Puttini, P. Fibromyalgia: One year in review 2022. Clin. Exp. Rheumatol. 2022, 40, 1065–1072. [Google Scholar] [CrossRef] [PubMed]
- Sarzi-Puttini, P.; Giorgi, V.; Marotto, D.; Atzeni, F. Fibromyalgia: An update on clinical characteristics, aetiopathogenesis and treatment. Nat. Rev. Rheumatol. 2020, 16, 645–660. [Google Scholar] [CrossRef]
- Wolfe, F.; Smythe, H.A.; Yunus, M.B.; Bennett, R.M.; Bombardier, C.; Goldenberg, D.L.; Tugwell, P.; Campbell, S.M.; Abeles, M.; Clark, P.; et al. The american college of rheumatology 1990 Criteria for the classification of fibromyalgia. Report of the Multicenter Criteria Committee. Arthritis Rheumatol. 1990, 33, 160–172. [Google Scholar] [CrossRef]
- Wolfe, F.; Clauw, D.J.; Fitzcharles, M.-A.; Goldenberg, D.L.; Katz, R.S.; Mease, P.; Russell, A.S.; Russell, I.J.; Winfield, J.B.; Yunus, M.B. The American College of Rheumatology Preliminary Diagnostic Criteria for Fibromyalgia and Measurement of Symptom Severity. Arthritis Care Res. 2010, 62, 600–610. [Google Scholar] [CrossRef]
- Wolfe, F.; Clauw, D.J.; Fitzcharles, M.A.; Goldenberg, D.L.; Häuser, W.; Katz, R.L.; Mease, P.J.; Russell, A.S.; Russell, I.J.; Walitt, B. 2016 Revisions to the 2010/2011 fibromyalgia diagnostic criteria. Semin. Arthritis. Rheum. 2016, 46, 319–329. [Google Scholar] [CrossRef]
- Treede, R.-D.; Rief, W.; Barke, A.; Aziz, Q.; Bennett, M.I.; Benoliel, R.; Cohen, M.; Evers, S.; Finnerup, N.B.; First, M.B.; et al. Chronic pain as a symptom or a disease: The IASP Classification of Chronic Pain for the International Classification of Diseases (ICD-11). Pain 2019, 160, 19–27. [Google Scholar] [CrossRef]
- Nicholas, M.; Vlaeyen, J.W.; Rief, W.; Barke, A.; Aziz, Q.; Benoliel, R.; Cohen, M.; Evers, S.; Giamberardino, M.A.; Goebel, A.; et al. The IASP classification of chronic pain for ICD-11: Chronic primary pain. Pain 2019, 160, 28–37. [Google Scholar] [CrossRef]
- Kosek, E.; Cohen, M.; Baron, R.; Gebhart, G.F.; Mico, J.-A.; Rice, A.S.C.; Rief, W.; Sluka, A.K. Do we need a third mechanistic descriptor for chronic pain states? Pain 2016, 157, 1382–1386. [Google Scholar] [CrossRef]
- Trouvin, A.-P.; Perrot, S. New concepts of pain. Best Pract. Res. Clin. Rheumatol. 2019, 33, 101415. [Google Scholar] [CrossRef]
- Fitzcharles, M.-A.; Cohen, S.P.; Clauw, D.J.; Littlejohn, G.; Usui, C.; Häuser, W. Nociplastic pain: Towards an understanding of prevalent pain conditions. Lancet 2021, 397, 2098–2110. [Google Scholar] [CrossRef] [PubMed]
- Üçeyler, N.; Zeller, D.; Kahn, A.-K.; Kewenig, S.; Kittel-Schneider, S.; Schmid, A.; Casanova-Molla, J.; Reiners, K.; Sommer, C. Small fibre pathology in patients with fibromyalgia syndrome. Brain 2013, 136, 1857–1867. [Google Scholar] [CrossRef]
- Caro, X.J.; Winter, E.F. Evidence of Abnormal Epidermal Nerve Fiber Density in Fibromyalgia: Clinical and Immunologic Implications. Arthritis Rheumatol. 2014, 66, 1945–1954. [Google Scholar] [CrossRef]
- Giannoccaro, M.P.; Donadio, V.; Incensi, A.; Avoni, P.; Liguori, R. Small nerve fiber involvement in patients referred for fibromyalgia. Muscle Nerve 2014, 49, 757–759. [Google Scholar] [CrossRef] [PubMed]
- Lawson, V.H.; Grewal, J.; Hackshaw, K.V.; Mongiovi, P.C.; Stino, A.M. Fibromyalgia syndrome and small fiber, early or mild sensory polyneuropathy. Muscle Nerve 2018, 58, 625–630. [Google Scholar] [CrossRef] [PubMed]
- Van Assche, D.C.F.; Plaghki, L.; Masquelier, E.; Hatem, S.M. Fibromyalgia syndrome—A laser-evoked potentials study unsupportive of small nerve fibre involvement. Eur. J. Pain 2019, 24, 448–456. [Google Scholar] [CrossRef]
- Fasolino, A.; Di Stefano, G.; Leone, C.; Galosi, E.; Gioia, C.; Lucchino, B.; Terracciano, A.; Di Franco, M.; Cruccu, G.; Truini, A. Small-fibre pathology has no impact on somatosensory system function in patients with fibromyalgia. Pain 2020, 161, 2385–2393. [Google Scholar] [CrossRef]
- de Tommaso, M.; Vecchio, E.; Nolano, M. The puzzle of fibromyalgia between central sensitization syndrome and small fiber neuropathy: A narrative review on neurophysiological and morphological evidence. Neurol. Sci. 2022, 43, 1667–1684. [Google Scholar] [CrossRef]
- Staud, R.; Rodriguez, M.E. Mechanisms of Disease: Pain in fibromyalgia syndrome. Nat. Clin. Pract. Rheumatol. 2006, 2, 90–98. [Google Scholar] [CrossRef]
- Schmidt-Wilcke, T.; Clauw, D.J. Fibromyalgia: From pathophysiology to therapy. Nat. Rev. Rheumatol. 2011, 7, 518–527. [Google Scholar] [CrossRef] [PubMed]
- Bradley, L.A. Pathophysiology of Fibromyalgia. Am. J. Med. 2009, 122 (Suppl. 12), S22–S30. [Google Scholar] [CrossRef]
- Staud, R.; Smitherman, M.L. Peripheral and central sensitization in fibromyalgia: Pathogenetic role. Curr. Pain Headache Rep. 2002, 6, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Yam, M.F.; Loh, Y.C.; Tan, C.S.; Adam, S.K.; Manan, N.A.; Basir, R. General Pathways of Pain Sensation and the Major Neurotransmitters Involved in Pain Regulation. Int. J. Mol. Sci. 2018, 19, 2164. [Google Scholar] [CrossRef] [PubMed]
- Todd, A.J. Neuronal circuitry for pain processing in the dorsal horn. Nat. Rev. Neurosci. 2010, 11, 823–836. [Google Scholar] [CrossRef]
- Heinricher, M.; Tavares, I.; Leith, J.; Lumb, B. Descending control of nociception: Specificity, recruitment and plasticity. Brain Res. Rev. 2009, 60, 214–225. [Google Scholar] [CrossRef]
- Di Maio, V.; Ventriglia, F.; Santillo, S. A model of cooperative effect of AMPA and NMDA receptors in glutamatergic synapses. Cogn. Neurodyn. 2016, 10, 315–325. [Google Scholar] [CrossRef]
- Liu, X.J.; Salter, M.W. Glutamate receptor phosphorylation and trafficking in pain plasticity in spinal cord dorsal horn. Eur. J. Neurosci. 2010, 32, 278–289. [Google Scholar] [CrossRef]
- Latremoliere, A.; Woolf, C.J. Central Sensitization: A Generator of Pain Hypersensitivity by Central Neural Plasticity. J. Pain 2009, 10, 895–926. [Google Scholar] [CrossRef]
- Wang, Z.; Yuan, M.; Xiao, J. Gray Matter Abnormalities in Patients with Chronic Primary Pain: A Coordinate-Based Meta-Analysis. Pain Physician 2022, 25, 1–13. [Google Scholar]
- Cagnie, B.; Coppieters, I.; Denecker, S.; Six, J.; Danneels, L.; Meeus, M. Central sensitization in fibromyalgia? A systematic review on structural and functional brain MRI. Semin. Arthritis Rheum. 2014, 44, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Siracusa, R.; Di Paola, R.; Cuzzocrea, S.; Impellizzeri, D. Fibromyalgia: Pathogenesis, Mechanisms, Diagnosis and Treatment Options Update. Int. J. Mol. Sci. 2021, 22, 3891. [Google Scholar] [CrossRef] [PubMed]
- Truini, A.; Tinelli, E.; Gerardi, M.C.; Calistri, V.; Iannuccelli, C.; La Cesa, S.; Tarsitani, L.; Mainero, C.; Sarzi-Puttini, P.; Cruccu, G.; et al. Abnormal resting state functional connectivity of the periaqueductal grey in patients with fibromyalgia. Clin. Exp. Rheumatol. 2016, 34 (Suppl. 36), S129–S133. [Google Scholar]
- Baraniuk, J.N.; Whalen, G.; Cunningham, J.; Clauw, D.J. Cerebrospinal fluid levels of opioid peptides in fibromyalgia and chronic low back pain. BMC Musculoskelet. Disord. 2004, 5, 48. [Google Scholar] [CrossRef] [PubMed]
- Sluka, K.A.; Clauw, D.J. Neurobiology of fibromyalgia and chronic widespread pain. Neuroscience 2016, 338, 114–129. [Google Scholar] [CrossRef]
- Cifre, I.; Sitges, C.; Fraiman, D.; Muñoz, M.Á.; Balenzuela, P.; González-Roldán, A.; Martínez-Jauand, M.; Birbaumer, N.; Chialvo, D.R.; Montoya, P. Disrupted Functional Connectivity of the Pain Network in Fibromyalgia. Psychosom. Med. 2012, 74, 55–62. [Google Scholar] [CrossRef]
- Ji, R.-R.; Chamessian, A.; Zhang, Y.-Q. Pain regulation by non-neuronal cells and inflammation. Science 2016, 354, 572–577. [Google Scholar] [CrossRef]
- Ji, R.-R.; Nackley, A.; Huh, B.Y.; Terrando, N.; Maixner, D.W. Neuroinflammation and Central Sensitization in Chronic and Widespread Pain. Anesthesiology 2018, 129, 343–366. [Google Scholar] [CrossRef]
- Mai, L.; Liu, Q.; Huang, F.; He, H.; Fan, W. Involvement of Mast Cells in the Pathophysiology of Pain. Front. Cell. Neurosci. 2021, 15, 665066. [Google Scholar] [CrossRef]
- Traina, G. Mast Cells in Gut and Brain and Their Potential Role as an Emerging Therapeutic Target for Neural Diseases. Front. Cell. Neurosci. 2019, 13, 345. [Google Scholar] [CrossRef]
- Skaper, S.D.; Facci, L. Mast cell–glia axis in neuroinflammation and therapeutic potential of the anandamide congener palmitoylethanolamide. Philos. Trans. R. Soc. B Biol. Sci. 2012, 367, 3312–3325. [Google Scholar] [CrossRef]
- Salcman, B.; Affleck, K.; Bulfone-Paus, S. P2X Receptor-Dependent Modulation of Mast Cell and Glial Cell Activities in Neuroinflammation. Cells 2021, 10, 2282. [Google Scholar] [CrossRef] [PubMed]
- Skaper, S.D.; Facci, L.; Zusso, M.; Giusti, P. Neuroinflammation, Mast Cells, and Glia: Dangerous Liaisons. Neuroscientist 2017, 23, 478–498. [Google Scholar] [CrossRef] [PubMed]
- Qian, Y.; Dong, H.; Zhang, X. Mast Cells and Neuroinflammation. Med. Sci. Monit. Basic Res. 2014, 20, 200–206. [Google Scholar] [CrossRef]
- Grace, P.M.; Hutchinson, M.R.; Maier, S.F.; Watkins, L.R. Pathological pain and the neuroimmune interface. Nat. Rev. Immunol. 2014, 14, 217–231. [Google Scholar] [CrossRef]
- Ji, R.-R.; Berta, T.; Nedergaard, M. Glia and pain: Is chronic pain a gliopathy? Pain 2013, 154, S10–S28. [Google Scholar] [CrossRef]
- Zhou, R.; Ji, B.; Kong, Y.; Qin, L.; Ren, W.; Guan, Y.; Ni, R. PET Imaging of Neuroinflammation in Alzheimer’s Disease. Front. Immunol. 2021, 12, 739130. [Google Scholar] [CrossRef]
- Zhang, P.-F.; Gao, F. Neuroinflammation in Parkinson’s disease: A meta-analysis of PET imaging studies. J. Neurol. 2021, 269, 2304–2314. [Google Scholar] [CrossRef]
- Meyer, J.H.; Cervenka, S.; Kim, M.-J.; Kreisl, W.C.; Henter, I.D.; Innis, R.B. Neuroinflammation in psychiatric disorders: PET imaging and promising new targets. Lancet Psychiatry 2020, 7, 1064–1074. [Google Scholar] [CrossRef]
- Gritti, D.; Delvecchio, G.; Ferro, A.; Bressi, C.; Brambilla, P. Neuroinflammation in Major Depressive Disorder: A Review of PET Imaging Studies Examining the 18-kDa Translocator Protein. J. Affect. Disord. 2021, 292, 642–651. [Google Scholar] [CrossRef] [PubMed]
- Albrecht, D.S.; Granziera, C.; Hooker, J.M.; Loggia, M.L. In Vivo Imaging of Human Neuroinflammation. ACS Chem. Neurosci. 2016, 7, 470–483. [Google Scholar] [CrossRef] [PubMed]
- Jung, C.; Ichesco, E.; Ratai, E.-M.; Gonzalez, R.G.; Burdo, T.; Loggia, M.L.; Harris, R.E.; Napadow, V. Magnetic resonance imaging of neuroinflammation in chronic pain: A role for astrogliosis? Pain 2020, 161, 1555–1564. [Google Scholar] [CrossRef]
- Petrou, M.; Harris, R.; Foerster, B.; McLean, S.; Sen, A.; Clauw, D.; Sundgren, P. Proton MR Spectroscopy in the Evaluation of Cerebral Metabolism in Patients with Fibromyalgia: Comparison with Healthy Controls and Correlation with Symptom Severity. Am. J. Neuroradiol. 2008, 29, 913–918. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, M.; Huh, Y.; Ji, R.-R. Roles of inflammation, neurogenic inflammation, and neuroinflammation in pain. J. Anesth. 2018, 33, 131–139. [Google Scholar] [CrossRef]
- Littlejohn, G. Neurogenic neuroinflammation in fibromyalgia and complex regional pain syndrome. Nat. Rev. Rheumatol. 2015, 11, 639–648. [Google Scholar] [CrossRef]
- Deussing, J.M.; Chen, A. The Corticotropin-Releasing Factor Family: Physiology of the Stress Response. Physiol. Rev. 2018, 98, 2225–2286. [Google Scholar] [CrossRef]
- Butler, R.K.; Finn, D.P. Stress-induced analgesia. Prog. Neurobiol. 2009, 88, 184–202. [Google Scholar] [CrossRef]
- Schlereth, T.; Birklein, F. The Sympathetic Nervous System and Pain. NeuroMol. Med. 2007, 10, 141–147. [Google Scholar] [CrossRef]
- Di Franco, M.; Iannuccelli, C.; Valesini, G. Neuroendocrine immunology of fibromyalgia. Ann. N. Y. Acad. Sci. 2010, 1193, 84–90. [Google Scholar] [CrossRef]
- Vachon-Presseau, E. Effects of stress on the corticolimbic system: Implications for chronic pain. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2018, 87, 216–223. [Google Scholar] [CrossRef]
- Timmers, I.; Quaedflieg, C.W.; Hsu, C.; Heathcote, L.C.; Rovnaghi, C.R.; Simons, L.E. The interaction between stress and chronic pain through the lens of threat learning. Neurosci. Biobehav. Rev. 2019, 107, 641–655. [Google Scholar] [CrossRef] [PubMed]
- Woda, A.; Picard, P.; Dutheil, F. Dysfunctional stress responses in chronic pain. Psychoneuroendocrinology 2016, 71, 127–135. [Google Scholar] [CrossRef]
- Singh, L.; Kaur, A.; Bhatti, M.S.; Bhatti, R. Possible Molecular Mediators Involved and Mechanistic Insight into Fibromyalgia and Associated Co-morbidities. Neurochem. Res. 2019, 44, 1517–1532. [Google Scholar] [CrossRef]
- D’agnelli, S.; Arendt-Nielsen, L.; Gerra, M.C.; Zatorri, K.; Boggiani, L.; Baciarello, M.; Bignami, E. Fibromyalgia: Genetics and epigenetics insights may provide the basis for the development of diagnostic biomarkers. Mol. Pain 2018, 15, 1744806918819944. [Google Scholar] [CrossRef]
- Zouikr, I.; Karshikoff, B. Lifetime Modulation of the Pain System via Neuroimmune and Neuroendocrine Interactions. Front. Immunol. 2017, 8, 276. [Google Scholar] [CrossRef] [PubMed]
- Park, D.-J.; Kang, J.-H.; Yim, Y.-R.; Kim, J.-E.; Lee, J.-W.; Lee, K.-E.; Wen, L.; Kim, T.-J.; Park, Y.-W.; Lee, S.-S. Exploring Genetic Susceptibility to Fibromyalgia. Chonnam Med. J. 2015, 51, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Park, D.-J.; Lee, S.-S. New insights into the genetics of fibromyalgia. Korean J. Intern. Med. 2017, 32, 984–995. [Google Scholar] [CrossRef] [PubMed]
- Tammimäki, A.; Männistö, P. Catechol-O-methyltransferase gene polymorphism and chronic human pain: A systematic re-view and meta-analysis. Pharmacogenet. Genom. 2012, 22, 673–691. [Google Scholar] [CrossRef]
- Lee, Y.H.; Kim, J.-H.; Song, G.G. Association between the COMT Val158Met polymorphism and fibromyalgia susceptibility and fibromyalgia impact questionnaire score: A meta-analysis. Rheumatol. Int. 2014, 35, 159–166. [Google Scholar] [CrossRef]
- Zhang, L.; Zhu, J.; Chen, Y.; Zhao, J. Meta-analysis reveals a lack of association between a common catechol-O-methyltransferase (COMT) polymorphism val¹⁵⁸met and fibromyalgia. Int. J. Clin. Exp. Pathol. 2014, 7, 8489–8497. [Google Scholar] [PubMed]
- Peek, A.L.; Rebbeck, T.; Puts, N.A.; Watson, J.; Aguila, M.-E.R.; Leaver, A.M. Brain GABA and glutamate levels across pain conditions: A systematic literature review and meta-analysis of 1H-MRS studies using the MRS-Q quality assessment tool. Neuroimage 2020, 210, 116532. [Google Scholar] [CrossRef] [PubMed]
- Lesnak, J.; Sluka, K.A. Chronic non-inflammatory muscle pain: Central and peripheral mediators. Curr. Opin. Physiol. 2019, 11, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Peres, M.; Zukerman, E.; Soares, C.S.; Alonso, E.; Santos, B.; Faulhaber, M. Cerebrospinal Fluid Glutamate Levels in Chronic Migraine. Cephalalgia 2004, 24, 735–739. [Google Scholar] [CrossRef] [PubMed]
- Sarchielli, P.; Mancini, M.L.; Floridi, A.; Coppola, F.; Rossi, C.; Nardi, K.; Acciarresi, M.; Pini, L.A.; Calabresi, P. Increased Levels of Neurotrophins Are Not Specific for Chronic Migraine: Evidence From Primary Fibromyalgia Syndrome. J. Pain 2007, 8, 737–745. [Google Scholar] [CrossRef]
- Harris, R.E.; Sundgren, P.; Craig, A.; Kirshenbaum, E.; Sen, A.; Napadow, V.; Clauw, D.J. Elevated insular glutamate in fibromyalgia is associated with experimental pain. Arthritis Rheum. 2009, 60, 3146–3152. [Google Scholar] [CrossRef]
- Harris, R.E.; Sundgren, P.; Pang, Y.; Hsu, M.; Petrou, M.; Kim, S.-H.; McLean, S.; Gracely, R.H.; Clauw, D.J. Dynamic levels of glutamate within the insula are associated with improvements in multiple pain domains in fibromyalgia. Arthritis Rheum. 2008, 58, 903–907. [Google Scholar] [CrossRef]
- Fayed, N.; Garcia-Campayo, J.; Magallón, R.; Andrés-Bergareche, H.; Luciano, J.V.; Andres, E.; Beltrán, J. Localized 1H-NMR spectroscopy in patients with fibromyalgia: A controlled study of changes in cerebral glutamate/glutamine, inositol, choline, and N-acetylaspartate. Thromb. Haemost. 2010, 12, R134. [Google Scholar] [CrossRef]
- Valdés, M.; Collado, A.; Bargalló, N.; Vázquez, M.; Rami, L.; Gómez, E.; Salamero, M. Increased glutamate/glutamine compounds in the brains of patients with fibromyalgia: A magnetic resonance spectroscopy study. Arthritis Rheum. 2010, 62, 1829–1836. [Google Scholar] [CrossRef]
- Feraco, P.; Bacci, A.; Pedrabissi, F.; Passamonti, L.; Zampogna, G.; Pedrabissi, F.; Malavolta, N.; Leonardi, M. Metabolic Abnormalities in Pain-Processing Regions of Patients with Fibromyalgia: A 3T MR Spectroscopy Study. Am. J. Neuroradiol. 2011, 32, 1585–1590. [Google Scholar] [CrossRef] [PubMed]
- Feraco, P.; Nigro, S.; Passamonti, L.; Grecucci, A.; Caligiuri, M.E.; Gagliardo, C.; Bacci, A. Neurochemical Correlates of Brain Atrophy in Fibromyalgia Syndrome: A Magnetic Resonance Spectroscopy and Cortical Thickness Study. Brain Sci. 2020, 10, 395. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Andronesi, O.C.; Torrado-Carvajal, A.; Ratai, E.; Loggia, M.L.; Weerasekera, A.; Berry, M.P.; Ellingsen, D.; Isaro, L.; Lazaridou, A.; et al. 3D magnetic resonance spectroscopic imaging reveals links between brain metabolites and multidimensional pain features in fibromyalgia. Eur. J. Pain 2021, 25, 2050–2064. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Yang, T.; Zhao, H.; Zhang, M.; Meng, F.; Fu, H.; Xie, Y.; Xu, H. Insular Cortex is Critical for the Perception, Modulation, and Chronification of Pain. Neurosci. Bull. 2016, 32, 191–201. [Google Scholar] [CrossRef]
- Harris, R.E. Elevated excitatory neurotransmitter levels in the fibromyalgia brain. Arthritis Res. Ther. 2010, 12, 141. [Google Scholar] [CrossRef]
- Steinhoff, M.S.; von Mentzer, B.; Geppetti, P.; Pothoulakis, C.; Bunnett, N.W.; Bakirtzi, K.; Law, I.K.M.; Fang, K.; Iliopoulos, D.; Dér, B.; et al. Tachykinins and Their Receptors: Contributions to Physiological Control and the Mechanisms of Disease. Physiol. Rev. 2014, 94, 265–301. [Google Scholar] [CrossRef]
- Almeida, T.A.; Rojo, J.; Nieto, P.M.; Pinto, F.M.; Hernandez, M.; Martín, J.D.; Candenas, M.L. Tachykinins and Tachykinin Receptors: Structure and Activity Relationships. Curr. Med. Chem. 2004, 11, 2045–2081. [Google Scholar] [CrossRef] [PubMed]
- Schank, J.R.; Heilig, M. Substance P and the Neurokinin-1 Receptor: The New CRF. Int. Rev. Neurobiol. 2017, 136, 151–175. [Google Scholar] [CrossRef] [PubMed]
- Seybold, V. The Role of Peptides in Central Sensitization. Handb. Exp. Pharmacol. 2009, 194, 451–491. [Google Scholar] [CrossRef]
- Pace, M.C.; Passavanti, M.B.; De Nardis, L.; Bosco, F.; Sansone, P.; Pota, V.; Barbarisi, M.; Palagiano, A.; Iannotti, F.A.; Panza, E.; et al. Nociceptor plasticity: A closer look. J. Cell. Physiol. 2017, 233, 2824–2838. [Google Scholar] [CrossRef]
- Navratilova, E.; Porreca, F. Substance P and Inflammatory Pain: Getting It Wrong and Right Simultaneously. Neuron 2019, 101, 353–355. [Google Scholar] [CrossRef]
- Green, D.P.; Limjunyawong, N.; Gour, N.; Pundir, P.; Dong, X. A Mast-Cell-Specific Receptor Mediates Neurogenic Inflammation and Pain. Neuron 2019, 101, 412–420.e3. [Google Scholar] [CrossRef]
- Vaerøy, H.; Helle, R.; Førre, Ø.; Kåss, E.; Terenius, L. Elevated CSF levels of substance P and high incidence of Raynaud phenomenon in patients with fibromyalgia: New features for diagnosis. Pain 1988, 32, 21–26. [Google Scholar] [CrossRef]
- Russell, I.J.; Orr, M.D.; Littman, B.; Vipraio, G.A.; Alboukrek, D.; Michalek, J.E.; Lopez, Y.; Mackillip, F. Elevated cerebrospinal fluid levels of substance p in patients with the fibromyalgia syndrome. Arthritis Rheum. 1994, 37, 1593–1601. [Google Scholar] [CrossRef]
- Russell, I.J. Neurochemical pathogenesis of fibromyalgia. Z. Rheumatol. 1998, 57, S63–S66. [Google Scholar] [CrossRef]
- Reynolds, W.J.; Chiu, B.; Inman, R.D. Plasma substance P levels in fibrositis. J. Rheumatol. 1988, 15, 1802–1803. [Google Scholar]
- Tsilioni, I.; Russell, I.J.; Stewart, J.M.; Gleason, R.M.; Theoharides, T.C. Neuropeptides CRH, SP, HK-1, and Inflammatory Cytokines IL-6 and TNF Are Increased in Serum of Patients with Fibromyalgia Syndrome, Implicating Mast Cells. J. Pharmacol. Exp. Ther. 2016, 356, 664–672. [Google Scholar] [CrossRef] [PubMed]
- Tsilioni, I.; Pipis, H.; Freitag, M.S.C.; Izquierdo, M.D.C.; Freitag, K.; Theoharides, T.C. Effects of an Extract of Salmon Milt on Symptoms and Serum TNF and Substance P in Patients With Fibromyalgia Syndrome. Clin. Ther. 2019, 41, 1564–1574.e2. [Google Scholar] [CrossRef] [PubMed]
- Field, T.; Diego, M.; Cullen, C.; Hernandez-Reif, M.; Sunshine, W.; Douglas, S. Fibromyalgia Pain and Substance P Decrease and Sleep Improves After Massage Therapy. Am. J. Clin. Oncol. 2002, 8, 72–76. [Google Scholar] [CrossRef] [PubMed]
- Karatay, S.; Okur, S.C.; Uzkeser, H.; Yildirim, K.; Akcay, F. Effects of Acupuncture Treatment on Fibromyalgia Symptoms, Serotonin, and Substance P Levels: A Randomized Sham and Placebo-Controlled Clinical Trial. Pain Med. 2017, 19, 615–628. [Google Scholar] [CrossRef]
- Sprott, H.; Franke, S.; Kluge, H.; Hein, G. Pain treatment of fibromyalgia by acupuncture. Rheumatol. Int. 1998, 18, 35–36. [Google Scholar] [CrossRef] [PubMed]
- Bjersing, J.L.; Dehlin, M.; Erlandsson, M.; Bokarewa, M.I.; Mannerkorpi, K. Changes in pain and insulin-like growth factor 1 in fibromyalgia during exercise: The involvement of cerebrospinal inflammatory factors and neuropeptides. Arthritis Res. Ther. 2012, 14, R162. [Google Scholar] [CrossRef] [PubMed]
- Denk, F.; Bennett, D.L.; McMahon, S.B. Nerve Growth Factor and Pain Mechanisms. Annu. Rev. Neurosci. 2017, 40, 307–325. [Google Scholar] [CrossRef]
- Svensson, P.; Cairns, B.E.; Wang, K.; Arendt-Nielsen, L. Injection of nerve growth factor into human masseter muscle evokes long-lasting mechanical allodynia and hyperalgesia. Pain 2003, 104, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Rukwied, R.; Mayer, A.; Kluschina, O.; Obreja, O.; Schley, M.; Schmelz, M. NGF induces non-inflammatory localized and lasting mechanical and thermal hypersensitivity in human skin. Pain 2010, 148, 407–413. [Google Scholar] [CrossRef]
- Weinkauf, B.; Obreja, O.; Schmelz, M.; Rukwied, R. Differential time course of NGF-induced hyperalgesia to heat versus mechanical and electrical stimulation in human skin. Eur. J. Pain 2014, 19, 789–796. [Google Scholar] [CrossRef]
- Andersen, H.; Arendt-Nielsen, L.; Svensson, P.; Danneskiold-Samsøe, B.; Graven-Nielsen, T. Spatial and temporal aspects of muscle hyperalgesia induced by nerve growth factor in humans. Exp. Brain Res. 2008, 191, 371–382. [Google Scholar] [CrossRef]
- Gerber, R.K.H.; Nie, H.; Arendt-Nielsen, L.; Curatolo, M.; Graven-Nielsen, T. Local Pain and Spreading Hyperalgesia Induced by Intramuscular Injection of Nerve Growth Factor Are Not Reduced by Local Anesthesia of the Muscle. Clin. J. Pain 2011, 27, 240–247. [Google Scholar] [CrossRef]
- Sørensen, L.B.; Boudreau, S.A.; Gazerani, P.; Graven-Nielsen, T. Enlarged Areas of Pain and Pressure Hypersensitivityby Spatially Distributed Intramuscular Injections ofLow-Dose Nerve Growth Factor. J. Pain 2018, 20, 566–576. [Google Scholar] [CrossRef] [PubMed]
- Kelleher, J.H.; Tewari, D.; McMahon, S.B. Neurotrophic factors and their inhibitors in chronic pain treatment. Neurobiol. Dis. 2017, 97, 127–138. [Google Scholar] [CrossRef]
- Bloom, A.P.; Jimenez-Andrade, J.M.; Taylor, R.N.; Castañeda-Corral, G.; Kaczmarska, M.J.; Freeman, K.T.; Coughlin, K.A.; Ghilardi, J.R.; Kuskowski, M.A.; Mantyh, P.W. Breast Cancer-Induced Bone Remodeling, Skeletal Pain, and Sprouting of Sensory Nerve Fibers. J. Pain 2011, 12, 698–711. [Google Scholar] [CrossRef]
- Barker, P.A.; Mantyh, P.; Arendt-Nielsen, L.; Viktrup, L.; Tive, L. Nerve Growth Factor Signaling and Its Contribution to Pain. J. Pain Res. 2020, 13, 1223–1241. [Google Scholar] [CrossRef]
- Skaper, S.D. Nerve growth factor: A neuroimmune crosstalk mediator for all seasons. Immunology 2017, 151, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Redegeld, F.A.; Yu, Y.; Kumari, S.; Charles, N.; Blank, U. Non-IgE mediated mast cell activation. Immunol. Rev. 2018, 282, 87–113. [Google Scholar] [CrossRef] [PubMed]
- Kritas, S.; Caraffa, A.; Antinolfi, P.; Saggini, A.; Pantalone, A.; Rosati, M.; Tei, M.; Speziali, A.; Pandolfi, F.; Cerulli, G.; et al. Nerve Growth Factor Interactions with Mast Cells. Int. J. Immunopathol. Pharmacol. 2014, 27, 15–19. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Shi, X.; Li, X.; Zou, J.; Zhou, C.; Liu, W.; Shao, H.; Chen, H.; Shi, L. Neurotransmitter and neuropeptide regulation of mast cell function: A systematic review. J. Neuroinflamm. 2020, 17, 356. [Google Scholar] [CrossRef] [PubMed]
- Cirulli, F.; Alleva, E. The NGF saga: From animal models of psychosocial stress to stress-related psychopathology. Front. Neuroendocr. 2009, 30, 379–395. [Google Scholar] [CrossRef] [PubMed]
- Mozafarihashjin, M.; Togha, M.; Ghorbani, Z.; Farbod, A.; Rafiee, P.; Martami, F. Assessment of peripheral biomarkers potentially involved in episodic and chronic migraine: A case-control study with a focus on NGF, BDNF, VEGF, and PGE2. J. Headache Pain 2022, 23, 3. [Google Scholar] [CrossRef]
- Montagnoli, C.; Tiribuzi, R.; Crispoltoni, L.; Pistilli, A.; Stabile, A.M.; Manfreda, F.; Placella, G.; Rende, M.; Cerulli, G.G. β-NGF and β-NGF receptor upregulation in blood and synovial fluid in osteoarthritis. Biol. Chem. 2017, 398, 1045–1054. [Google Scholar] [CrossRef]
- Giovengo, S.L.; Russell, I.J.; Larson, A.A. Increased concentrations of nerve growth factor in cerebrospinal fluid of patients with fibromyalgia. J. Rheumatol. 1999, 26, 1564–1569. [Google Scholar]
- Baumeister, D.; Eich, W.; Saft, S.; Geisel, O.; Hellweg, R.; Finn, A.; Svensson, C.I.; Tesarz, J. No evidence for altered plasma NGF and BDNF levels in fibromyalgia patients. Sci. Rep. 2019, 9, 13667. [Google Scholar] [CrossRef]
- Jablochkova, A.; Bäckryd, E.; Kosek, E.; Mannerkorpi, K.; Ernberg, M.; Gerdle, B.; Ghafouri, B. Unaltered low nerve growth factor and high brain-derived neurotrophic factor levels in plasma from patients with fibromyalgia after a 15-week progressive resistance exercise. J. Rehabil. Med. 2019, 51, 779–787. [Google Scholar] [CrossRef]
- Binder, D.K.; Scharfman, H.E. Brain-derived Neurotrophic Factor. Growth Factors 2004, 22, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Malfait, A.-M.; Miller, R.E.; Block, J.A. Targeting neurotrophic factors: Novel approaches to musculoskeletal pain. Pharmacol. Ther. 2020, 211, 107553. [Google Scholar] [CrossRef]
- Ferrini, F.; Salio, C.; Boggio, E.M.; Merighi, A. Interplay of BDNF and GDNF in the Mature Spinal Somatosensory System and Its Potential Therapeutic Relevance. Curr. Neuropharmacol. 2021, 19, 1225–1245. [Google Scholar] [CrossRef]
- Garraway, S.M.; Huie, J.R. Spinal Plasticity and Behavior: BDNF-Induced Neuromodulation in Uninjured and Injured Spinal Cord. Neural Plast. 2016, 2016, 9857201. [Google Scholar] [CrossRef]
- Brigadski, T.; Leßmann, V. The physiology of regulated BDNF release. Cell Tissue Res. 2020, 382, 15–45. [Google Scholar] [CrossRef]
- Taves, S.; Berta, T.; Chen, G.; Ji, R.-R. Microglia and Spinal Cord Synaptic Plasticity in Persistent Pain. Neural Plast. 2013, 2013, 753656. [Google Scholar] [CrossRef]
- Obata, K.; Noguchi, K. BDNF in sensory neurons and chronic pain. Neurosci. Res. 2006, 55, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Beggs, S.; Salter, M.W. The known knowns of microglia–neuronal signalling in neuropathic pain. Neurosci. Lett. 2013, 557, 37–42. [Google Scholar] [CrossRef]
- Price, T.J.; Inyang, K.E. Commonalities Between Pain and Memory Mechanisms and Their Meaning for Understanding Chronic Pain. Prog. Mol. Biol. Transl. Sci. 2015, 131, 409–434. [Google Scholar] [CrossRef] [PubMed]
- Haas, L.; Portela, L.V.C.; Böhmer, A.E.; Oses, J.P.; Lara, D.R. Increased Plasma Levels of Brain Derived Neurotrophic Factor (BDNF) in Patients with Fibromyalgia. Neurochem. Res. 2010, 35, 830–834. [Google Scholar] [CrossRef]
- Laske, C.; Stransky, E.; Eschweiler, G.W.; Klein, R.; Wittorf, A.; Leyhe, T.; Richartz, E.; Köhler, N.; Bartels, M.; Buchkremer, G.; et al. Increased BDNF serum concentration in fibromyalgia with or without depression or antidepressants. J. Psychiatr. Res. 2007, 41, 600–605. [Google Scholar] [CrossRef] [PubMed]
- Stefani, L.C.; Leite, F.M.; Tarragó, M.D.G.L.; Zanette, S.A.; de Souza, A.; Castro, S.M.; Caumo, W. BDNF and serum S100B levels according the spectrum of structural pathology in chronic pain patients. Neurosci. Lett. 2019, 706, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Nugraha, B.; Korallus, C.; Gutenbrunner, C. Serum level of brain-derived neurotrophic factor in fibromyalgia syndrome correlates with depression but not anxiety. Neurochem. Int. 2013, 62, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Nugraha, B.; Karst, M.; Engeli, S.; Gutenbrunner, C. Brain-derived neurotrophic factor and exercise in fibromyalgia syndrome patients: A mini review. Rheumatol. Int. 2012, 32, 2593–2599. [Google Scholar] [CrossRef]
- Ribeiro, V.; Mendonça, V.; Souza, A.; Fonseca, S.; Camargos, A.; Lage, V.; Neves, C.; Santos, J.; Teixeira, A.L.; Vieira, E.; et al. Inflammatory biomarkers responses after acute whole body vibration in fibromyalgia. Braz. J. Med. Biol. Res. 2018, 51, e6775. [Google Scholar] [CrossRef]
- Ranzolin, A.; Duarte, A.L.B.P.; Bredemeier, M.; Neto, C.A.D.C.; Ascoli, B.M.; Wollenhaupt-Aguiar, B.; Kapczinski, F.; Xavier, R.M. Evaluation of cytokines, oxidative stress markers and brain-derived neurotrophic factor in patients with fibromyalgia—A controlled cross-sectional study. Cytokine 2016, 84, 25–28. [Google Scholar] [CrossRef]
- Iannuccelli, C.; Lucchino, B.; Gioia, C.; Dolcini, G.; Rabasco, J.; Venditto, T.; Ioppolo, F.; Santilli, V.; Conti, F.; Di Franco, M. Gender influence on clinical manifestations, depressive symptoms and brain-derived neurotrophic factor (BDNF) serum levels in patients affected by fibromyalgia. Clin. Rheumatol. 2022, 41, 2171–2178. [Google Scholar] [CrossRef]
- Rasmussen, P.; Brassard, P.; Adser, H.; Pedersen, M.V.; Leick, L.; Hart, E.; Secher, N.H.; Pedersen, B.K.; Pilegaard, H. Evidence for a release of brain-derived neurotrophic factor from the brain during exercise. Exp. Physiol. 2009, 94, 1062–1069. [Google Scholar] [CrossRef]
- Fujimura, H.; Chen, R.; Nakamura, T.; Nakahashi, T.; Kambayashi, J.-I.; Sun, B.; Altar, C.; Tandon, N.N. Brain-derived Neurotrophic Factor Is Stored in Human Platelets and Released by Agonist Stimulation. Thromb. Haemost. 2002, 87, 728–734. [Google Scholar] [CrossRef]
- Matthews, V.B.; Åström, M.-B.; Chan, M.H.S.; Bruce, C.R.; Krabbe, K.S.; Prelovsek, O.; Åkerström, T.; Yfanti, C.; Broholm, C.; Mortensen, O.H.; et al. Brain-derived neurotrophic factor is produced by skeletal muscle cells in response to contraction and enhances fat oxidation via activation of AMP-activated protein kinase. Diabetologia 2009, 52, 1409–1418. [Google Scholar] [CrossRef] [PubMed]
- Björkholm, C.; Monteggia, L.M. BDNF—A Key Transducer of Antidepressant Effects. Neuropharmacology 2016, 102, 72–79. [Google Scholar] [CrossRef]
- Bidari, A.; Ghavidel-Parsa, B.; Gharibpoor, F. Comparison of the serum brain-derived neurotrophic factor (BDNF) between fibromyalgia and nociceptive pain groups; and effect of duloxetine on the BDNF level. BMC Musculoskelet. Disord. 2022, 23, 411. [Google Scholar] [CrossRef] [PubMed]
- Zanette, S.A.; Dussan-Sarria, J.A.; Souza, A.; Deitos, A.; Torres, I.L.S.; Caumo, W. Higher Serum S100B and BDNF Levels are Correlated with a Lower Pressure-Pain Threshold in Fibromyalgia. Mol. Pain 2014, 10, 46. [Google Scholar] [CrossRef]
- Caumo, W.; Deitos, A.; Carvalho, S.; Leite, J.; Carvalho, F.; Dussán-Sarria, J.A.; Tarragó, M.D.G.L.; Souza, A.; Torres, I.L.D.S.; Fregni, F. Motor Cortex Excitability and BDNF Levels in Chronic Musculoskeletal Pain According to Structural Pathology. Front. Hum. Neurosci. 2016, 10, 357. [Google Scholar] [CrossRef] [PubMed]
- Soldatelli, M.D.; Siepmann, T.; Illigens, B.M.-W.; dos Santos, V.S.; Torres, I.L.d.S.; Fregni, F.; Caumo, W. Mapping of predictors of the disengagement of the descending inhibitory pain modulation system in fibromyalgia: An exploratory study. Br. J. Pain 2020, 15, 221–233. [Google Scholar] [CrossRef]
- Tzadok, R.; Ablin, J.N. Current and Emerging Pharmacotherapy for Fibromyalgia. Pain Res. Manag. 2020, 2020, 6541798. [Google Scholar] [CrossRef]
- Peng, X.; Robinson, R.L.; Mease, P.; Kroenke, K.; Williams, D.A.; Chen, Y.; Faries, D.; Wohlreich, M.; McCarberg, B.; Hann, D. Long-term Evaluation of Opioid Treatment in Fibromyalgia. Clin. J. Pain 2015, 31, 7–13. [Google Scholar] [CrossRef]
- Hwang, J.-M.; Lee, B.-J.; Oh, T.H.; Park, D.; Kim, C.-H. Association between initial opioid use and response to a brief interdisciplinary treatment program in fibromyalgia. Medicine 2019, 98, e13913. [Google Scholar] [CrossRef]
- Younger, J.; Mackey, S. Fibromyalgia Symptoms Are Reduced by Low-Dose Naltrexone: A Pilot Study. Pain Med. 2009, 10, 663–672. [Google Scholar] [CrossRef]
- Younger, J.; Noor, N.; McCue, R.; Mackey, S. Low-dose naltrexone for the treatment of fibromyalgia: Findings of a small, randomized, double-blind, placebo-controlled, counterbalanced, crossover trial assessing daily pain levels. Arthritis Rheum. 2013, 65, 529–538. [Google Scholar] [CrossRef]
- Parkitny, L.; Younger, J. Reduced Pro-Inflammatory Cytokines after Eight Weeks of Low-Dose Naltrexone for Fibromyalgia. Biomedicines 2017, 5, 16. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.E.; Clauw, D.J.; Scott, D.J.; McLean, S.A.; Gracely, R.H.; Zubieta, J.-K. Decreased Central μ-Opioid Receptor Availability in Fibromyalgia. J. Neurosci. 2007, 27, 10000–10006. [Google Scholar] [CrossRef] [PubMed]
- Schrepf, A.; Harper, D.E.; Harte, S.E.; Wang, H.; Ichesco, E.; Hampson, J.P.; Zubieta, J.-K.; Clauw, D.J.; Harris, R.E. Endogenous opioidergic dysregulation of pain in fibromyalgia: A PET and fMRI study. Pain 2016, 157, 2217–2225. [Google Scholar] [CrossRef] [PubMed]
- Üçeyler, N.; Buchholz, H.-G.; Kewenig, S.; Ament, S.-J.; Birklein, F.; Schreckenberger, M.; Sommer, C. Cortical Binding Potential of Opioid Receptors in Patients With Fibromyalgia Syndrome and Reduced Systemic Interleukin-4 Levels—A Pilot Study. Front. Neurosci. 2020, 14, 512. [Google Scholar] [CrossRef]
- Brejchova, J.; Holan, V.; Svoboda, P. Expression of Opioid Receptors in Cells of the Immune System. Int. J. Mol. Sci. 2020, 22, 315. [Google Scholar] [CrossRef]
- Celik, M.Ö.; Labuz, D.; Henning, K.; Busch-Dienstfertig, M.; Gaveriaux-Ruff, C.; Kieffer, B.L.; Zimmer, A.; Machelska, H. Leukocyte opioid receptors mediate analgesia via Ca2+-regulated release of opioid peptides. Brain Behav. Immun. 2016, 57, 227–242. [Google Scholar] [CrossRef]
- Machelska, H.; Celik, M.Ö. Opioid Receptors in Immune and Glial Cells—Implications for Pain Control. Front. Immunol. 2020, 11, 300. [Google Scholar] [CrossRef]
- Raffaeli, W.; Malafoglia, V.; Bonci, A.; Tenti, M.; Ilari, S.; Gremigni, P.; Iannuccelli, C.; Gioia, C.; Di Franco, M.; Mollace, V.; et al. Identification of MOR-Positive B Cell as Possible Innovative Biomarker (Mu Lympho-Marker) for Chronic Pain Diagnosis in Patients with Fibromyalgia and Osteoarthritis Diseases. Int. J. Mol. Sci. 2020, 21, 1499. [Google Scholar] [CrossRef]
- Malafoglia, V.; Ilari, S.; Gioia, C.; Vitiello, L.; Tenti, M.; Iannuccelli, C.; Cristiani, C.M.; Garofalo, C.; Passacatini, L.C.; Viglietto, G.; et al. An Observational Study on Chronic Pain Biomarkers in Fibromyalgia and Osteoarthritis Patients: Which Role for Mu Opioid Receptor’s Expression on NK Cells? Biomedicines 2023, 11, 931. [Google Scholar] [CrossRef]
- Conti, P.; Gallenga, C.E.; Caraffa, A.; Ronconi, G.; Kritas, S.K. Impact of mast cells in fibromyalgia and low-grade chronic inflammation: Can IL-37 play a role? Dermatol. Ther. 2020, 33, e13191. [Google Scholar] [CrossRef]
- Eller-Smith, O.C.; Nicol, A.L.; Christianson, J.A. Potential Mechanisms Underlying Centralized Pain and Emerging Therapeutic Interventions. Front. Cell. Neurosci. 2018, 12, 35. [Google Scholar] [CrossRef]
- Enestrbm, S.; Bengtsson, A.; Frddin, T. Dermal IgG Deposits and Increase of Mast Cells in Patients with Fibromyalgia—Relevant Findings or Epiphenomena? Scand. J. Rheumatol. 1997, 26, 308–313. [Google Scholar] [CrossRef]
- Blanco, I.; Béritze, N.; Argüelles, M.; Cárcaba, V.; Fernández, F.; Janciauskiene, S.; Oikonomopoulou, K.; de Serres, F.J.; Fernández-Bustillo, E.; Hollenberg, M.D. Abnormal overexpression of mastocytes in skin biopsies of fibromyalgia patients. Clin. Rheumatol. 2010, 29, 1403–1412. [Google Scholar] [CrossRef]
- Ang, D.C.; Hilligoss, J.; Stump, T. Mast Cell Stabilizer (Ketotifen) in Fibromyalgia: Phase 1 Randomized Controlled Clinical Trial. Clin. J. Pain 2015, 31, 836–842. [Google Scholar] [CrossRef]
- Salemi, S.; Rethage, J.; Wollina, U.; Michel, B.A.; Gay, R.E.; Gay, S.; Sprott, H. Detection of interleukin 1beta (IL-1beta), IL-6, and tumor necrosis factor-alpha in skin of patients with fibromyalgia. J. Rheumatol. 2003, 30, 146–150. [Google Scholar]
- Üçeyler, N.; Kewenig, S.; Kafke, W.; Kittel-Schneider, S.; Sommer, C. Skin cytokine expression in patients with fibromyalgia syndrome is not different from controls. BMC Neurol. 2014, 14, 185. [Google Scholar] [CrossRef]
- Üçeyler, N.; Häuser, W.; Sommer, C. Systematic review with meta-analysis: Cytokines in fibromyalgia syndrome. BMC Musculoskelet. Disord. 2011, 12, 245. [Google Scholar] [CrossRef] [PubMed]
- O’mahony, L.F.; Srivastava, A.; Mehta, P.; Ciurtin, C. Is fibromyalgia associated with a unique cytokine profile? A systematic review and meta-analysis. Rheumatology 2021, 60, 2602–2614. [Google Scholar] [CrossRef] [PubMed]
- Kumbhare, D.; Hassan, S.; Diep, D.; Duarte, F.C.K.; Hung, J.; Damodara, S.; West, D.W.; Selvaganapathy, P.R. Potential role of blood biomarkers in patients with fibromyalgia: A systematic review with meta-analysis. Pain 2021, 163, 1232–1253. [Google Scholar] [CrossRef] [PubMed]
- Ang, D.C.; Moore, M.N.; Hilligoss, J.; Tabbey, R. MCP-1 and IL-8 as Pain Biomarkers in Fibromyalgia: A Pilot Study. Pain Med. 2011, 12, 1154–1161. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Buchner, M.; Moser, M.T.; Daniel, V.; Schiltenwolf, M. The Role of IL-8 in Patients With Fibromyalgia: A prospective longitudinal study of 6 months. Clin. J. Pain 2009, 25, 1–4. [Google Scholar] [CrossRef]
- Kadetoff, D.; Lampa, J.; Westman, M.; Andersson, M.; Kosek, E. Evidence of central inflammation in fibromyalgia—Increased cerebrospinal fluid interleukin-8 levels. J. Neuroimmunol. 2012, 242, 33–38. [Google Scholar] [CrossRef]
- Kosek, E.; Altawil, R.; Kadetoff, D.; Finn, A.; Westman, M.; Le Maître, E.; Andersson, M.; Jensen-Urstad, M.; Lampa, J. Evidence of different mediators of central inflammation in dysfunctional and inflammatory pain—Interleukin-8 in fibromyalgia and interleukin-1 β in rheumatoid arthritis. J. Neuroimmunol. 2015, 280, 49–55. [Google Scholar] [CrossRef]
- Theoharides, T.C.; Tsilioni, I.; Bawazeer, M. Mast Cells, Neuroinflammation and Pain in Fibromyalgia Syndrome. Front. Cell. Neurosci. 2019, 13, 353. [Google Scholar] [CrossRef]
- Kunes, P.; Holubcova, Z.; Kolackova, M.; Krejsek, J. Pentraxin 3(PTX 3): An Endogenous Modulator of the Inflammatory Response. Mediat. Inflamm. 2012, 2012, 920517. [Google Scholar] [CrossRef]
- CieŚlik, P.; Hrycek, A. Long pentraxin 3 (PTX3) in the light of its structure, mechanism of action and clinical implications. Autoimmunity 2012, 45, 119–128. [Google Scholar] [CrossRef]
- Daigo, K.; Mantovani, A.; Bottazzi, B. The yin-yang of long pentraxin PTX3 in inflammation and immunity. Immunol. Lett. 2014, 161, 38–43. [Google Scholar] [CrossRef]
- Skare, T.L.; Plawiak, A.C.; Veronese, G.; Paiva, E.S.; Messias-Reason, I.; Nisihara, R. Pentraxin 3 levels in women with fibromyalgia. Clin. Exp. Rheumatol. 2015, 33, S142. [Google Scholar]
- Skare, T.L.; Plawiak, A.C.; Veronese, G.; Paiva, E.S.; Messias-Reason, I.; Nisihara, R. Reply to ‘PTX-3 release is increased by monocytes from patients with primary fibromyalgia without major depression’ by J.J. Garcia et al. Ann. Rheum. Dis. 2016, 34, S151. [Google Scholar]
- García, J.J.; Herrero-Oléa, A.; Carvajal-Gil, J.; Gómez-Galán, R. PTX-3 release is increased by monocytes from patients with primary fibromyalgia without major depression. Ann. Rheum. Dis. 2015, 34, S150. [Google Scholar]
- Shende, P.; Desai, D. Physiological and Therapeutic Roles of Neuropeptide Y on Biological Functions. Adv. Exp. Med. Biol. 2020, 1237, 37–47. [Google Scholar] [CrossRef]
- Brothers, S.P.; Wahlestedt, C. Therapeutic potential of neuropeptide Y (NPY) receptor ligands. EMBO Mol. Med. 2010, 2, 429–439. [Google Scholar] [CrossRef]
- Diaz-Delcastillo, M.; Woldbye, D.P.; Heegaard, A.M. Neuropeptide Y and its Involvement in Chronic Pain. Neuroscience 2018, 387, 162–169. [Google Scholar] [CrossRef]
- Nelson, T.S.; Taylor, B.K. Targeting spinal neuropeptide Y1 receptor-expressing interneurons to alleviate chronic pain and itch. Prog. Neurobiol. 2020, 196, 101894. [Google Scholar] [CrossRef]
- Kask, A.; Harro, J.; von Hörsten, S.; Redrobe, J.P.; Dumont, Y.; Quirion, R. The neurocircuitry and receptor subtypes mediating anxiolytic-like effects of neuropeptide Y. Neurosci. Biobehav. Rev. 2002, 26, 259–283. [Google Scholar] [CrossRef]
- Reichmann, F.; Holzer, P. Neuropeptide Y: A stressful review. Neuropeptides 2016, 55, 99–109. [Google Scholar] [CrossRef]
- Iannuccelli, C.; Di Franco, M.; Alessandri, C.; Guzzo, M.; Croia, C.; Di Sabato, F.; Foti, M.; Valesini, G. Pathophysiology of fibromyalgia: A comparison with the tension-type headache, a localized pain syndrome. Ann. N. Y. Acad. Sci. 2010, 1193, 78–83. [Google Scholar] [CrossRef]
- Chen, W.-C.; Liu, Y.-B.; Liu, W.-F.; Zhou, Y.-Y.; He, H.-F.; Lin, S. Neuropeptide Y Is an Immunomodulatory Factor: Direct and Indirect. Front. Immunol. 2020, 11, 580378. [Google Scholar] [CrossRef]
- Crofford, L.J.; Pillemer, S.R.; Kalogeras, K.T.; Cash, J.M.; Michelson, D.; Kling, M.A.; Sternberg, E.M.; Gold, P.W.; Chrousos, G.P.; Wilder, R.L. Hypothalamic–pituitary–adrenal axis perturbations in patients with fibromyalgia. Arthritis Rheum. 1994, 37, 1583–1592. [Google Scholar] [CrossRef]
- Claw, D.J.; Sabol, M. Serum neuropeptides in patients with both fibromyalgia (FMS) and chronic fatigue syndrome (CFS). J. Musculoskelet. Pain 1995, 79, R28. [Google Scholar]
- Eisinger, J. Dysautonomia, fibromyalgia and reflex dystrophy. Arthritis Res. Ther. 2007, 9, 105. [Google Scholar] [CrossRef]
- Di Franco, M.; Iannuccelli, C.; Alessandri, C.; Paradiso, M.; Riccieri, V.; Libri, F.; Valesini, G. Autonomic dysfunction and neuropeptide Y in fibromyalgia. Ann. Rheum. Dis. 2010, 27, S75–S78. [Google Scholar]
- Anderberg, U.M.; Liu, Z.; Berglund, L.; Nyberg, F. Elevated plasma levels of neuropeptide Y in female fibromyalgia patients. Eur. J. Pain 1999, 3, 19–30. [Google Scholar] [CrossRef]
- Shipton, E.A.; Shipton, E.E. Vitamin D and Pain: Vitamin D and Its Role in the Aetiology and Maintenance of Chronic Pain States and Associated Comorbidities. Pain Res. Treat. 2015, 2015, 904967. [Google Scholar] [CrossRef] [PubMed]
- Charoenngam, N.; Shirvani, A.; Holick, M.F. Vitamin D for skeletal and non-skeletal health: What we should know. J. Clin. Orthop. Trauma 2019, 10, 1082–1093. [Google Scholar] [CrossRef] [PubMed]
- Karras, S.; Rapti, E.; Matsoukas, S.; Kotsa, K. Vitamin D in Fibromyalgia: A Causative or Confounding Biological Interplay? Nutrients 2016, 8, 343. [Google Scholar] [CrossRef]
- Habib, A.M.; Nagi, K.; Thillaiappan, N.B.; Sukumaran, V.; Akhtar, S. Vitamin D and Its Potential Interplay With Pain Signaling Pathways. Front. Immunol. 2020, 11, 820. [Google Scholar] [CrossRef]
- Ellis, S.D.; Kelly, S.T.; Shurlock, J.H.; Hepburn, A.L.N. The role of vitamin D testing and replacement in fibromyalgia: A systematic literature review. BMC Rheumatol. 2018, 2, 28. [Google Scholar] [CrossRef] [PubMed]
- Makrani, A.H.; Afshari, M.; Ghajar, M.; Foroughi, Z.; Moosazadeh, M. Vitamin D and fibromyalgia: A meta-analysis. Korean J. Pain 2017, 30, 250–257. [Google Scholar] [CrossRef]
- Hsiao, M.-Y.; Hung, C.-Y.; Chang, K.-V.; Han, D.-S.; Wang, T.-G. Is Serum Hypovitaminosis D Associated with Chronic Widespread Pain Including Fibromyalgia? A Meta-analysis of Observational Studies. Pain Physician 2015, 18, E877–E887. [Google Scholar] [PubMed]
- Joustra, M.L.; Minovic, I.; Janssens, K.A.M.; Bakker, S.J.L.; Rosmalen, J.G.M. Vitamin and mineral status in chronic fatigue syndrome and fibromyalgia syndrome: A systematic review and meta-analysis. PLoS ONE 2017, 12, e0176631. [Google Scholar] [CrossRef] [PubMed]
- Martins, Y.A.; Cardinali, C.A.E.; Ravanelli, M.I.; Brunaldi, K. Is hypovitaminosis D associated with fibromyalgia? A systematic review. Nutr. Rev. 2020, 78, 115–133. [Google Scholar] [CrossRef]
- Ali, O.M.E. Prevalence of Vitamin D Deficiency and Its Relationship with Clinical Outcomes in Patients with Fibromyalgia: A Systematic Review of the Literature. SN Compr. Clin. Med. 2022, 4, 38. [Google Scholar] [CrossRef]
- Haddad, H.W.; Jumonville, A.C.; Stark, K.J.; Temple, S.N.; Dike, C.C.; Cornett, E.M.; Kaye, A.D. The Role of Vitamin D in the Management of Chronic Pain in Fibromyalgia: A Narrative Review. Health Psychol. Res. 2021, 9, 25208. [Google Scholar] [CrossRef]
- Mohajeri, M.H.; La Fata, G.; Steinert, R.E.; Weber, P. Relationship between the gut microbiome and brain function. Nutr. Rev. 2018, 76, 481–496. [Google Scholar] [CrossRef]
- Ghaisas, S.; Maher, J.; Kanthasamy, A. Gut microbiome in health and disease: Linking the microbiome–gut–brain axis and environmental factors in the pathogenesis of systemic and neurodegenerative diseases. Pharmacol. Ther. 2015, 158, 52–62. [Google Scholar] [CrossRef]
- Cryan, J.F.; O’Riordan, K.J.; Cowan, C.S.M.; Sandhu, K.V.; Bastiaanssen, T.F.S.; Boehme, M.; Codagnone, M.G.; Cussotto, S.; Fulling, C.; Golubeva, A.V.; et al. The Microbiota-Gut-Brain Axis. Physiol. Rev. 2019, 99, 1877–2013. [Google Scholar] [CrossRef]
- Guo, R.; Chen, L.-H.; Xing, C.; Liu, T. Pain regulation by gut microbiota: Molecular mechanisms and therapeutic potential. Br. J. Anaesth. 2019, 123, 637–654. [Google Scholar] [CrossRef]
- Clos-Garcia, M.; Andrés-Marin, N.; Fernández-Eulate, G.; Abecia, L.; Lavín, J.L.; van Liempd, S.; Cabrera, D.; Royo, F.; Valero, A.; Errazquin, N.; et al. Gut microbiome and serum metabolome analyses identify molecular biomarkers and altered glutamate metabolism in fibromyalgia. Ebiomedicine 2019, 46, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Minerbi, A.; Fitzcharles, M.-A. Gut microbiome: Pertinence in fibromyalgia. Ann. Rheum. Dis. 2020, 38, 99–104. [Google Scholar]
- Erdrich, S.; Hawrelak, J.A.; Myers, S.P.; Harnett, J.E. Determining the association between fibromyalgia, the gut microbiome and its biomarkers: A systematic review. BMC Musculoskelet. Disord. 2020, 21, 181. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Population Sample | Specimen | Results | Correlation with Pain | |
|---|---|---|---|---|
| Peres MF et al., 2004 [74] | 8 CM pts, 12 FM pts and 20 HC | CSF Glu levels measured by HPLC | Higher Glu levels in FM than CM (p < 0.04) and HC (p < 0.001) | Positive correlation of mean pain scores with Glu levels (r 0.551 p < 0.012) |
| Sarchielli P et al., 2007 [75] | 20 FM pts, 20 CM pts and 20 HC | CSF Glu levels measured by HPLC | Higher Glu levels in FM (p < 0.003) and CM (p < 0.001) than HC | Absence of correlation between Glu levels and VAS values and pain intensity, pressure pain threshold and TPC |
| Harris RE et al., 2009 [76] | 19 FM pts and 14 HC | Posterior right insula Glu levels assessed by H-MRS | Higher Glu levels in FM pts (p < 0.009) than HC | Inverse correlation of low (r −0.53 p < 0.002) medium (r −0.43 p < 0.012) and high (r −0.38 p < 0.03) pressure pain thresholds with posterior insula Glu levels |
| Fayed N et al., 2010 [78] | 10 FM pts and 10 HC | Posterior cingulate gyrus Glx and Glx/Cr levels assessed by MRS | Higher Glx (p < 0.049) and Glx/Cr levels in FM pts (p < 0.034) than HC | Inverse correlation of pain threshold assessed by sphygmomanometer with posterior cingulate Glx (r −0.45 p < 0.047) and Glx/Cr levels (r −0.50 p < 0.024) |
| Valdés et al., 2010 [79] | 28 FM pts and 24 HC | Right amygdala Glx and Glx/Cr levels assessed by H-MRS | Higher Glx (p < 0.03) and Glx/Cr levels in FM pts (p < 0.04) than HC | Absence of correlation with pain |
| Feraco T et al., 2011 [80] | 12 FM pts and 12 HC | Right VLPC and left thalamus Glu/Cr levels assessed by MRS | Higher Glu/Cr levels in FM pts (p < 0.01) than HC | Positive correlation of VAS pain (r 0.73 p < 0.007) with left thalamus Glu/Cr levels |
| Population Sample | Specimen | Results | Correlation with Pain | |
|---|---|---|---|---|
| Vaerøy H, et al., 1988 [92] | 30 FM pts | CSF SP levels measured by RIA technique | Higher SP levels in FM pts (p < 0.001) compared to normal value | Not evaluated |
| Reynolds WJ et al., 1988 [95] | 32 FM pts and 26 HC | Plasma SP levels measured by RIA technique | No significant difference between FM pts and HC | Not evaluated |
| Russel IJ et al., 1994 [93] | 32 FM pts and 30 HC | CSF SP levels measured by RIA technique | Higher SP levels in FM pts (p < 0.001) than HC | Absence of correlation between SP levels and pain |
| Tsilioni I et al., 2016 [96] | 84 FM pts and 20 HC | Serum SP levels measured by ELISA technique | Higher SP levels in FM pts (p < 0.0001) than HC | Not evaluated |
| Population Sample | Specimen | Results | Correlation with Pain | |
|---|---|---|---|---|
| Sarchielli P et al., 2007 [75] | 20 FM patients, 20 CM patients and 20 HC | CSF NGF levels measured by ELISA technique | Higher NGF levels in FM pts (p < 0.001) and CM pts (p < 0.0005) than HC | Positive correlation of NGF levels with duration of chronic pain (r 0.66 p < 0.003); absence of correlation between NGF levels and VAS values and pain intensity, pressure pain threshold and TPC |
| Baumeister D et al., 2019 [120] | 97 FM patients and 35 HC | Serum NGF levels measured by ECL assay technique | No significant difference between FM pts and HC | Not evaluated |
| Jablochkova A et al., 2019 [121] | 75 FM patients and 25 HC | Plasma NGF levels measured by ECL assay technique | Lower NGF levels in FM pts (p < 0.001) than HC | Absence of correlation between NGF levels and global pain intensity, pressure pain threshold and TPC |
| Population Sample | Specimen | Results | Correlation with Pain | |
|---|---|---|---|---|
| Sarchielli P et al., 2007 [75] | 20 FM pts, 20 CM pts and 20 HC | CSF BDNF levels measured by ELISA technique | Higher BDNF levels in FM pts (p < 0.001) and CM pts (p < 0.0001) than HC | Positive correlation of BDNF levels with duration of chronic pain (r 0.57 p < 0.01); absence of correlation between BDNF levels and VAS values and pain intensity, pressure pain threshold and TPC |
| Laske C et al., 2007 [132] | 41 FM pts and 45 HC | Plasma BDNF levels measured by ELISA technique | Higher BDNF levels in FM pts (p < 0.0001) than HC | Not evaluated |
| Haas L et al., 2010 [131] | 30 FM pts and 30 HC | Plasma BDNF levels measured by ELISA technique | Higher BDNF levels in FM pts (p < 0.049) than HC | Absence of correlation between BDNF levels and VAS values and TPC |
| Nugraha B et al., 2013 [134] | 28 FM pts and 27 HC | Serum BDNF levels measured by ELISA technique | Higher BDNF levels in FM pts (p < 0.05) than HC | Absence of correlation between BDNF levels and pain and TPC |
| Ranzolin A et al., 2016 [137] | 69 FM pts and 61 HC | Serum BDNF levels measured by ELISA technique | No significant difference between FM pts and HC | Not evaluated |
| Baumeister D et al., 2019 [121] | 97 FM pts and 35 HC | Serum BDNF levels measured by ELISA technique | No significant difference between FM pts and HC | Not evaluated |
| Jablochkova A et al., 2019 [121] | 75 FM pts and 25 HC | Plasma BDNF levels measured by ECL assay technique | Higher BDNF levels in FM pts (p < 0.001) than HC | Absence of correlation between BDNF levels and global pain intensity, pressure pain threshold and TPC |
| Iannuccelli C et al., 2022 [138] | 40 FM pts and 40 HC | Serum BDNF levels measured by ELISA technique | Lower BDNF levels in FM pts (p < 0.0001) than in HC | Absence of correlation between BDNF levels and TPC |
| Bidari A et al., 2022 [143] | 53 FM pts and 23 non-FM chronic nociceptive pain pts | Serum BDNF levels measured by ELISA technique | No significant difference between FM pts and non-FM chronic nociceptive pain pts | Negative correlation of BDNF levels with VAS pain (r −0.32, p < 0.05) |
| Population Sample | Specimen | Results | Correlation with Pain | |
|---|---|---|---|---|
| Skare TL et al., 2015 [176] | 94 FM pts and 94 HC | Plasma PTX-3 levels measured by ELISA technique | Higher PTX-3 levels in FM pts (p < 0.005) than HC | Absence of correlation between PTX-3 levels and pain |
| Garcia JJ et al., 2016 [178] | 10 FM pts and 10 HC | PTX-3 release by phagocytes, measured by ELISA technique | Higher PTX-3 constitutive release in FM pts (p < 0.05) than HC | Not evaluated |
| Population Sample | Specimen | Results | Correlation with Pain | |
|---|---|---|---|---|
| Crofford LJ et al., 1994 [187] | 12 FM pts and 10 HC | Plasma NPY levels measured by RIA technique | Lower NPY levels in FM pts (p < 0.001) than HC | Not evaluated |
| Anderberg UM et al., 1999 [191] | 24 FM pts and 17 HC | Plasma NPY levels measured by RIA technique | Higher NPY levels in FM pts (p < 0.002) than HC | Absence of correlation between NPY levels and pain |
| Di Franco M et al., 2009 [190] | 51 FM pts, 25 SSc pts and 15 HC | Serum NPY levels measured by immunoenzymatic assay technique | Higher NPY levels in FM pts (p < 0.0001) than SSc patients and HC | Absence of correlation between NPY levels and pain |
| Iannuccelli C et al., 2010 [185] | 51 FM pts, 25 TTH patients and 15 HC | Serum NPY levels measured by immunoenzymatic assay technique | Higher NPY levels in FM pts (p < 0.0001) than HC | Absence of correlation between NPY levels and pain |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Favretti, M.; Iannuccelli, C.; Di Franco, M. Pain Biomarkers in Fibromyalgia Syndrome: Current Understanding and Future Directions. Int. J. Mol. Sci. 2023, 24, 10443. https://doi.org/10.3390/ijms241310443
Favretti M, Iannuccelli C, Di Franco M. Pain Biomarkers in Fibromyalgia Syndrome: Current Understanding and Future Directions. International Journal of Molecular Sciences. 2023; 24(13):10443. https://doi.org/10.3390/ijms241310443
Chicago/Turabian StyleFavretti, Martina, Cristina Iannuccelli, and Manuela Di Franco. 2023. "Pain Biomarkers in Fibromyalgia Syndrome: Current Understanding and Future Directions" International Journal of Molecular Sciences 24, no. 13: 10443. https://doi.org/10.3390/ijms241310443
APA StyleFavretti, M., Iannuccelli, C., & Di Franco, M. (2023). Pain Biomarkers in Fibromyalgia Syndrome: Current Understanding and Future Directions. International Journal of Molecular Sciences, 24(13), 10443. https://doi.org/10.3390/ijms241310443