
Novel 2-alkythio-4-chloro-N-[imino(heteroaryl)methyl]benzenesulfonamide Derivatives: Synthesis, Molecular Structure, Anticancer Activity and Metabolic Stability

 ,
,  , ,
, ,  and
and 
Abstract
1. Introduction
2. Results and Discussion
2.1. Chemistry
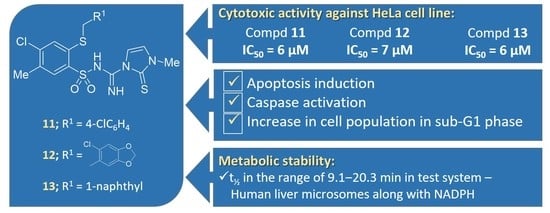
2.2. Screening for Anticancer Agents
2.3. Apoptosis
2.3.1. Cytotoxic Activity
2.3.2. Apoptosis Induction
2.3.3. Caspase Activation
2.3.4. Cell Cycle Distribution
2.4. In Vitro Metabolic Stability Assay
3. Materials and Methods
3.1. Synthesis
3.2. X-ray Structure Determination
3.3. Cell Culture and Cell Viability Assay
3.4. In Vitro Metabolic Stability Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ferlay, J.; Ervik, M.; Lam, F.; Colombet, M.; Mery, L.; Piñeros, M. Global Cancer Observatory: Cancer Today; International Agency for Research on Cancer: Lyon, France, 2020; Available online: https://gco.iarc.fr/today (accessed on 9 May 2023).
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Barreca, M.; Ingarra, A.M.; Raimondi, M.V.; Spanò, V.; Piccionello, A.P.; De Franco, M.; Menilli, L.; Gandin, V.; Miolo, G.; Barraja, P.; et al. New tricyclic systems as photosensitizers towards triple negative breast cancer cells. Arch. Pharmacal Res. 2022, 45, 806–821. [Google Scholar] [CrossRef]
- Liu, J.; Zhu, X.; Yu, L.; Mao, M. Discovery of novel sulphonamide hybrids that inhibit LSD1 against bladder cancer cells. J. Enzym. Inhib. Med. Chem. 2022, 37, 866–875. [Google Scholar] [CrossRef] [PubMed]
- AL-Ghulikah, H.A.; El-Sebaey, S.A.; Bass, A.K.A.; El-Zoghbi, M.S. New Pyrimidine-5-Carbonitriles as COX-2 Inhibitors: Design, Synthesis, Anticancer Screening, Molec-ular Docking, and In Silico ADME Profile Studies. Molecules 2022, 27, 7485. [Google Scholar] [CrossRef]
- Grillone, K.; Riillo, C.; Rocca, R.; Ascrizzi, S.; Spanò, V.; Scionti, F.; Polerà, N.; Maruca, A.; Barreca, M.; Juli, G.; et al. The New Microtubule-Targeting Agent SIX2G In-duces Immunogenic Cell Death in Multiple Myeloma. Int. J. Mol. Sci. 2022, 23, 10222. [Google Scholar] [CrossRef] [PubMed]
- Fortin, S.; Bérubé, G. Advances in the development of hybrid anticancer drugs. Expert Opin. Drug Discov. 2013, 8, 1029–1047. [Google Scholar] [CrossRef]
- Nepali, K.; Sharma, S.; Sharma, M.; Bedi, P.M.; Dhar, K.L. Rational approaches, design strategies, structure activity relationship and mechanistic insights for anticancer hybrids. Eur. J. Med. Chem. 2014, 77, 422–487. [Google Scholar] [CrossRef]
- Kleczkowska, P.; Kowalczyk, A.; Lesniak, A.; Bujalska-Zadrozny, M. The Discovery and Development of Drug Combinations for the Treatment of Various Diseases from Patent Literature (1980–Present). Curr. Top. Med. Chem. 2017, 17, 875–894. [Google Scholar] [CrossRef]
- Kleczkowska, P. Chimeric Structures in Mental Illnesses—“Magic” Molecules Specified for Complex Disorders. Int. J. Mol. Sci. 2022, 23, 3739. [Google Scholar] [CrossRef]
- Morphy, R.; Rankovic, Z. Designed Multiple Ligands. An Emerging Drug Discovery Paradigm. J. Med. Chem. 2005, 48, 6523–6543. [Google Scholar] [CrossRef]
- Fujii, H. Twin and Triplet Drugs in Opioid Research. In Chemistry of Opioids; Topics in Current Chemistry; Nagase, H., Ed.; Springer: Berlin/Heidelberg, Germany, 2010; pp. 239–275. [Google Scholar]
- Abdolmaleki, A.; Ghasemi, J.B. Dual-acting of hybrid compounds—A new dawn in the discovery of multi-target drugs: Lead generation approaches. Curr. Top. Med. Chem. 2017, 17, 1096–1114. [Google Scholar] [CrossRef] [PubMed]
- Qingjie, Z.; Guozheng, H. Anticancer Hybrids. In Design of Hybrid Molecules for Drug Development; Decker, M., Ed.; Elsevier: Amsterdam, The Netherlands, 2017; pp. 193–218. [Google Scholar]
- Nagaraju, K.; Lalitha, G.; Suresh, M.; Kranthi, K.G.; Sreekantha, B.J. A Review on Recent Advances in Nitrogen-Containing Molecules and Their Biological Applications. Molecules 2020, 25, 1909. [Google Scholar]
- Ali, I.; Lone, M.N.; Aboul-Enein, H.Y. Imidazoles as potential anticancer agents. Med. Chem. Commun. 2017, 8, 1742–1773. [Google Scholar] [CrossRef]
- Rani, N.; Sharma, A.; Singh, R. Imidazoles as promising scaffolds for antibacterial activity: A review. Mini Rev. Med. Chem. 2013, 13, 1812–1835. [Google Scholar] [CrossRef] [PubMed]
- Zhan, P.; Liu, X.; Zhu, J.; Fang, Z.; Li, Z.; Pannecouque, C.; de Clercq, E. Synthesis and biological evaluation of imidazole thioacetanilides as novel non-nucleoside HIV-1 reverse transcriptase inhibitors. Bioorg. Med. Chem. 2009, 17, 5775–5781. [Google Scholar] [CrossRef] [PubMed]
- Mishra, R.; Ganguly, S. Imidazole as an anti-epileptic: An overview. Med. Chem. Res. 2012, 21, 3929–3939. [Google Scholar] [CrossRef]
- Fan, Y.L.; Jin, X.H.; Huang, Z.P.; Yu, H.F.; Zeng, Z.G.; Gao, T.; Feng, L.S. Recent advances of imidazole-containing derivatives as anti-tubercular agents. Eur. J. Med. Chem. 2018, 150, 347–365. [Google Scholar] [CrossRef]
- Rani, N.; Sharma, A.; Gupta, G.K.; Singh, R. Imidazoles as potential antifungal agents: A review. Mini Rev. Med. Chem. 2013, 13, 1626–1655. [Google Scholar] [CrossRef]
- Meng, L.; Zhao, P.; Hu, Z.; Ma, W.; Niu, Y.; Su, J.; Zhang, Y. Nilotinib, A Tyrosine Kinase Inhibitor, Suppresses the Cell Growth and Triggers Autophagy in Papillary Thyroid Cancer. Anticancer Agents Med. Chem. 2022, 22, 596–602. [Google Scholar] [CrossRef]
- Amatu, A.; Sartore-Bianchi, A.; Moutinho, C.; Belotti, A.; Bencardino, K.; Chirico, G.; Cassingena, A.; Rusconi, F.; Esposito, A.; Nichelatti, M.; et al. Promoter CpG island hypermethylation of the DNA repair enzyme MGMT predicts clinical response to dacarbazine in a phase II study for metastatic colorectal cancer. Clin. Cancer Res. 2015, 19, 2265–2272. [Google Scholar] [CrossRef]
- Moulin, A.; Bibian, M.; Blayo, A.L.; El Habnouni, S.; Martinez, J.; Fehrentz, J.A. Synthesis of 3,4,5-trisubstituted-1,2,4-triazoles. Chem. Rev. 2010, 110, 1809–1827. [Google Scholar] [CrossRef]
- Gupta, D.; Jain, D.K. Synthesis, antifungal and antibacterial activity of novel 1,2,4-triazole derivatives. J. Adv. Pharm. Technol. Res. 2015, 6, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Sarigol, D.; Uzgoren-Baran, A.; Tel, B.C.; Somuncuoglu, E.I.; Kazkayasi, I.; Ozadali-Sari, K.; Unsal-Tan, O.; Okay, G.; Ertan, M.; Tozkoparan, B. Novel thiazolo[3,2-b]-1,2,4-triazoles derived from naproxen with analgesic/anti-inflammatory properties: Synthesis, biological evaluation and molecular modeling studies. Bioorg. Med. Chem. 2015, 23, 2518–2528. [Google Scholar] [CrossRef] [PubMed]
- Karczmarzyk, Z.; Swatko-Ossor, M.; Wysocki, W.; Drozd, M.; Ginalska, G.; Pachuta-Stec, A.; Pitucha, M. New Application of 1,2,4-Triazole Derivatives as Antitubercular Agents. Structure, In Vitro Screening and Docking Studies. Molecules 2020, 25, 6033. [Google Scholar] [CrossRef] [PubMed]
- El-Sebaey, S.A. Recent Advances in 1,2,4-Triazole Scaffolds as Antiviral Agents. ChemistrySelect 2020, 5, 11654–11680. [Google Scholar] [CrossRef]
- Azim, T.; Wasim, M.; Akhtar, M.S.; Akram, I. An in vivo evaluation of anti-inflammatory, analgesic and anti-pyretic activities of newly synthesized 1,2,4 Triazole derivatives. BMC Complement. Med. Ther. 2021, 21, 304. [Google Scholar] [CrossRef] [PubMed]
- Ramandeep, K.; Ashish, D.R.; Bhupinder, K.; Vinod, K. Recent Developments on 1,2,4-Triazole Nucleus in Anticancer Compounds: A Review. Anticancer Agents Med. Chem. 2016, 16, 465–489. [Google Scholar]
- Chumsri, S. Clinical utilities of aromatase inhibitors in breast cancer. Int. J. Womens Health 2015, 7, 493–499. [Google Scholar] [CrossRef]
- Talpaz, M.; Kiladjian, J.J. Fedratinib, a newly approved treatment for patients with myeloproliferative neoplasm-associated myelofibrosis. Leukemia 2021, 35, 1–17. [Google Scholar] [CrossRef]
- Kumar, R.; Knick, V.B.; Rudolph, S.K.; Johnson, J.H.; Crosby, R.M.; Crouthamel, M.C.; Hopper, T.M.; Miller, C.G.; Harrington, L.E.; Onori, J.A.; et al. Pharmacokinetic-pharmacodynamic correlation from mouse to human with pazopanib, a multikinase angiogenesis inhibitor with potent antitumor and antiangiogenic activity. Mol. Cancer Ther. 2007, 6, 2012–2021. [Google Scholar] [CrossRef]
- Long, G.V.; Hauschild, A.; Santinami, M.; Atkinson, V.; Mandala, M.; Chiarion-Sileni, V.; Larkin, J.; Nyakas, M.; Dutriaux, C.; Haydon, A.; et al. Adjuvant Dabrafenib plus Trametinib in Stage III BRAF-Mutated Melanoma. N. Engl. J. Med. 2017, 377, 1813–1823. [Google Scholar] [CrossRef] [PubMed]
- Fisher, R.; Larkin, J. Vemurafenib: A new treatment for BRAF-V600 mutated advanced melanoma. Cancer Manag. Res. 2012, 4, 243–252. [Google Scholar] [PubMed]
- Guerra, V.A.; DiNardo, C.; Konopleva, M. Venetoclax-based therapies for acute myeloid leukemia. Best Pract. Res. Clin. Haematol. 2019, 32, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Mohamad Anuar, N.N.; Nor Hisam, N.S.; Liew, S.L.; Ugusman, A. Clinical Review: Navitoclax as a Pro-Apoptotic and Anti-Fibrotic Agent. Front. Pharmacol. 2020, 11, 564108. [Google Scholar] [CrossRef]
- Meier, T.; Uhlik, M.; Chintharlapalli, S.; Dowless, M.; Van Horn, R.; Stewart, J.; Blosser, W.; Cook, J.; Young, D.; Ye, X.; et al. Tasisulam sodium, an antitumor agent that inhibits mitotic progression and induces vascular normalization. Mol. Cancer Ther. 2011, 10, 2168–2178. [Google Scholar] [CrossRef]
- Fulda, S. Tumor resistance to apoptosis. Int. J. Cancer 2009, 124, 511–515. [Google Scholar] [CrossRef]
- Fulda, S. Evasion of apoptosis as a cellular stress response in cancer. Int. J. Cell Biol. 2010, 2010, 370835. [Google Scholar] [CrossRef]
- Sławiński, J. Syntheses and some reactions of 3-amino-6-chloro-7-methyl-1,1-dioxo-1,4,2-benzodithiazine. Polish J. Chem. 2001, 75, 1309–1316. [Google Scholar]
- Sławiński, J.; Żołnowska, B.; Orlewska, C.; Chojnacki, J. Synthesis and molecular structure of novel 2-(alkylthio)-4-chloro-N-(4,5-dihydro-5-oxo-1H-1,2,4-triazol-3-yl)-5-methylbenzenesulfonamides with potential anticancer activity. Mon. Für Chem. 2012, 143, 1705–1718. [Google Scholar] [CrossRef]
- Sławiński, J.; Pogorzelska, A.; Żołnowska, B.; Kędzia, A.; Ziółkowska-Klinkosz, M.; Kwapisz, E. Synthesis and anti-yeast evaluation of novel 2-alkylthio-4-chloro-5-methyl-N-[imino-(1-oxo-(1H)-phthalazin-2-yl)methyl]benzenesulfonamide derivatives. Molecules 2014, 19, 13704–13723. [Google Scholar] [CrossRef]
- Żołnowska, B.; Sławiński, J.; Pogorzelska, A.; Chojnacki, J.; Vullo, D.; Supuran, C.T. Carbonic anhydrase inhibitors. Synthesis, and molecular structure of novel series N-substituted N’-(2-arylmethylthio-4-chloro-5-methylbenzenesulfonyl)guanidines and their inhibition of human cytosolic isozymes I and II and the transmembrane tumor-associated isozymes IX and XII. Eur. J. Med. Chem. 2014, 17, 135–147. [Google Scholar]
- Zaretzki, J.; Matlock, M.; Swamidass, S.J. XenoSite: Accurately Predicting CYP-Mediated Sites of Metabolism with Neural Networks. J. Chem. Inf. Model. 2013, 53, 3373–3383. [Google Scholar] [CrossRef] [PubMed]
- Henderson, M.C.; Siddens, L.K.; Krueger, S.K.; Stevens, J.F.; Kedzie, K.; Fang, W.K.; Heidelbaugh, T.; Nguyen, P.; Chow, K.; Garst, M.; et al. Flavin-containing monooxygenase S-oxygenation of a series of thioureas and thiones. Toxicol. Appl. Pharmacol. 2014, 278, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, M.; Shimizu, M.; Uno, Y.; Yamazaki, H. Drug oxygenation activities mediated by liver microsomal flavin-containing monooxygenases 1 and 3 in humans, monkeys, rats, and minipigs. Biochem. Pharmacol. 2014, 90, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. 2015, A71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. 2015, C71, 3–8. [Google Scholar]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Obach, R.S. Prediction of Human Clearance of Twenty-Nine Drugs from Hepatic Microsomal Intrinsic Clearance Data: An Examination of In Vitro Half-Life Approach and Nonspecific Binding to Microsomes. Drug Metab. Dispos. 1999, 27, 1350–1359. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | IC50 [µM] | |||
|---|---|---|---|---|
| MCF-7 | HeLa | HCT-116 | HaCaT | |
| 11 | 330 ± 10 | 6 ± 0.1 | 11 ± 0.3 | 18 ± 0.5 |
| 12 | * | 7 ± 0.3 | 180 ± 4.5 | 20 ± 1 |
| 13 | * | 6 ± 0.1 | 73 ± 2 | 18 ± 0.5 |
| 19 | 82 ± 4 | 60 ± 3 | 36 ± 1 | NT |
| 20 | 49 ± 1 | 43 ± 1 | 17 ± 0.5 | NT |
| 21 | 62 ± 3 | 52 ± 1 | 23 ± 0.2 | NT |
| 22 | 73 ± 2 | 49 ± 2 | 27 ± 0.5 | NT |
| 23 | 50 ± 2 | 57 ± 1 | 34 ± 0.3 | NT |
| 24 | 56 ± 2 | 33 ± 1 | 21 ± 0.6 | NT |
| Cisplatin | 3.0 ± 0.1 | 2.2 ± 0.1 | 3.8 ± 0.2 | 7.7 ± 0.2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Żołnowska, B.; Sławiński, J.; Belka, M.; Bączek, T.; Chojnacki, J.; Kawiak, A. Novel 2-alkythio-4-chloro-N-[imino(heteroaryl)methyl]benzenesulfonamide Derivatives: Synthesis, Molecular Structure, Anticancer Activity and Metabolic Stability. Int. J. Mol. Sci. 2023, 24, 9768. https://doi.org/10.3390/ijms24119768
Żołnowska B, Sławiński J, Belka M, Bączek T, Chojnacki J, Kawiak A. Novel 2-alkythio-4-chloro-N-[imino(heteroaryl)methyl]benzenesulfonamide Derivatives: Synthesis, Molecular Structure, Anticancer Activity and Metabolic Stability. International Journal of Molecular Sciences. 2023; 24(11):9768. https://doi.org/10.3390/ijms24119768
Chicago/Turabian StyleŻołnowska, Beata, Jarosław Sławiński, Mariusz Belka, Tomasz Bączek, Jarosław Chojnacki, and Anna Kawiak. 2023. "Novel 2-alkythio-4-chloro-N-[imino(heteroaryl)methyl]benzenesulfonamide Derivatives: Synthesis, Molecular Structure, Anticancer Activity and Metabolic Stability" International Journal of Molecular Sciences 24, no. 11: 9768. https://doi.org/10.3390/ijms24119768
APA StyleŻołnowska, B., Sławiński, J., Belka, M., Bączek, T., Chojnacki, J., & Kawiak, A. (2023). Novel 2-alkythio-4-chloro-N-[imino(heteroaryl)methyl]benzenesulfonamide Derivatives: Synthesis, Molecular Structure, Anticancer Activity and Metabolic Stability. International Journal of Molecular Sciences, 24(11), 9768. https://doi.org/10.3390/ijms24119768