
How to Set Up Genetic Counselling for Inherited Macular Dystrophies: Focus on Genetic Characterization

 , , , , ,
, , , , ,  , ,
, , 
Abstract
1. Introduction
1.1. Most Common Types of IMDs
1.2. Transmission

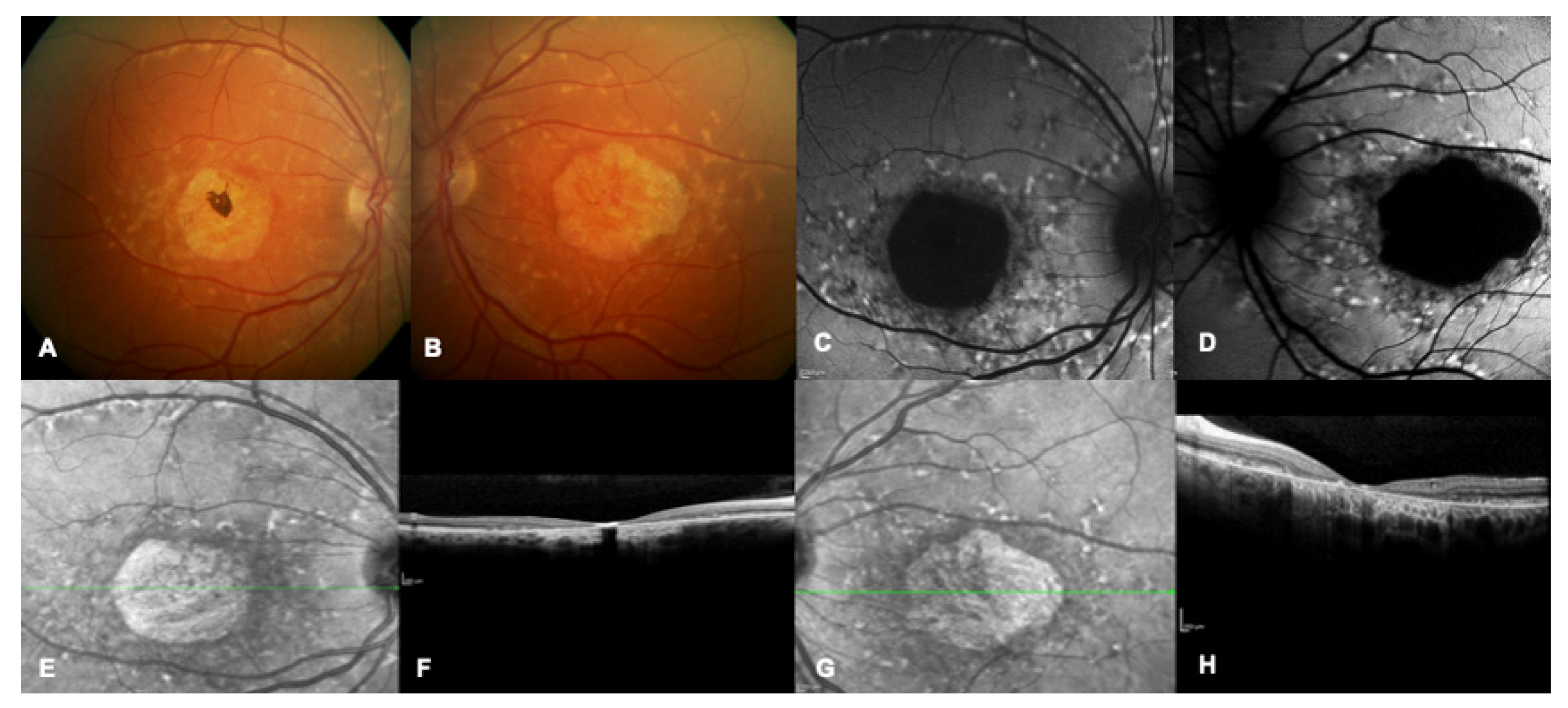{kind=link}
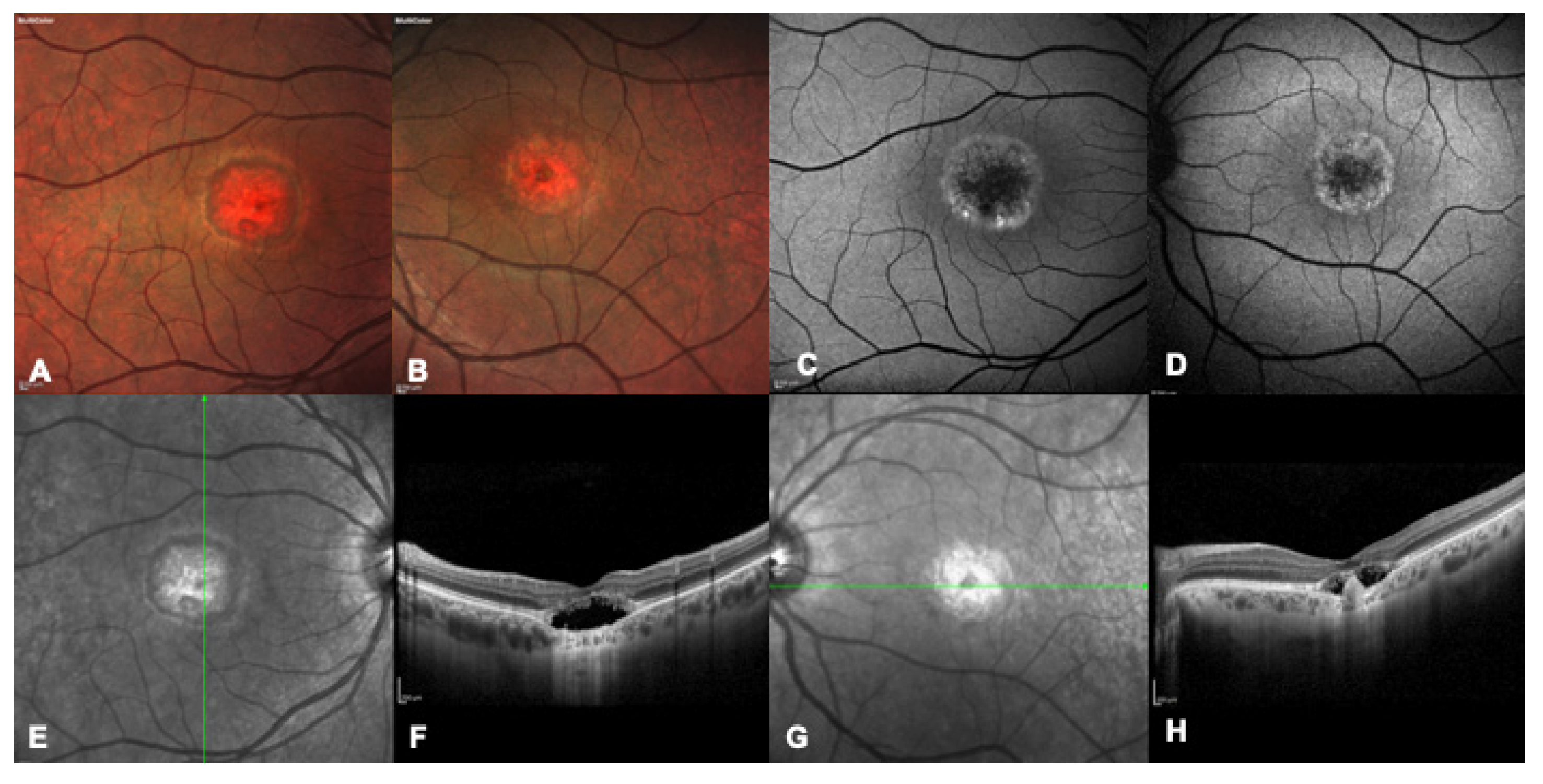{kind=link}
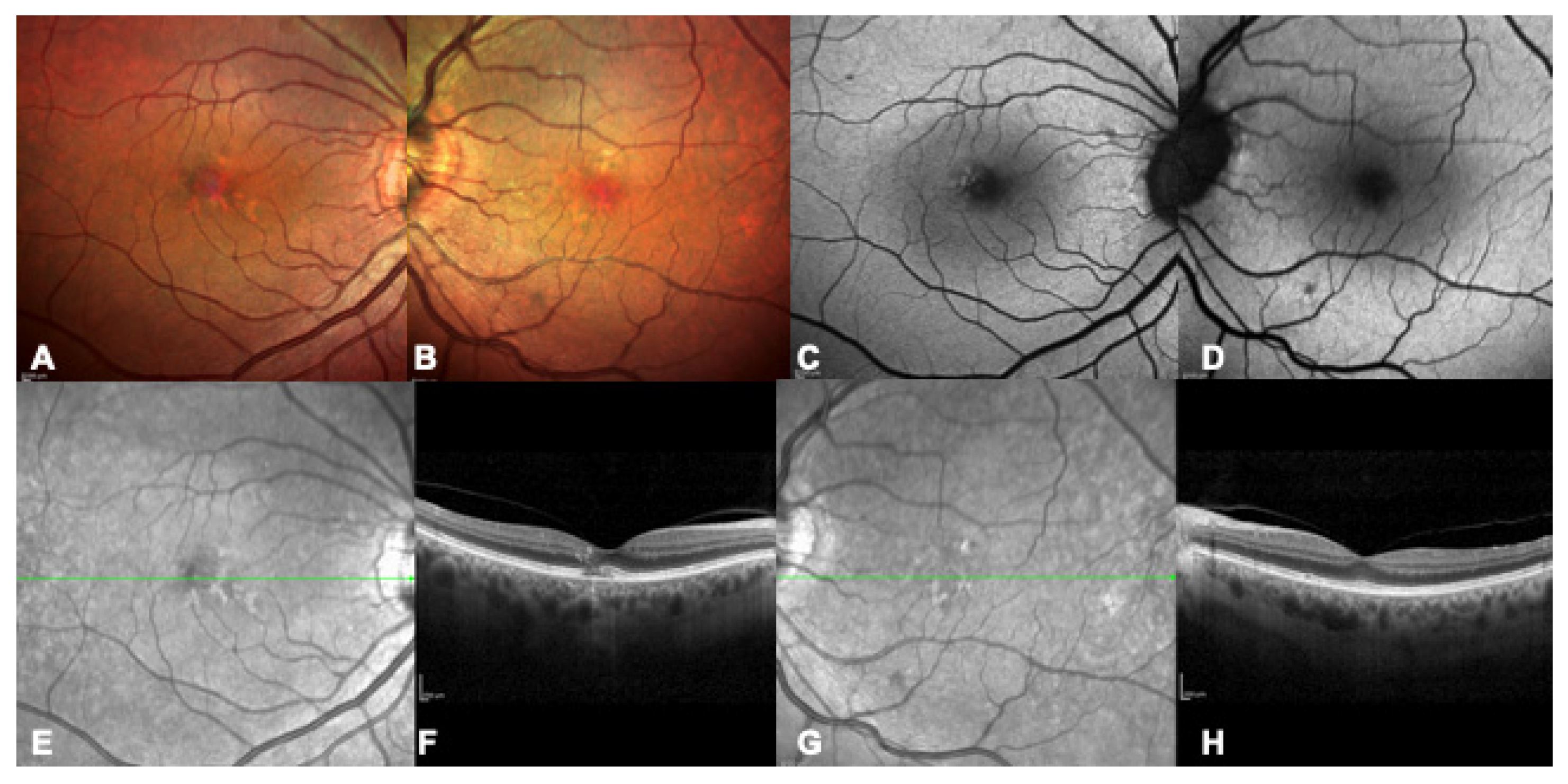{kind=link}
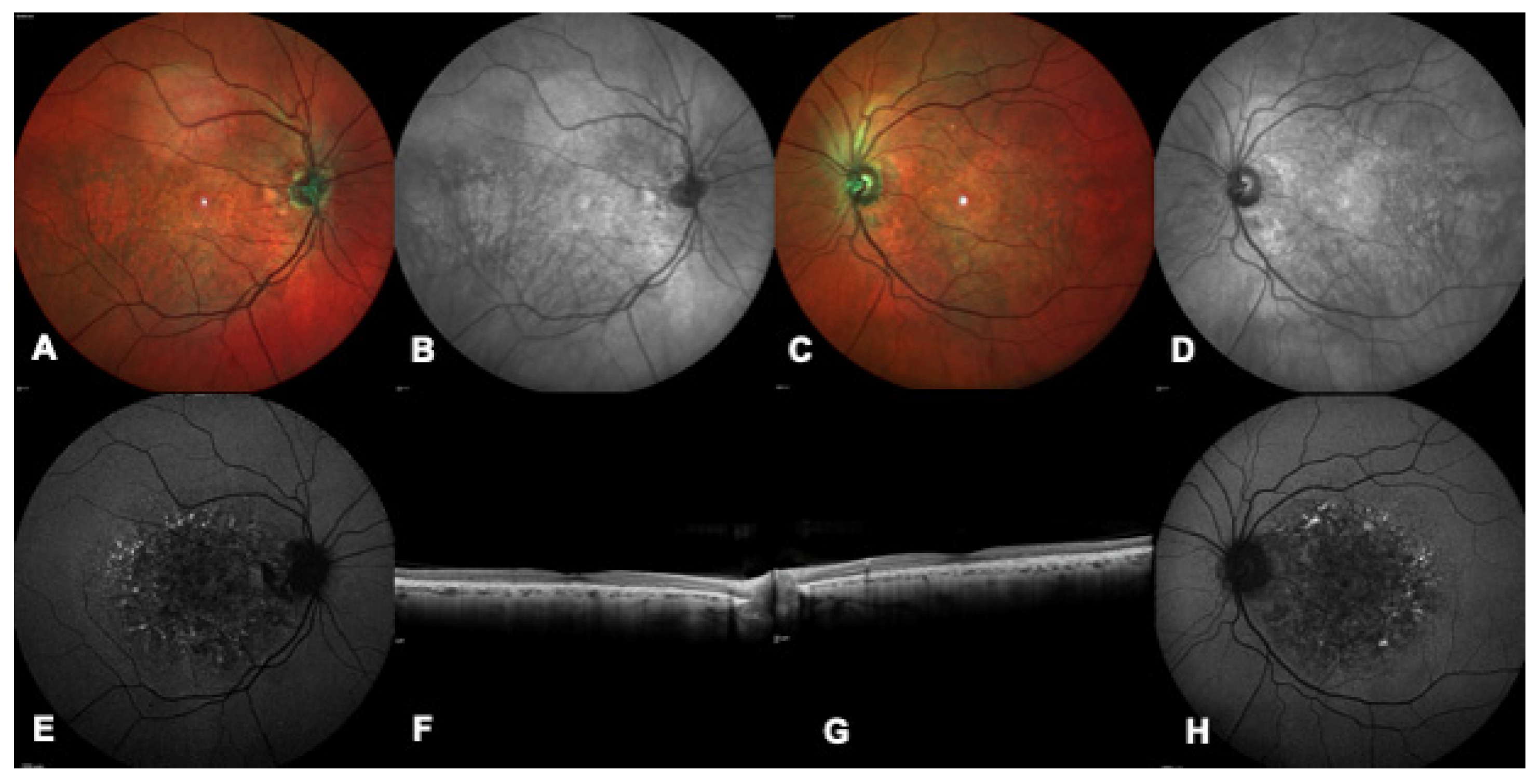{kind=link}
{kind=link}
| IMD | Clinical Presentation | Imaging Presentation | Therapeutic Options | Ongoing Trials | Ref. |
|---|---|---|---|---|---|
| Stargardt and Stargardt-like disease | Decreased central vision FO: foveal atrophy, bull’s eye pattern, yellow macular pisciform flecks (fundus flavimaculatus) | OCT: photoreceptor layer and RPE atrophy OCT-A: choriocapillaris anomalies FAG: paramacular hyperfluorescence associated with flecks (bull’s-eye aspect) and dark (or ‘silent’) choroid (screen effect on normal choroidal fluorescence) FAF: elevated background autofluorescence, central macular hypoautofluorescence, hyperautofluorescent flecks interleaved with hypoautofluorescent areas, peripapillary sparing of the RPE changes. | Patients with Stargardt disease should avoid supplementation of vitamin A and exposition to bright light. | Drug therapies to reduce lipofuscin accumulation, gene therapies, and stem cell treatments. A recent trial explored the use of a lentivirus, equine infectious anaemia virus (EIAV) to carry the ABCA4 gene. | [4,6,9,26,28,31,32,33,34,35,36] |
| Best disease/Adult Vitelliform Macular Dystrophy | Decreased vision, hyperopia (BEST1) FO: Egg yolk-like lesion (stage I-II), evolving to pseudohypopyon (stage III), reabsorption (stage IV) and geographic atrophy (stage V) | OCT: hyperreflective vitelliform lesion between the EZ and the RPE, cysts in the neurosensory retina FAF: hyperautofluorescent zones due to the lipofuscin accumulation, hypoautofluorescent zones due to the RPE atrophy | No therapeutic options are available. Choroidal neovascularization (CNV) can be treated with antiVEGF intravitreal therapy. | [11,37] | |
| PRPH2 (more common), CTNNA1 | Often asymptomatic, patients may develop metamorphopsia, a slight decrease in vision and a delayed recovery from exposure to bright light. Rarely, patients may present with rapid vision loss due to development of CNV. FO: various pattern of lipofuscin accumulation due to RPE defects. Often intra-familial variability | OCT: subretinal hyperreflective lesions FAF: areas of hyperfluorescence or hypofluorescence corresponding to lipofuscin contents in the RPE | No therapeutic options are available, CNV can be treated with antiVEGF therapy. | [13,38] | |
| X-linked retinoschisis | FO: Small superficial cysts arranged in a stellate pattern radiating from the fovea, evolving to non-specific atrophy in late stages. The peripheral retina may show RPE alterations and schisis | OCT: inner retinal layer schisis FAF: modification of normal foveal autofluorescence with a radial pattern | Oral acetazolamide or topic inhibitors of carbonic anhydrase (CAIs) may reduce cystic spaces, vitreoretinal surgery in patients who develop vitreous hemorrhage or rhegmatogenous retinal detachment | Ongoing trials about RS1 gene therapy that demonstrated a good profile of security in animal models | [14,15,39,40,41] |
| North-Carolina macular dystrophy | Normal visual acuity in grade 1 and grade 2, central visual loss in grade 3. | FO: drusen (grade1), macular yellowish-white atrophic lesions (grade 2), colobomatous macular defects (grade 3) | No therapeutic options are avaliable | [17,18] | |
| Doyne macular dystrophy | Asymptomatic until the 4th or the 5th decade, metamorphopsis and visual acuity reduction. | OCT: hyperreflective deposits between the RPE and Bruch’s membrane start as focal dome-shaped, saw-tooth, or diffuse elevations. As time passes, they tend to merge and become more confluent. | No therapeutic options are available. Choroidal neovascularization (CNV) can be treated with antiVEGF intravitreal therapy. | [20] | |
| Sorsby fundus dystrophy | Around the ages of 40 to 60, decline in visual acuity and onset of metamorphopsias. | OCT: diffused drusenoid deposits, possible reticular pseudodrusen | No therapeutic options are available. Choroidal neovascularization (CNV) can be treated with antiVEGF intravitreal therapy. | [21] |
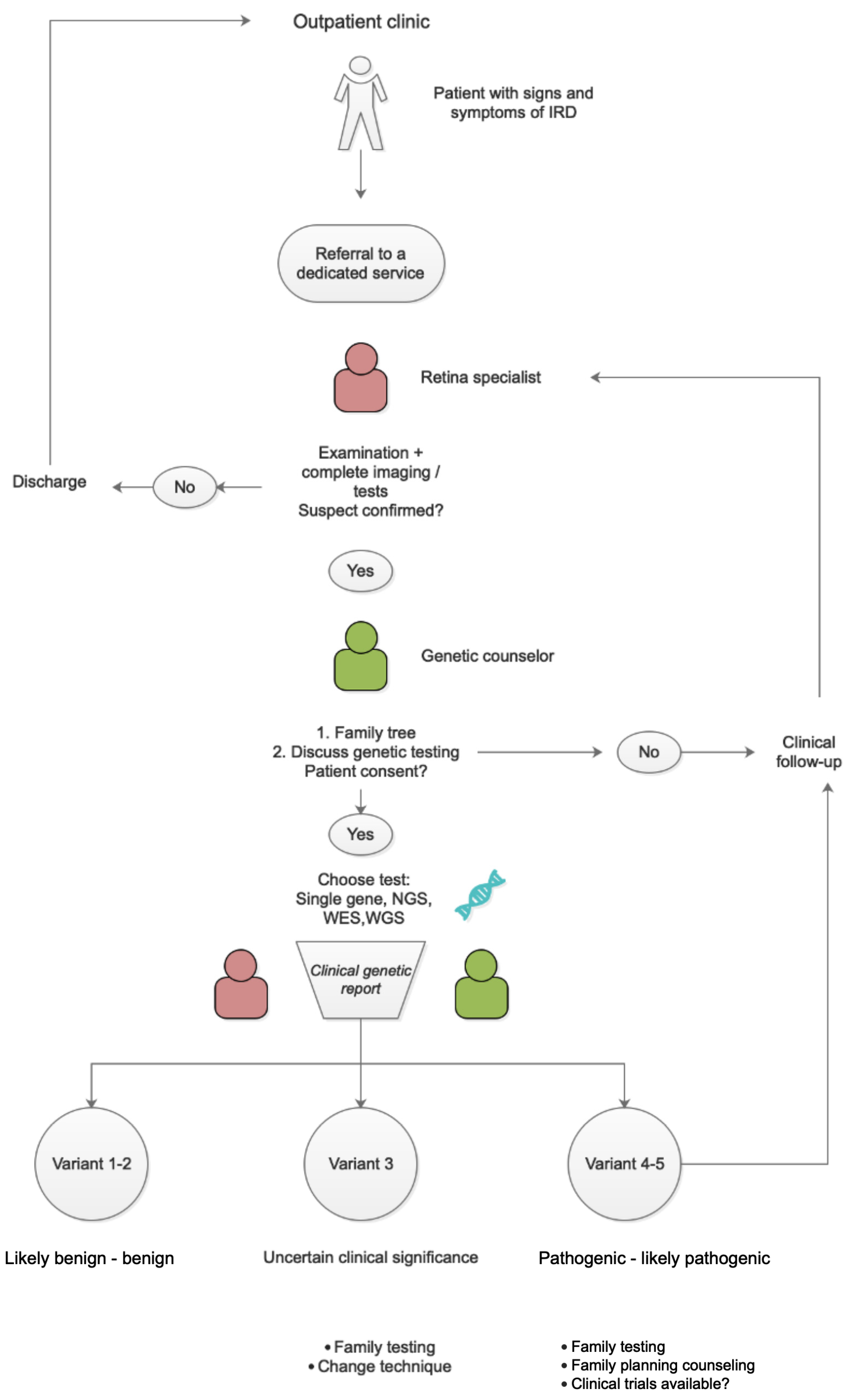
2. Pattern of Care
2.1. Interviewing—Data Collection about Age at Onset, Symptoms, and Evolution
2.2. Phenotyping
2.2.1. Imaging
2.2.2. Electrodiagnostic Tests (EDTS)
3. Genetic Testing
4. Discussion
5. Therapeutic Options
5.1. Gene Therapy
5.2. Gene Editing
5.3. Optogenetics
5.4. Drug Therapies
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rahman, N.; Georgiou, M.; Khan, K.N.; Michaelides, M. Macular dystrophies: Clinical and imaging features, molecular genetics and therapeutic options. Br. J. Ophthalmol. 2020, 104, 451–460. [Google Scholar] [CrossRef] [PubMed]
- Yla-Herttuala, S. Gene therapy in age related macular degeneration and hereditary macular disorders. Front. Biosci. 2012, E4, 565. [Google Scholar] [CrossRef] [PubMed]
- Baig, A.; Buckley, D.; Codina, C. Behavioural adaptation to hereditary macular dystrophy: A systematic review on the effect of early onset central field loss on peripheral visual abilities. Br. Ir. Orthopt. J. 2021, 17, 104–118. [Google Scholar] [CrossRef] [PubMed]
- Lois, N.; Holder, G.E.; Bunce, C.; Fitzke, F.W.; Bird, A.C. Phenotypic subtypes of Stargardt macular dystrophy-fundus flavimaculatus. Arch. Ophthalmol. 2001, 119, 359–369. [Google Scholar] [CrossRef] [PubMed]
- ABCA4 Gene. Available online: www.lovd.nl/ABCA4 (accessed on 5 May 2023).
- Zernant, J.; Lee, W.; Nagasaki, T.; Collison, F.T.; Fishman, G.A.; Bertelsen, M.; Rosenberg, T.; Gouras, P.; Tsang, S.H.; Allikmets, R. Extremely hypomorphic and severe deep intronic variants in the ABCA4 locus result in varying Stargardt disease phenotypes. Cold Spring Harb. Mol. Case Stud. 2018, 4, a002733. [Google Scholar] [CrossRef]
- Méjécase, C.; Malka, S.; Guan, Z.; Slater, A.; Arno, G.; Moosajee, M. Practical guide to genetic screening for inherited eye diseases. Ther. Adv. Ophthalmol. 2020, 12, 2515841420954592. [Google Scholar] [CrossRef]
- Garg, A.; Lee, W.; Sengillo, J.D.; Allikmets, R.; Garg, K.; Tsang, S.H. Peripapillary sparing in RDH12-associated Leber congenital amaurosis. Ophthalmic Genet. 2017, 38, 575–579. [Google Scholar] [CrossRef]
- Amaral, R.A.S.; Zin, O.A.; Salles, M.V.; Motta, F.L.; Sallum, J.M.F. Macular dystrophies associated with Stargardt-like phenotypes. Arq. Bras. Oftalmol. 2023; S0004-27492023005002303, ahead-of-print. [Google Scholar] [CrossRef]
- Dalvin, L.A.; Pulido, J.S.; Marmorstein, A.D. Vitelliform dystrophies: Prevalence in Olmsted County, Minnesota, United States. Ophthalmic Genet. 2017, 38, 143–147. [Google Scholar] [CrossRef]
- Johnson, A.A.; Guziewicz, K.E.; Lee, C.J.; Kalathur, R.C.; Pulido, J.S.; Marmorstein, L.Y.; Marmorstein, A.D. Bestrophin 1 and retinal disease. Prog. Retin. Eye Res. 2017, 58, 45–69. [Google Scholar] [CrossRef]
- Souied, E.H.; Querques, G. Macular Dystrophies, 1st ed.; Springer: Cham, Switzerland, 2016. [Google Scholar]
- Renner, A.B.; Fiebig, B.S.; Weber, B.H.F.; Wissinger, B.; Andreasson, S.; Gal, A.; Cropp, E.; Kohl, S.; Kellner, U. Phenotypic variability and long-term follow-up of patients with known and novel PRPH2/RDS gene mutations. Am. J. Ophthalmol. 2009, 147, 518–530. [Google Scholar] [CrossRef]
- Audo, I.; Sahel, J.; Mohand-Said, S.; Holder, G.; Moore, A. X-Linked Retinoschisis. In Macular Dystophies; Springer: Cham, Switzerland, 2016; pp. 71–82. [Google Scholar]
- Sikkink, S.K.; Biswas, S.; Parry, N.R.A.; Stanga, P.E.; Trump, D. X-linked retinoschisis: An update. J. Med. Genet. 2007, 44, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Vijayasarathy, C.; Ziccardi, L.; Sieving, P.A. Biology of retinoschisin. Adv. Exp. Med. Biol. 2012, 723, 513–518. [Google Scholar] [PubMed]
- Small, K.W.; DeLuca, A.P.; Whitmore, S.S.; Rosenberg, T.; Silva-Garcia, R.; Udar, N.; Puech, B.; Garcia, C.A.; Rice, T.A.; Fishman, G.A.; et al. North Carolina Macular Dystrophy Is Caused by Dysregulation of the Retinal Transcription Factor PRDM13. Ophthalmology 2016, 123, 9–18. [Google Scholar] [CrossRef]
- Bowne, S.J.; Sullivan, L.S.; Wheaton, D.K.; Locke, K.G.; Jones, K.D.; Koboldt, D.C.; Fulton, R.S.; Wilson, R.K.; Blanton, S.H.; Birch, D.G.; et al. North Carolina macular dystrophy (MCDR1) caused by a novel tandem duplication of the PRDM13 gene. Mol. Vis. 2016, 22, 1239–1247. [Google Scholar]
- Vaclavik, V.; Munier, F. Malattia Leventinese (Autosomal Dominant Drusen). In Macular Dystophies; Springer: Cham, Switzerland, 2016; pp. 39–52. [Google Scholar]
- Tsang, S.H.; Sharma, T. Doyne Honeycomb Retinal Dystrophy (Malattia Leventinese, Autosomal Dominant Drusen). Adv. Exp. Med. Biol. 2018, 1085, 97–102. [Google Scholar] [CrossRef]
- Tsokolas, G. Sorsby fundus dystrophy (SFD): A narrative review. Medicine 2022, 101, e30595. [Google Scholar] [CrossRef]
- Davidson, A.E.; Sergouniotis, P.I.; Mackay, D.S.; Wright, G.A.; Waseem, N.H.; Michaelides, M.; Holder, G.E.; Robson, A.G.; Moore, A.T.; Plagnol, V.; et al. RP1L1 Variants are Associated with a Spectrum of Inherited Retinal Diseases Including Retinitis Pigmentosa and Occult Macular Dystrophy. Hum. Mutat. 2013, 34, 506–514. [Google Scholar] [CrossRef]
- Bianco, L.; Arrigo, A.; Antropoli, A.; Carrera, P.; Spiga, I.; Patricelli, M.G.; Bandello, F.; Battaglia Parodi, M. Multimodal imaging evaluation of occult macular dystrophy associated with a novel RP1L1 variant. Am. J. Ophthalmol. Case Rep. 2022, 26, 101550. [Google Scholar] [CrossRef]
- Oliveira-Ferreira, C.; Leuzinger-Dias, M.; Tavares-Ferreira, J.; Silva, S.E.; Brandão, E.; Falcão-Reis, F.; Rocha-Sousa, A. Hypotrichosis with juvenile macular dystrophy. Ophthalmic Genet. 2019, 40, 574–577. [Google Scholar] [CrossRef] [PubMed]
- Puech, B.; De Laey, J.J.; Holder, G.E. Inherited Chorioretinal Dystrophies; Springer: Berlin/Heidelberg, Germany,, 2014. [Google Scholar]
- D’Esposito, F.; Cennamo, G.; de Crecchio, G.; Maltese, P.E.; Cecchin, S.; Bertelli, M.; Ziccardi, L.; Veneruso, P.E.; Magli, A.; Cennamo, G.; et al. Multimodal Imaging in Autosomal Dominant Cone-Rod Dystrophy Caused by Novel CRX Variant. Ophthalmic Res. 2018, 60, 169–175. [Google Scholar] [CrossRef]
- Marino, V.; Dal Cortivo, G.; Oppici, E.; Maltese, P.E.; D’Esposito, F.; Manara, E.; Ziccardi, L.; Falsini, B.; Magli, A.; Bertelli, M.; et al. A novel p.(Glu111Val) missense mutation in GUCA1A associated with cone-rod dystrophy leads to impaired calcium sensing and perturbed second messenger homeostasis in photoreceptors. Hum. Mol. Gen. 2018, 27, 4204–4217. [Google Scholar] [CrossRef] [PubMed]
- Hsu, S.T.; Ponugoti, A.; Deaner, J.D.; Vajzovic, L. Update on Retinal Drug Toxicities. Curr. Ophthalmol. Rep. 2021, 9, 168–177. [Google Scholar] [CrossRef]
- Retinal Information Network. Available online: https://web.sph.uth.edu/RetNet/ (accessed on 12 March 2023).
- Forrester, J.V.; Dick, A.D.; McMenamin, P.G.; Roberts, F.; Pearlman, E. The Eye, Basic Sciences in Practice, 5th ed.; Elvesier: Amsterdam, The Netherlands, 2021; pp. 519–520. [Google Scholar]
- Fujinami, K.; Lois, N.; Davidson, A.E.; Mackey, D.S.; Hogg, C.R.; Stone, E.M.; Tsunoda, K.; Tsubota, K.; Bunce, C.; Robson, A.G.; et al. A longitudinal study of stargardt disease: Clinical and electrophysiologic assessment, progression, and genotype correlations. Am. J. Ophthalmol. 2013, 155, 1075–1088.e13. [Google Scholar] [CrossRef]
- Trapani, I. Dual AAV Vectors for Stargardt Disease. In Retinal Gene Therapy. Methods in Molecular Biology; Boon, C., Wijnholds, J., Eds.; Humana Press: New York, NY, USA, 2018; Volume 1715. [Google Scholar]
- Binley, K.; Widdowson, P.; Loader, J.; Kelleher, M.; Iqball, S.; Ferrige, G.; de Belin, J.; Carlucci, M.; Angell-Manning, D.; Hurst, F.; et al. Transduction of photoreceptors with equine infectious anemia virus lentiviral vectors: Safety and biodistribution of StarGen for Stargardt disease. Investig. Ophthalmol. Vis. Sci. 2013, 54, 4061–4071. [Google Scholar] [CrossRef]
- Huang, D.; Jeffery, R.C.H.; Aung-Htut, M.T.; McLenachan, S.; Fletcher, S.; Wilton, S.D.; Chen, F.K. Stargardt disease and progress in therapeutic strategies. Ophthalmic Genet. 2022, 43, 1–26. [Google Scholar] [CrossRef]
- Lu, L.J.; Liu, J.; Adelman, R.A. Novel therapeutics for Stargardt disease. Graefe’s Arch. Clin. Exp. Ophthalmol. 2017, 255, 1057–1062. [Google Scholar] [CrossRef]
- Prokopiou, E.; Kolovos, P.; Kalogerou, M.; Neokleous, A.; Nicolaou, O.; Sokratous, K.; Kyriacou, K.; Georgiou, T. Omega-3 Fatty Acids Supplementation: Therapeutic Potential in a Mouse Model of Stargardt Disease. Investig. Ophthalmol. Vis. Sci. 2018, 59, 2757–2767. [Google Scholar] [CrossRef] [PubMed]
- Guduru, A.; Gupta, A.; Tyagi, M.; Jalali, S.; Chhablani, J. Optical coherence tomography angiography characterisation of Best disease and associated choroidal neovascularisation. Br. J. Ophthalmol. 2018, 102, 444–447. [Google Scholar] [CrossRef] [PubMed]
- Michaelides, M.; Hunt, D.M.; Moore, A.T. The genetics of inherited macular dystrophies. J. Med. Genet. 2003, 40, 641–650. [Google Scholar] [CrossRef]
- Gerth, C.; Zawadzki, R.J.; Werner, J.S.; Héon, E. Retinal morphological changes of patients with X-linked retinoschisis evaluated by Fourier-domain optical coherence tomography. Arch. Ophthalmol. 2008, 126, 807–811. [Google Scholar] [CrossRef]
- Sieving, P.A.; Bingham, E.L.; Kemp, J.; Richards, J.; Hiriyanna, K. Juvenile X-linked retinoschisis from XLRS1 Arg213Trp mutation with preservation of the electroretinogram scotopic b-wave. Am. J. Ophthalmol. 1999, 128, 179–184. [Google Scholar] [CrossRef]
- Hoffman, D.R.; Hughbanks-Wheaton, D.K.; Spencer, R.; Fish, G.E.; Pearson, N.S.; Wang, Y.-Z.; Klein, M.; Takacs, A.; Locke, K.G.; Birch, D.G. Docosahexaenoic Acid Slows Visual Field Progression in X-Linked Retinitis Pigmentosa: Ancillary Outcomes of the DHAX Trial. Investig. Ophthalmol. Vis. Sci. 2015, 56, 6646–6653. [Google Scholar] [CrossRef] [PubMed]
- Chowers, I.; Boon, C. The Pattern Dystrophies. In Macular Dystophies; Springer: Cham, Switzerland, 2016; pp. 11–24. [Google Scholar]
- Boon, C.J.F.; Jeroen Klevering, B.; Keunen, J.E.E.; Hoyng, C.B.; Theelen, T. Fundus autofluorescence imaging of retinal dystrophies. Vis. Res. 2008, 48, 2569–2577. [Google Scholar] [CrossRef]
- Querques, G.; Regenbogen, M.; Quijano, C.; Delphin, N.; Soubrane, G.; Souied, E.H. High-definition optical coherence tomography features in vitelliform macular dystrophy. Am. J. Ophthalmol. 2008, 146, 501–507. [Google Scholar] [CrossRef] [PubMed]
- Nõupuu, K.; Lee, W.; Zernant, J.; Tsang, S.H.; Allikmets, R. Structural and genetic assessment of the ABCA4-associated optical gap phenotype. Investig. Ophthalmol. Vis. Sci. 2014, 55, 7217–7226. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.; Nõupuu, K.; Oll, M.; Duncker, T.; Burke, T.; Zernant, J.; Bearelly, S.; Tsang, S.T.H.; Sparrow, J.R.; Allikmets, R. The external limiting membrane in early-onset Stargardt disease. Investig. Ophthalmol. Vis. Sci. 2014, 55, 6139–6149. [Google Scholar] [CrossRef]
- Mastropasqua, R.; Toto, L.; Borrelli, E.; Di Antonio, L.; Mattei, P.A.; Senatore, A.; Di Nicola, M.; Mariotti, C. Optical Coherence Tomography Angiography Findings in Stargardt Disease. PLoS ONE 2017, 12, e0170343. [Google Scholar] [CrossRef]
- Chiang, T.K.; Yu, M. Electrophysiological Evaluation of Macular Dystrophies. J. Clin. Med. 2023, 12, 1430. [Google Scholar] [CrossRef]
- Padhy, S.K.; Parameswarappa, D.C.; Agarwal, K.; Takkar, B.; Behera, S.; Panchal, B.; Ramappa, M.; Padhi, T.R.; Jalali, S. Clinical and visual electrophysiological characteristics of vitelliform macular dystrophies in the first decade of life. Indian J. Ophthalmol. 2022, 70, 2516–2525. [Google Scholar]
- Stradiotto, E.; Allegrini, D.; Fossati, G.; Raimondi, R.; Sorrentino, T.; Tripepi, D.; Barone, G.; Inforzato, A.; Romano, M.R. Genetic Aspects of Age-Related Macular Degeneration and Their Therapeutic Potential. Int. J. Mol. Sci. 2022, 23, 13280. [Google Scholar] [CrossRef]
- Hohman, T.C. Hereditary Retinal Dystrophy. Handb. Exp. Pharmacol. 2017, 242, 337–367. [Google Scholar]
- Verma, I.C.; Paliwal, P.; Singh, K. Genetic Testing in Pediatric Ophthalmology. Indian J. Pediatr. 2018, 85, 228–236. [Google Scholar] [CrossRef]
- Tsang, S.H.; Sharma, T. Genetic Testing for Inherited Retinal Dystrophy: Basic Understanding. Adv. Exp. Med. Biol. 2018, 1085, 261–268. [Google Scholar]
- Khan, M.; Cornelis, S.S.; Pozo-Valero, M.D.; Whelan, L.; Runhart, E.H.; Mishra, K.; Bults, F.; AlSwaiti, Y.; AlTalbishi, A.; De Baere, E.; et al. Resolving the dark matter of ABCA4 for 1054 Stargardt disease probands through integrated genomics and transcriptomics. Genet. Med. 2020, 22, 1235–1246. [Google Scholar] [CrossRef]
- Mc Clinton, B.; Corradi, Z.; McKibbin, M.; Panneman, D.M.; Roosing, S.; Boonen, E.G.M.; Ali, M.; Watson, C.M.; Steel, D.H.; Cremers, F.P.M.; et al. Effective smMIPs-Based Sequencing of Maculopathy-Associated Genes in Stargardt Disease Cases and Allied Maculopathies from the UK. Genes 2023, 14, 191. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hedge, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. Off. J. Am. Coll. Med. Genet. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Neiweem, A.E.; Hariprasad, S.M.; Ciulla, T.A. Genetic Testing Prevalence, Guidelines, and Pitfalls in Large, University-Based Medical Systems. Ophthalmic Surg. Lasers Imaging Retina 2021, 52, 6–10. [Google Scholar] [CrossRef] [PubMed]
- Stone, E.M.; Aldave, A.J.; Drack, A.V.; Maccumber, M.W.; Sheffield, V.C.; Traboulsi, E.; Weleber, R.G. Recommendations for genetic testing of inherited eye diseases: Report of the American Academy of Ophthalmology task force on genetic testing. Ophthalmology 2012, 119, 2408–2410. [Google Scholar] [CrossRef]
- Al-Khuzaei, S.; Broadgate, S.; Foster, C.R.; Shah, M.; Yu, J.; Downes, S.M.; Halford, S. An Overview of the Genetics of ABCA4 Retinopathies, an Evolving Story. Genes 2021, 12, 1241. [Google Scholar] [CrossRef] [PubMed]
- Black, G.C.; Sergouniotis, P.; Sodi, A.; Leroy, B.P.; Van Cauwenbergh, C.; Liskova, P.; Grønskov, K.; Klett, A.; Kohl, S.; Taurina, G.; et al. The need for widely available genomic testing in rare eye diseases: An ERN-EYE position statement. Orphanet. J. Rare Dis. 2021, 16, 142. [Google Scholar] [CrossRef] [PubMed]
- Kingdom, R.; Wright, C.F. Incomplete Penetrance and Variable Expressivity: From Clinical Studies to Population Cohorts. Front. Genet. 2022, 13, 920390. [Google Scholar] [CrossRef]
- Gerasimavicius, L.; Livesey, B.J.; Marsh, J.A. Loss-of-function, gain-of-function and dominant-negative mutations have profoundly different effects on protein structure. Nat Commun. 2022, 13, 3895. [Google Scholar] [CrossRef]
- Cevik, S.; Biswas, S.B.; Biswas-Fiss, E.E. Structural and Pathogenic Impacts of ABCA4 Variants in Retinal Degenerations-An In-Silico Study. Int. J. Mol. Sci. 2023, 24, 7280. [Google Scholar] [CrossRef] [PubMed]
- Chiu, W.; Lin, T.-Y.; Chang, Y.-C.; Lai, H.I.-A.M.; Lin, S.-C.; Ma, C.; Yarmishyn, A.A.; Lin, S.-C.; Chang, K.-J.; Chou, Y.-B.; et al. An Update on Gene Therapy for Inherited Retinal Dystrophy: Experience in Leber Congenital Amaurosis Clinical Trials. Int. J. Mol. Sci. 2021, 22, 4534. [Google Scholar] [CrossRef] [PubMed]
- Surace, E.M.; Auricchio, A. Versatility of AAV vectors for retinal gene transfer. Vis. Res. 2008, 48, 353–359. [Google Scholar] [CrossRef] [PubMed]
- Moore, N.A.; Morral, N.; Ciulla, T.A.; Bracha, P. Gene therapy for inherited retinal and optic nerve degenerations. Expert Opin. Biol. Ther. 2018, 18, 37–49. [Google Scholar] [CrossRef]
- Ochakovski, G.A.; Bartz-Schmidt, K.U.; Fischer, M.D. Retinal Gene Therapy: Surgical Vector Delivery in the Translation to Clinical Trials. Front. Neurosci. 2017, 11, 174. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.L.; Gregori, N.Z.; MacLaren, R.E.; Lam, B.L. Surgical Technique for Subretinal Gene Therapy in Humans with Inherited Retinal Degeneration. Retina 2019, 39 (Suppl. S1), S2–S8. [Google Scholar] [CrossRef]
- Scherbakova, I.; Ragi, S.D.; Sharma, T. Ocular Injection Techniques for Retinitis Pigmentosa: Intravitreal, Subretinal, and Suprachoroidal. Methods Mol. Biol. 2023, 2560, 375–392. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration. Voretigene Neparvovec-rzyl BL125610/0. 2017. Available online: https://www.fda.gov/media/109487/download (accessed on 26 September 2020).
- Russell, S.; Bennett, J.; Wellman, J.A.; Chung, D.C.; Yu, Z.-F.; Tillman, A.; Wittes, J.; Pappas, J.; Elci, O.; McCague, S.; et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: A randomised, controlled, open-label, phase 3 trial. Lancet 2017, 390, 849–860. [Google Scholar] [CrossRef]
- Tamboli, V.; Mishra, G.P.; Mitrat, A.K. Polymeric vectors for ocular gene delivery. Ther. Deliv. 2011, 2, 523–536. [Google Scholar] [CrossRef] [PubMed]
- Clinicaltrials.gov Webpage. Available online: clinicaltrials.gov (accessed on 9 December 2022).
- Benati, D.; Patrizi, C.; Recchia, A. Gene editing prospects for treating inherited retinal diseases. J. Med. Genet. 2020, 57, 437–444. Available online: https://jmg.bmj.com/content/57/7/437 (accessed on 9 December 2022). [CrossRef] [PubMed]
- John, M.C.; Quinn, J.; Hu, M.L.; Cehajic-Kapetanovic, J.; Xue, K. Gene-agnostic therapeutic approaches for inherited retinal degenerations. Front. Mol. Neurosci. 2023, 15, 1068185. [Google Scholar] [CrossRef]
- Jurgensmeier, C.; Bhosale, P.; Bernstein, P.S. Evaluation of 4-methylpyrazole as a potential therapeutic dark adaptation inhibitor. Curr. Eye Res. 2007, 32, 911–915. [Google Scholar] [CrossRef]
- Han, J.; Li, Y.; Liu, X.; Zhou, T.; Sun, H.; Edwards, P.; Gao, H.; Yu, F.-S.; Qiao, X. Metformin suppresses retinal angiogenesis and inflammation in vitro and in vivo. PLoS ONE 2018, 13, e0193031. [Google Scholar] [CrossRef]
- Kim, N.; Priefer, R. Retinol binding protein 4 antagonists and protein synthesis inhibitors: Potential for therapeutic development. Eur. J. Med. Chem. 2021, 226, 113856. [Google Scholar] [CrossRef]
- Kubota, R.; Birch, D.G.; Gregory, J.K.; Koester, J.M. Randomised study evaluating the pharmacodynamics of emixustat hydrochloride in subjects with macular atrophy secondary to Stargardt disease. Br. J. Ophthalmol. 2022, 106, 403–408. [Google Scholar] [CrossRef] [PubMed]
- Piccardi, M.; Fadda, A.; Martelli, F.; Marangoni, D.; Magli, A.; Minnella, A.M.; Bertelli, M.; Di Marco, S.; Bisti, S.; Falsini, B. Antioxidant Saffron and Central Retinal Function in ABCA4-Related Stargardt Macular Dystrophy. Nutrients 2019, 11, 2461. [Google Scholar] [CrossRef]





| Medical History | ||||
|---|---|---|---|---|
| Age at birth | ||||
| Connatal/perinatal infections | ||||
| Onset (age at first symptoms appearance/first diagnosis) | ||||
| Progression | ||||
| Family History | ||||
| Number, sex (M/F) | Affected (Y/N) | Age of onset | Progression | |
| Children | ||||
| Parents | ||||
| Brothers-sisters | ||||
| Nephews-nieces | ||||
| Maternal grandparents | ||||
| Maternal uncles/aunts | ||||
| Maternal cousins | ||||
| Paternal grandparents | ||||
| Paternal uncles/aunts | ||||
| Paternal cousins | ||||
| Family Tree | ||||
 | ||||
| Inheritance Patterns Hints | ||||
| Both sex equally affected? | Is there consanguinity? | → Autosomal recessive | ||
| Both sex equally affected? | Are parents affected? | → Autosomal dominant | ||
| Primarily affecting men? | No male-to-male transmission? | → X linked recessive | ||
| Males with more severe symptoms? | No male-to-male transmission? | → X linked dominant | ||
| Only maternal inheritance but both sexes affected? | → Mitochondrial | |||
| Multiples affected siblings from healthy parents? | → Germline mosaicism | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Raimondi, R.; D’Esposito, F.; Sorrentino, T.; Tsoutsanis, P.; De Rosa, F.P.; Stradiotto, E.; Barone, G.; Rizzato, A.; Allegrini, D.; Costagliola, C.; et al. How to Set Up Genetic Counselling for Inherited Macular Dystrophies: Focus on Genetic Characterization. Int. J. Mol. Sci. 2023, 24, 9722. https://doi.org/10.3390/ijms24119722
Raimondi R, D’Esposito F, Sorrentino T, Tsoutsanis P, De Rosa FP, Stradiotto E, Barone G, Rizzato A, Allegrini D, Costagliola C, et al. How to Set Up Genetic Counselling for Inherited Macular Dystrophies: Focus on Genetic Characterization. International Journal of Molecular Sciences. 2023; 24(11):9722. https://doi.org/10.3390/ijms24119722
Chicago/Turabian StyleRaimondi, Raffaele, Fabiana D’Esposito, Tania Sorrentino, Panos Tsoutsanis, Francesco Paolo De Rosa, Elisa Stradiotto, Gianmaria Barone, Angelica Rizzato, Davide Allegrini, Ciro Costagliola, and et al. 2023. "How to Set Up Genetic Counselling for Inherited Macular Dystrophies: Focus on Genetic Characterization" International Journal of Molecular Sciences 24, no. 11: 9722. https://doi.org/10.3390/ijms24119722
APA StyleRaimondi, R., D’Esposito, F., Sorrentino, T., Tsoutsanis, P., De Rosa, F. P., Stradiotto, E., Barone, G., Rizzato, A., Allegrini, D., Costagliola, C., & Romano, M. R. (2023). How to Set Up Genetic Counselling for Inherited Macular Dystrophies: Focus on Genetic Characterization. International Journal of Molecular Sciences, 24(11), 9722. https://doi.org/10.3390/ijms24119722