
Next-Generation Sequencing and Triple-Negative Breast Cancer: Insights and Applications
 ,
,  ,
,  ,
, 
Abstract
1. Introduction
2. Next-Generation Sequencing
2.1. First-Generation Sequencing
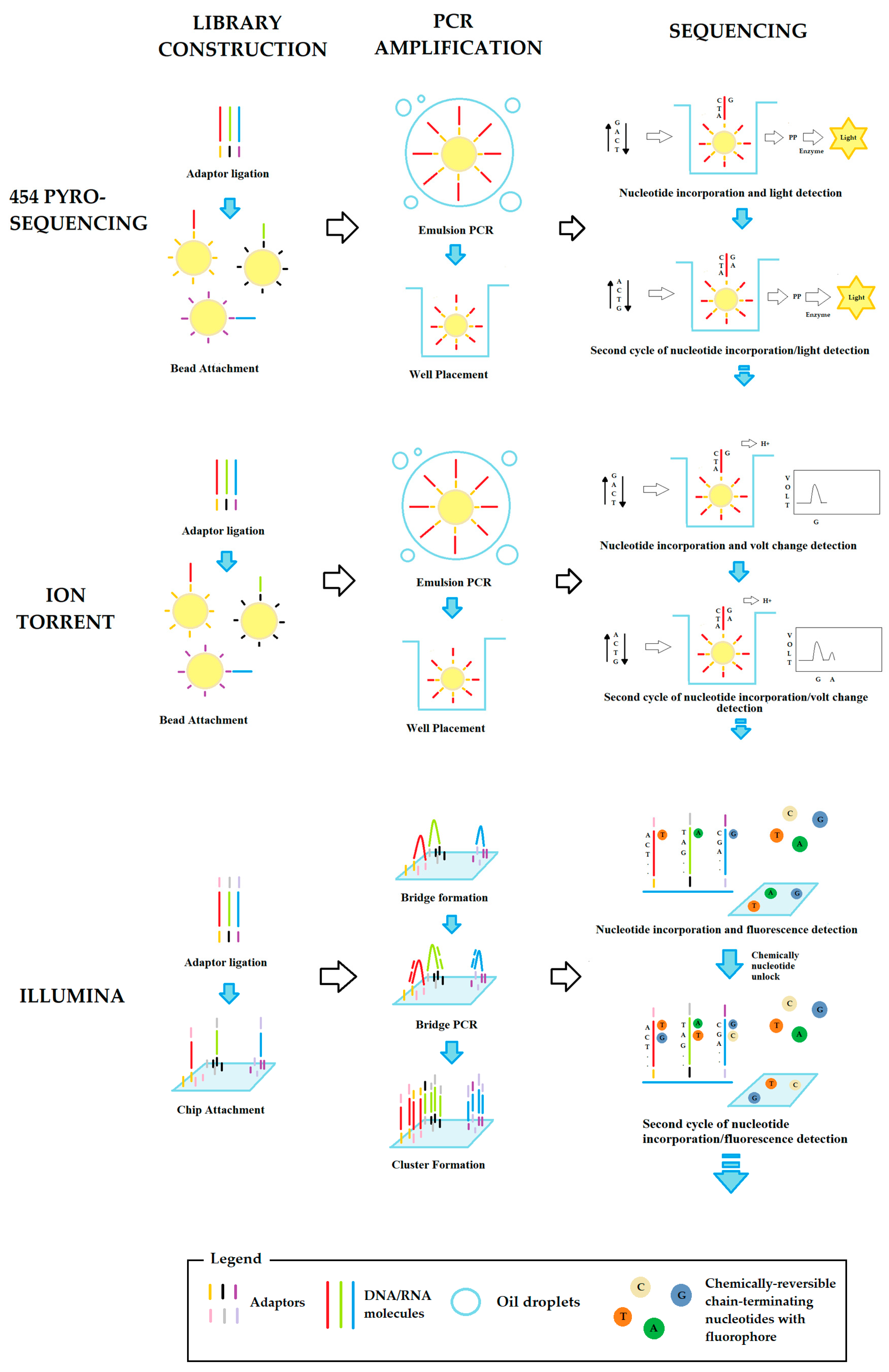
2.2. Second-Generation Sequencing
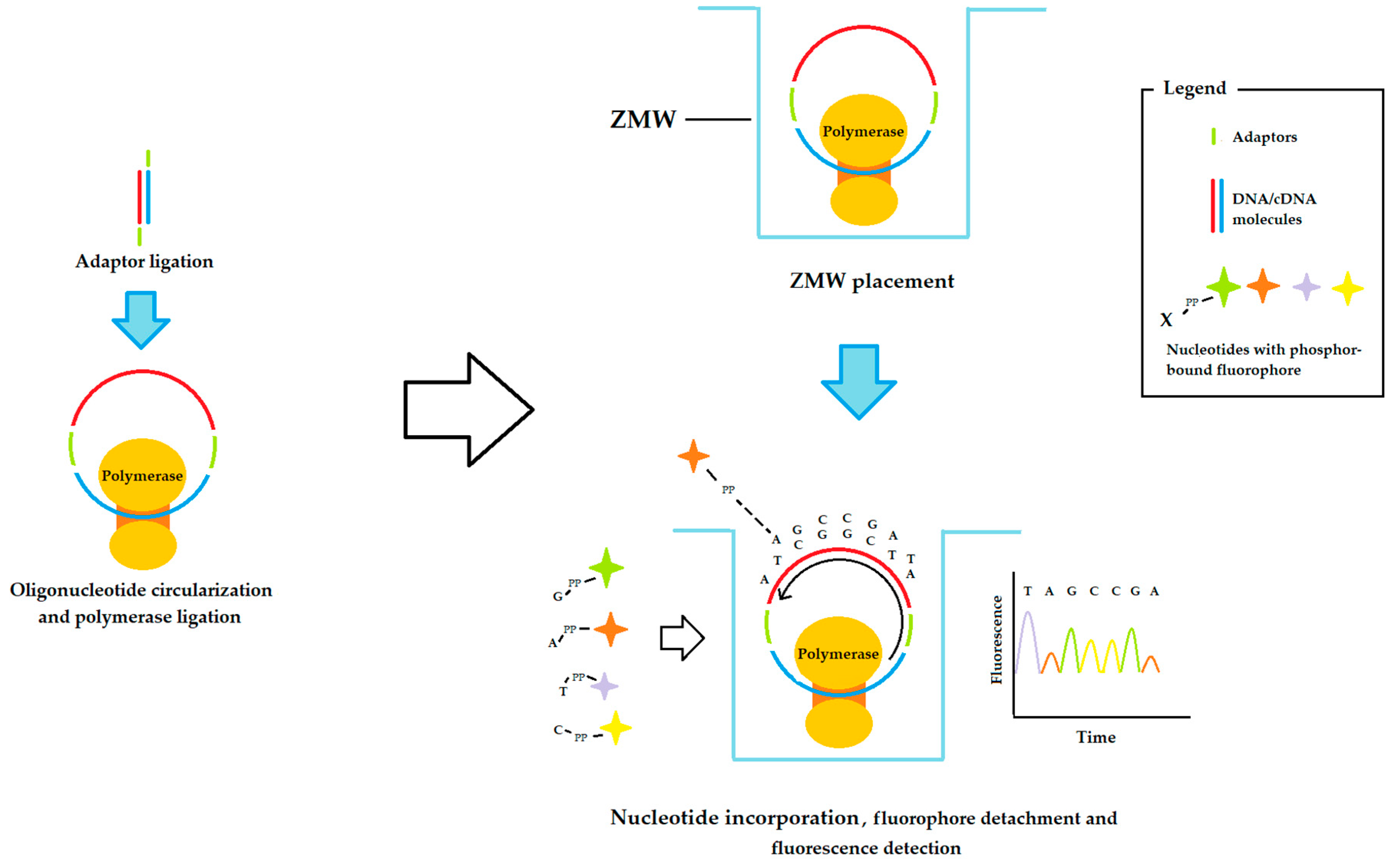
2.3. Third-Generation Sequencing
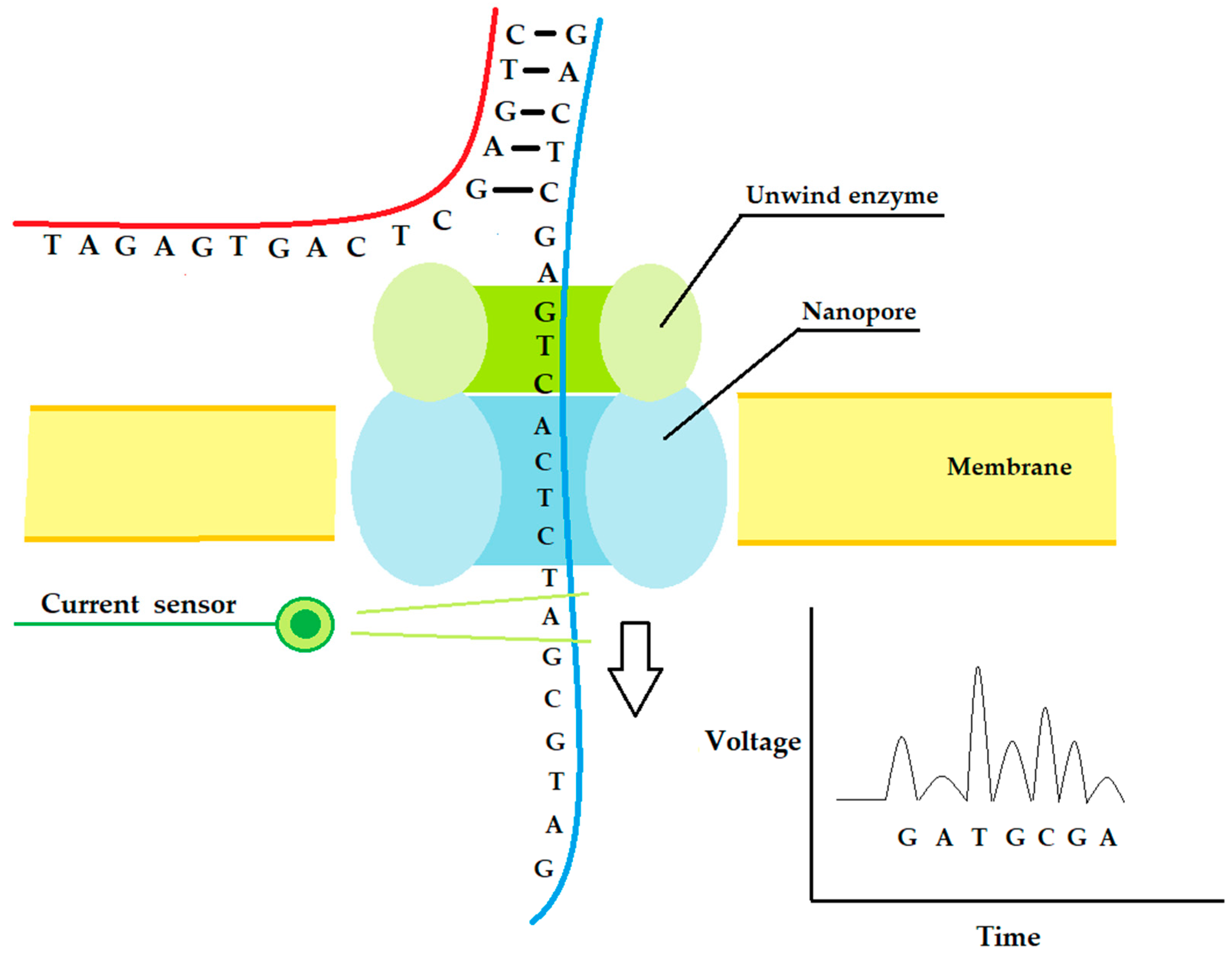
2.4. Fourth-Generation Sequencing
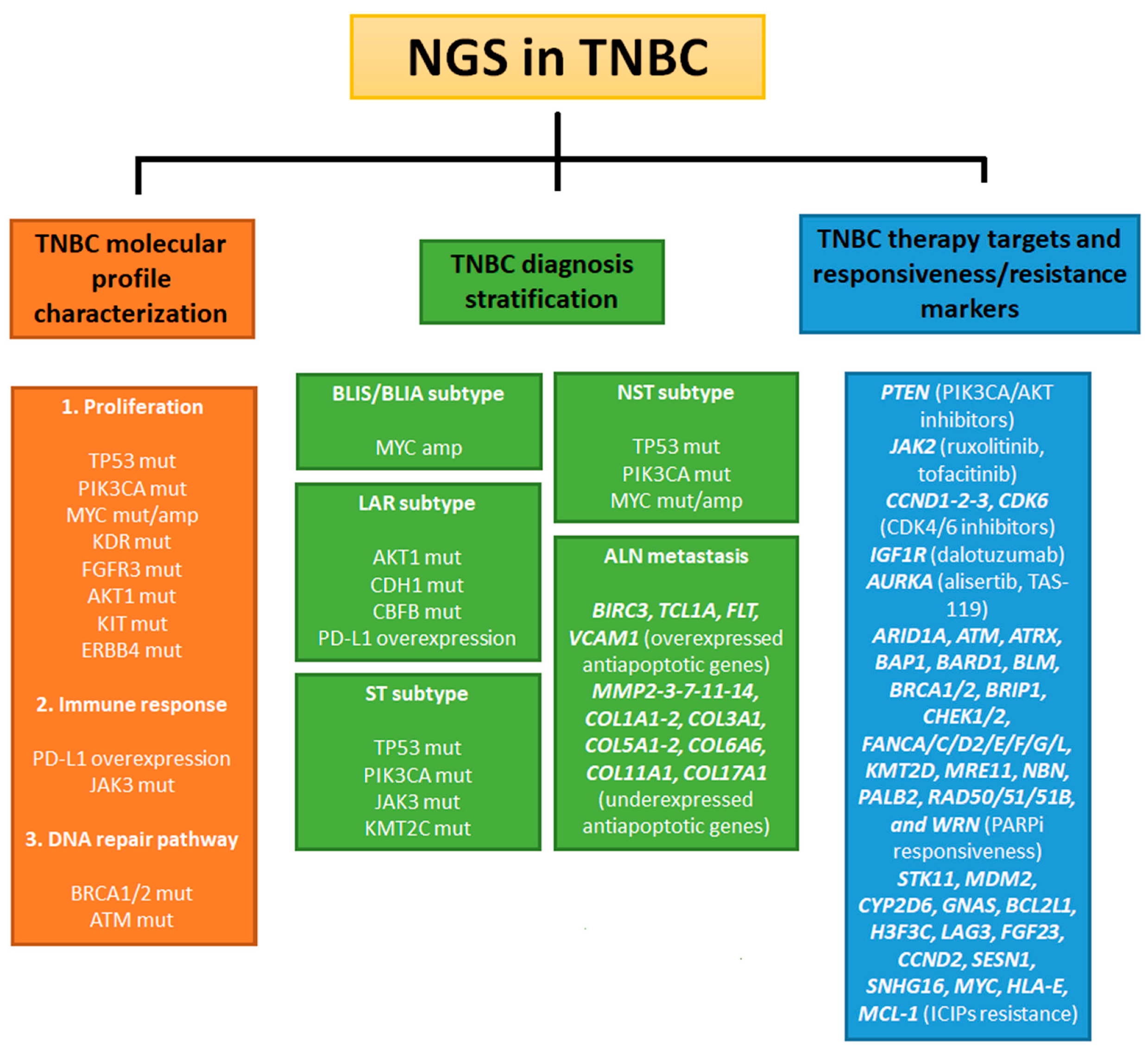
3. NGS Technologies in TNBC Research
3.1. NGS Analysis of Markers for TNBC

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient Population | Median Age (Min-Max) | Source of Sample | Target | NGS Platform | Main Finding | Ref. |
|---|---|---|---|---|---|---|
| 156 IBC patients (51 TNBC) | 53 yrs (23–84) | Fresh or frozen biopsy | Mutations of 91 breast cancer-related genes | Illumina | DNA repair, NOTCH, RAS and PIK3CA signaling pathways were more frequently altered in IBC patients (especially TNBC) than in non-IBC patients | [47] |
| 14 ER+ patients with paired metastasis/primary samples, 17 TNBC patients with paired metastasis/primary samples | ER+ patients: 61 yrs (35–79); TNBC patients: 62 yrs (30–88) | FFPE | Expression level of 2567 cancer-associated genes | Illumina | 97 upregulated genes and 115 downregulated genes in metastasis compared to primary site were common between ER+ and TNBC patients. Anti-apoptotic genes were overexpressed, and microenvironment-regulating genes were downregulated in TNBC metastasis as paired primary sites | [48,49] |
| 20 TNBC patients | 57 yrs (40–91) | FFPE | Pathogenic variants in 358 cancer-related genes | Illumina | MYC was amplified in 75% of TNBC cases. TP53, AURKA, and KDR were mutated in 30% of patients | [50] |
| 72 NST TNBC patients, 17 ST TNBC patients | 49 yrs (22–81) | Plasma | Mutations in 520 cancer-related genes | Illumina | The most frequently mutated genes in NST-TNBC were TP53 (88.7%), PIK3CA (26.8%), and MYC (18.3%), and in ST TNBC were TP53 (68.8%), PIK3CA (50%), JAK3 (18.8%), and KMT2c (18.8%) | [57] |
| 96 TNBC patients | 55 yrs (36–71) | FFPE | Most frequent mutations in 46 genes with a well-defined role in cancer | Ion Torrent | TP53, KDR, PIK3CA, ATM, AKT1, KIT, ERBB4, FGFR3, and MET had high mutation frequency in TNBC. AKT1 rs3730358, KDR rs34231037 (c.1444T > 5), KIT rs3822214 (c.1621A-C), TP53 rs28934576 (c.818G > A), and BRCA1/2 class 5 mutations were associated with poor survival | [58] |
| 85 TNBC patients post-NAC | 48 yrs (24–78) | FFPE | Mutations in 182 cancer-related genes | Illumina | In recurrent post- NAC TNBC, there was a high mutation frequency of genes available for targeted therapies: PTEN (PIK3CA/AKT pathway inhibitors), JAK2 (ruxolitinib or tofacitinib), CDK6, CCDN 1/2/3 (CDK4/6 inhibitors), and IGF1R (dalotuzumab) | [59] |
| 56 TNBC patients before-NAC | 40 yrs (23–74) | FFPE, plasma | Pathogenic variants in 1977 tumorigenesis-related genes | SOLiD 5500xL | No significant difference was found in the genomic profile of treatment-responsive and resistance TNBCs. PIK3CA mutations occur exclusively in BRCA1 wild-type patients | [61] |
| 7 TNBC patients post NAC with short-DFS (less than 1 yrs); 7 TNBC patients with long-DFS (more than 1 year) | 53 yrs (37–71) | FFPE, plasma | Pathogenic variants in 422 cancer-related genes | Illumina | Higher TMB was found in patients with short DFS than in patients with long DFS. Mutations of PTPN13 and JARID2 occurred only in the short DFS group | [62] |
| 4647 BC patients (2183 ER/PR+ patients, 237 ER/PR+ and HER2+ patients, 217 HER2+ patients, and 1568 TNBC patients) | NR | FFPE | Mutations in 592 cancer-associated genes | Illumina | 18.2% of TNBC had at least one mutation in genes involved in the HR DNA repair pathway. TNBC with HRD had a higher rate of TMB and PDL1 positivity and a higher frequency of alterations in the PIK3CA pathway than patients without HRD | [63] |
| 158 primary and/or metastatic TNBC patients | 51 yrs (21–72) | FFPE | Mutations in 468 key cancer-related genes | Illumina | 47.4% of TNBC are PD-L1 positive. CBFB mutations were more common in PDL1-negative patients than in positive patients. CBFB mutations frequently occurred together with AKT1 and CDH1 mutations in LAR TNBC | [66] |
| 11 ICPI-treated TNBC patients | 50 yrs (NR) | Plasma | Pathogenic alterations in 457 cancer-related genes | Illumina | TNBC patients with CYP2D6 loss and gain in CNV of GNAS, BCL2L1, H3F3C, LAG3, FGF23, CCND2, SESN1, SNHG16, MYC, HLA-E, and MCL-1 had a shorter PFS after ICPI treatment | [68] |
| 1237 ER+ patients, 1953 HER2+ patients, and 641 TNBC | ER+ patients: 55 yrs (23–89); HER2+ patients: 55 yrs (20–89); TNBC patients: 53 yrs (20–85) | FFPE | TMB, MSI and mutations in 324 cancer-related genes | Illumina | 48.6% of ER+, 12.1% of HER2+, and 56.4% of TNBC patients had at least one mutation in ICPI response-associated genes. 2–3% of TNBC patients had mutations in ICPI resistance-associated genes STK11 and MDM2 | [78] |
| 305 TNBC patients | 49 yrs (NR) | FFPE | Fusion events in NRTK gene | Illumina | IHC and FISH indicated 11.15% of TNBC cases with NRTK fusion events, but NGS validation did not show positivity | [82] |
3.2. NGS in Ethnic-Specific Molecular Profiling of TNBC
| Patient Population | Median Age (Min-Max) | Source Sample | Target | NGS Platform | Main Finding | Ref. |
|---|---|---|---|---|---|---|
| 11 Ghanaian BC patients (9 TNBC patients) | 48 yrs (38–64) | FFPE | CNAs in 130 cancer-related genes | Ion Torrent | 13 of the 17 genes with CNAs were detected (involved in cell proliferation, apoptosis, and PIK3CA signaling pathways). These genes were associated with overexpression of EZH2, an epigenetic regulator frequently altered in Ghanaian TNBCs | [83] |
| 110 Tunisian BC patients (26 TNBC patients) | 48 yrs (30–71) | Plasma | Mutations in BRCA1 and BRCA2 | Illumina | Mutations c.1310-1313 del AAGA in BRCA2 and c.5030_5033 in BRCA1 were found in 4% of BC patients. 5 of 26 TNBC patients had BRCA alterations, including 4 with BRCA1 mutations | [86] |
| 30 early-onset Moroccan TNBC patients | 38 yrs (NR) | Plasma | Pathogenic variants in 63 cancer-related genes | Illumina | 17% of TNBC cases had BRCA alterations. Among them, mutation c.1310_1313 in BRCA2 was reported. The analysis result showed 42 VUSs | [89] |
3.3. NGS of Third and Fourth Generation in TNBC Research
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
List of Abbreviations
| ALN | Axillary Lymph Node metastasis |
| ARS | Artificial Reference Sequence |
| BLIA | Basal Like Immune Activated |
| BLIS | Basal Like Immune Suppressed |
| CAN | Copy Number Alterations |
| FFPE | Formalin-Fixed Paraffin-Embedded |
| HRD | Homologous Recombination Deficiency |
| IBC | Imflammatory Breast Cancer |
| ICPI | Immune Checkpoint Inhibitors |
| LAR | Luminal Androgen Receptor |
| MES | Mesenchymal-like |
| MGC | Maxam-Gilbert Chemical Cleavage |
| NAC | Neo Adjuvant Chemotherapy |
| NGS | Next-Generation Sequencing |
| NGSH | Next-Generation Sequencing by Hybridization |
| NGSS | Next-Generation Sequencing by Synthesis |
| NST | Ductal Carcinoma of Not Special Type |
| ONT | Oxford Nanopore Technology |
| pCR | Pathological Complete Response |
| SBS | Sanger Sequencing By Synthesis |
| SMRT | Single Molecule Real Time |
| ST | Ductal Carcinoma of Special Type |
| TMB | Tumor Mutation Burden |
| TNBC | Triple-Negative Breast Cancer |
| VUS | Variations of Unknown/Uncertain Significance |
| ZMW | Zero-Mode Waveguide |
References
- Burstein, M.D.; Tsimelzon, A.; Poage, G.M.; Covington, K.R.; Contreras, A.; Fuqua, S.A.; Savage, M.I.; Osborne, C.K.; Hilsenbeck, S.G.; Chang, J.C.; et al. Comprehensive genomic analysis identifies novel subtypes and targets of triple-negative breast cancer. Clin. Cancer Res. 2015, 21, 1688–1698. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, F.; Kyriakides, S.; Ohno, S.; Penault-Llorca, F.; Poortmans, P.; Rubio, I.T.; Zackrisson, S.; Senkus, E.; ESMO Guidelines Committee. Early breast cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2019, 30, 1194–1220. [Google Scholar] [CrossRef] [PubMed]
- Giaquinto, A.N.; Sung, H.; Miller, K.D.; Kramer, J.L.; Newman, L.A.; Minihan, A.; Jemal, A.; Siegel, R.L. Breast Cancer Statistics, 2022. CA A Cancer J. Clin. 2022, 72, 524–541. [Google Scholar] [CrossRef]
- Nwagu, G.C.; Bhattarai, S.; Swahn, M.; Ahmed, S.; Aneja, R. Prevalence and Mortality of Triple-Negative Breast Cancer in West Africa: Biologic and Sociocultural Factors. JCO Glob. Oncol. 2021, 7, 1129–1140. [Google Scholar] [CrossRef]
- Bergin, A.R.T.; Loi, S. Triple-negative breast cancer: Recent treatment advances. F1000Research 2019, 8, 1342. [Google Scholar] [CrossRef] [PubMed]
- Borri, F.; Granaglia, A. Pathology of triple negative breast cancer. Semin. Cancer Biol. 2021, 72, 136–145. [Google Scholar] [CrossRef]
- Derakhshan, F.; Reis-Filho, J.S. Pathogenesis of Triple-Negative Breast Cancer. Annu. Rev. Pathol. Mech. Dis. 2021, 17, 181–204. [Google Scholar] [CrossRef]
- Vagia, E.; Mahalingam, D.; Cristofanilli, M. The landscape of targeted therapies in TNBC. Cancers 2020, 12, 916. [Google Scholar] [CrossRef]
- Weisman, P.S.; Ng, C.K.Y.; Brogi, E.; Eisenberg, R.E.; Won, H.H.; Piscuoglio, S.; De Filippo, M.R.; Ioris, R.; Akram, M.; Norton, L.; et al. Genetic alterations of triple negative breast cancer by targeted next-generation sequencing and correlation with tumor morphology. Mod. Pathol. 2016, 29, 476–488. [Google Scholar] [CrossRef]
- Nik-Zainal, S.; Davies, H.; Staaf, J.; Ramakrishna, M.; Glodzik, D.; Zou, X.; Martincorena, I.; Alexandrov, L.B.; Martin, S.; Wedge, D.C.; et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature 2016, 534, 47–54. [Google Scholar] [CrossRef]
- Turner, N.; Lambros, M.B.; Horlings, H.M.; Pearson, A.; Sharpe, R.; Natrajan, R.; Geyer, F.C.; van Kouwenhove, M.; Kreike, B.; Mackay, A.; et al. Integrative molecular profiling of triple negative breast cancers identifies amplicon drivers and potential therapeutic targets. Oncogene 2010, 29, 2013–2023. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Garrido-Castro, A.C.; Lin, N.U.; Polyak, K. Insights into molecular classifications of triple-negative breast cancer: Improving patient selection for treatment. Cancer Discov. 2019, 9, 176–198. [Google Scholar] [CrossRef] [PubMed]
- Bareche, Y.; Venet, D.; Ignatiadis, M.; Aftimos, P.; Piccart, M.; Rothe, F.; Sotiriou, C. Unravelling triple-negative breast cancer molecular heterogeneity using an integrative multiomic analysis. Ann. Oncol. 2018, 29, 895–902. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.-R.; Jiang, Y.-Z.; Xu, X.-E.; Yu, K.-D.; Jin, X.; Hu, X.; Zuo, W.-J.; Hao, S.; Wu, J.; Liu, G.-Y.; et al. Comprehensive transcriptome analysis identifies novel molecular subtypes and subtype-specific RNAs of triple-negative breast cancer. Breast Cancer Res. 2016, 18, 33. [Google Scholar] [CrossRef] [PubMed]
- Yin, L.; Duan, J.-J.; Bian, X.-W.; Yu, S.-C. Triple-negative breast cancer molecular subtyping and treatment progress. Breast Cancer Res. 2020, 22, 61. [Google Scholar] [CrossRef] [PubMed]
- Geenen, J.J.J.; Linn, S.C.; Beijnen, J.H.; Schellens, J.H.M. PARP Inhibitors in the Treatment of Triple-Negative Breast Cancer. Clin. Pharmacokinet. 2018, 57, 427–437. [Google Scholar] [CrossRef] [PubMed]
- Robson, M.; Im, S.A.; Senkus, E.; Xu, B.; Domchek, S.M.; Masuda, N.; Delaloge, S.; Li, W.; Tung, N.; Armstrong, A.; et al. Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. N. Engl. J. Med. 2017, 377, 523–533. [Google Scholar] [CrossRef]
- Kaufman, B.; Shapira-Frommer, R.; Schmutzler, R.K.; Audeh, M.W.; Friedlander, M.; Balmaña, J.; Mitchell, G.; Fried, G.; Stemmer, S.M.; Hubert, A.; et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J. Clin. Oncol. 2015, 33, 244–250. [Google Scholar] [CrossRef]
- Lei, Q.; Wang, D.; Sun, K.; Wang, L.; Zhang, Y. Resistance Mechanisms of Anti-PD1/PDL1 Therapy in Solid Tumors. Front. Cell Dev. Biol. 2020, 8, 672. [Google Scholar] [CrossRef]
- Heimes, A.S.; Schmidt, M. Atezolizumab for the treatment of triple-negative breast cancer. Expert Opin. Investig. Drugs 2019, 28, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Emens, L.A.; Adams, S.; Barrios, C.H.; Diéras, V.; Iwata, H.; Loi, S.; Rugo, H.S.; Schneeweiss, A.; Winer, E.P.; Patel, S.; et al. First-line atezolizumab plus nab-paclitaxel for unresectable, locally advanced, or metastatic triple-negative breast cancer: IMpassion130 final overall survival analysis. Ann. Oncol. 2021, 32, 983–993. [Google Scholar] [CrossRef] [PubMed]
- Schmid, P.; Adams, S.; Rugo, H.S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Diéras, V.; Hegg, R.; Im, S.-A.; Shaw Wright, G.; et al. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N. Engl. J. Med. 2018, 379, 2108–2121. [Google Scholar] [CrossRef] [PubMed]
- Cortes, J.; Cescon, D.W.; Rugo, H.S.; Nowecki, Z.; Im, S.-A.; Yusof, M.M.; Gallardo, C.; Lipatov, O.; Barrios, C.H.; Holgado, E.; et al. Pembrolizumab plus chemotherapy versus placebo plus chemotherapy for previously untreated locally recurrent inoperable or metastatic triple-negative breast cancer (KEYNOTE-355): A randomised, placebo-controlled, double-blind, phase 3 clinical trial. Lancet 2020, 396, 1817–1828. [Google Scholar] [CrossRef]
- Miles, D.; Gligorov, J.; André, F.; Cameron, D.; Schneeweiss, A.; Barrios, C.; Xu, B.; Wardley, A.; Kaen, D.; Andrade, L.; et al. LBA15 Primary results from IMpassion131, a double-blind placebo-controlled randomised phase III trial of first-line paclitaxel (PAC) ± atezolizumab (atezo) for unresectable locally advanced/metastatic triple-negative breast cancer (mTNBC). Ann. Oncol. 2020, 31, S1147–S1148. [Google Scholar] [CrossRef]
- Okazaki, S.; Sasaki, T.; Yasuda, S.; Abe, M.; Yoshida, N.; Yoshida, R.; Ishibashi, K.; Minami, Y.; Okumura, S.; Chiba, S.; et al. The feasibility of circulating tumor DNA analysis as a marker of recurrence in triple-negative breast cancer. Oncol. Lett. 2021, 21, 420. [Google Scholar] [CrossRef]
- Pascual, J.; Turner, N.C. Targeting the PI3-kinase pathway in triple-negative breast cancer. Ann. Oncol. 2019, 30, 1051–1060. [Google Scholar] [CrossRef]
- Wongchenko, M.J.; Kim, S.-B.; Saura, C.; Oliveira, M.; Lipson, D.; Kennedy, M.; Greene, M.; Breese, V.; Mani, A.; Xu, N.; et al. Circulating Tumor DNA and Biomarker Analyses From the LOTUS Randomized Trial of First-Line Ipatasertib and Paclitaxel for Metastatic Triple-Negative Breast Cancer. JCO Precis. Oncol. 2020, 4, 1012–1024. [Google Scholar] [CrossRef]
- McCombie, W.R.; McPherson, J.D.; Mardis, E.R. Next-generation sequencing technologies. Cold Spring Harb. Perspect. Med. 2019, 9, a036798. [Google Scholar] [CrossRef]
- Slatko, B.E.; Gardner, A.F.; Ausubel, F.M. Overview of Next-Generation Sequencing Technologies. Curr. Protoc. Mol. Biol. 2018, 122, e59. [Google Scholar] [CrossRef]
- Belikov, S.; Wieslander, L. Express protocol for generating Gi-A sequencing ladders. Nucleic Acids Res. 1995, 23, 310. [Google Scholar] [CrossRef] [PubMed]
- Crossley, B.M.; Bai, J.; Glaser, A.; Maes, R.; Porter, E.; Killian, M.L.; Clement, T.; Toohey-Kurth, K. Guidelines for Sanger sequencing and molecular assay monitoring. J. Veter-Diagn. Investig. 2020, 32, 767–775. [Google Scholar] [CrossRef]
- Greenough, L.; Schermerhorn, K.M.; Mazzola, L.; Bybee, J.; Rivizzigno, D.; Cantin, E.; Slatko, B.E.; Gardner, A.F. Adapting capillary gel electrophoresis as a sensitive, high-throughput method to accelerate characterization of nucleic acid metabolic enzymes. Nucleic Acids Res. 2016, 44, e15. [Google Scholar] [CrossRef]
- Schermerhorn, K.M.; Gardner, A.F. Pre-steady-state Kinetic Analysis of a Family D DNA Polymerase from Thermococcus sp. 9°N Reveals Mechanisms for Archaeal Genomic Replication and Maintenance. J. Biol. Chem. 2015, 290, 21800–21810. [Google Scholar] [CrossRef]
- Drmanac, R.; Drmanac, S.; Chui, G.; Diaz, R.; Hou, A.; Jin, H.; Jin, P.; Kwon, S.; Lacy, S.; Moeur, B.; et al. Sequencing by Hybridization (SBH): Advantages, Achievements, and Opportunities. Chip Technol. 2002, 77, 75–101. [Google Scholar] [CrossRef]
- Qin, Y.; Schneider, T.M.; Brenner, M.P. Sequencing by Hybridization of Long Targets. PLoS ONE 2012, 7, e35819. [Google Scholar] [CrossRef] [PubMed]
- Hu, T.; Chitnis, N.; Monos, D.; Dinh, A. Next-generation sequencing technologies: An overview. Hum. Immunol. 2021, 82, 801–811. [Google Scholar] [CrossRef]
- Harrington, C.T.; Lin, E.I.; Olson, M.T.; Eshleman, J.R. Fundamentals of Pyrosequencing. Arch. Pathol. Lab. Med. 2013, 137, 1296–1303. [Google Scholar] [CrossRef]
- Rothberg, J.M.; Hinz, W.; Rearick, T.M.; Schultz, J.; Mileski, W.; Davey, M.; Leamon, J.H.; Johnson, K.; Milgrew, M.J.; Edwards, M.; et al. An integrated semiconductor device enabling non-optical genome sequencing. Nature 2011, 475, 348–352. [Google Scholar] [CrossRef]
- Xu, Z.; Langie, S.A.S.; De Boever, P.; Taylor, J.A.; Niu, L. RELIC: A novel dye-bias correction method for Illumina Methylation BeadChip. BMC Genom. 2017, 18, 4. [Google Scholar] [CrossRef]
- Rhoads, A.; Au, K.F. PacBio Sequencing and Its Applications. Genom. Proteom. Bioinform. 2015, 13, 278–289. [Google Scholar] [CrossRef]
- Zhang, W.; Sun, Z.; Menghe, B.; Zhang, H. Short communication: Single molecule, real-time sequencing technology revealed species- and strain-specific methylation patterns of 2 Lactobacillus strains. J. Dairy Sci. 2015, 98, 3020–3024. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Wang, Y.; Deng, T.; Chen, Q. Solid-State Nanopore-Based DNA Sequencing Technology. J. Nanomater. 2016, 2016, 5284786. [Google Scholar] [CrossRef]
- Chen, Q.; Liu, Z. Fabrication and Applications of Solid-State Nanopores. Sensors 2019, 19, 1886. [Google Scholar] [CrossRef] [PubMed]
- Cao, M.D.; Ganesamoorthy, D.; Elliott, A.G.; Zhang, H.; Cooper, M.A.; Coin, L.J. Streaming algorithms for identification of pathogens and antibiotic resistance potential from real-time MinIONTM sequencing. Gigascience 2016, 5, 32. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Giordano, F.; Ning, Z. Oxford Nanopore MinION Sequencing and Genome Assembly. Genom. Proteom. Bioinform. 2016, 14, 265–279. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Vacher, S.; Boulai, A.; Bernard, V.; Baulande, S.; Bohec, M.; Bièche, I.; Lerebours, F.; Callens, C. Targeted next-generation sequencing identifies clinically relevant somatic mutations in a large cohort of inflammatory breast cancer. Breast Cancer Res. 2018, 20, 88. [Google Scholar] [CrossRef] [PubMed]
- Srour, M.K.; Qu, Y.; Deng, N.; Carlson, K.; Mirocha, J.; Gao, B.; Dadmanesh, F.; Cui, X.; Giuliano, A.E. Gene expression comparison between primary estrogen receptor-positive and triple-negative breast cancer with paired axillary lymph node metastasis. Breast J. 2021, 27, 432–440. [Google Scholar] [CrossRef]
- Srour, M.K.; Gao, B.; Dadmanesh, F.; Carlson, K.; Qu, Y.; Deng, N.; Cui, X.; Giuliano, A.E. Gene expression comparison between primary triple-negative breast cancer and paired axillary and sentinel lymph node metastasis. Breast J. 2020, 26, 904–910. [Google Scholar] [CrossRef]
- Dillon, J.; Mockus, S.; Ananda, G.; Spotlow, V.; Wells, W.; Tsongalis, G.; Marotti, J. Somatic gene mutation analysis of triple negative breast cancers. Breast 2016, 29, 202–207. [Google Scholar] [CrossRef]
- Xu, J.; Chen, Y.; Olopade, O.I. MYC and Breast Cancer. Genes Cancer 2010, 1, 629–640. [Google Scholar] [CrossRef] [PubMed]
- Badve, S.; Dabbs, D.J.; Schnitt, S.J.; Baehner, F.L.; Decker, T.; Eusebi, V.; Fox, S.B.; Ichihara, S.; Jacquemier, J.; Lakhani, S.R.; et al. Basal-like and triple-negative breast cancers: A critical review with an emphasis on the implications for pathologists and oncologists. Mod. Pathol. 2011, 24, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Zheng, F.; Yue, C.; Li, G.; He, B.; Cheng, W.; Wang, X.; Yan, M.; Long, Z.; Qiu, W.; Yuan, Z.; et al. Nuclear AURKA acquires kinase-independent transactivating function to enhance breast cancer stem cell phenotype. Nat. Commun. 2016, 7, 10180. [Google Scholar] [CrossRef] [PubMed]
- O’shaughnessy, J.; McIntyre, K.; Wilks, S.; Ma, L.; Block, M.; Andorsky, D.; Danso, M.; Locke, T.; Scales, A.; Wang, Y. Efficacy and Safety of Weekly Paclitaxel with or without Oral Alisertib in Patients with Metastatic Breast Cancer: A Randomized Clinical Trial. JAMA Netw. Open 2021, 4, e214103. [Google Scholar] [CrossRef] [PubMed]
- Robbrecht, D.G.J.; Lopez, J.; Calvo, E.; He, X.; Hiroshi, H.; Soni, N.; Cook, N.; Dowlati, A.; Fasolo, A.; Moreno, V.; et al. A first-in-human phase 1 and pharmacological study of TAS-119, a novel selective Aurora A kinase inhibitor in patients with advanced solid tumours. Br. J. Cancer 2021, 124, 391–398. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Ma, D.; Xiao, Y.; Jiang, Y.-Z.; Shao, Z.-M. Clinicopathologic features and prognoses of different histologic types of triple-negative breast cancer: A large population-based analysis. Eur. J. Surg. Oncol. 2018, 44, 420–428. [Google Scholar] [CrossRef]
- Li, Y.-Z.; Chen, B.; Lin, X.-Y.; Zhang, G.-C.; Lai, J.-G.; Li, C.; Lin, J.-L.; Guo, L.-P.; Xiao, W.-K.; Mok, H.; et al. Clinicopathologic and Genomic Features in Triple-Negative Breast Cancer Between Special and No-Special Morphologic Pattern. Front. Oncol. 2022, 12, 1124. [Google Scholar] [CrossRef]
- Pop, L.-A.; Cojocneanu-Petric, R.-M.; Pileczki, V.; Morar-Bolba, G.; Irimie, A.; Lazar, V.; Lombardo, C.; Paradiso, A.; Berindan-Neagoe, I. Genetic alterations in sporadic triple negative breast cancer. Breast 2018, 38, 30–38. [Google Scholar] [CrossRef]
- Balko, J.M.; Giltnane, J.M.; Wang, K.; Schwarz, L.J.; Young, C.D.; Cook, R.S.; Owens, P.; Sanders, M.E.; Kuba, M.G.; Sánchez, V.; et al. Molecular profiling of the residual disease of triple-negative breast cancers after neoadjuvant chemotherapy identifies actionable therapeutic targets. Cancer Discov. 2014, 4, 232–245. [Google Scholar] [CrossRef]
- Roeh, S.; Weber, P.; Rex-Haffner, M.; Deussing, J.M.; Binder, E.B.; Jakovcevski, M. Sequencing on the SOLiD 5500xl System—In-depth characterization of the GC bias. Nucleus 2017, 8, 370–380. [Google Scholar] [CrossRef]
- Lips, E.H.; Michaut, M.; Michaut, M.; Hoogstraat, M.; Mulder, L.; Besselink, N.J.; Koudijs, M.J.; Cuppen, E.; Voest, E.E.; Bernards, R.; et al. Next generation sequencing of triple negative breast cancer to find predictors for chemotherapy response. Breast Cancer Res. 2015, 17, 134. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Li, J.; Yang, Q.; Wang, Y.; Li, X.; Liu, Y.; Shan, B. Tumor mutation burden and JARID2 gene alteration are associated with short disease-free survival in locally advanced triple-negative breast cancer. Ann. Transl. Med. 2020, 8, 1052. [Google Scholar] [CrossRef] [PubMed]
- Heeke, A.L.; Xiu, J.; Elliott, A.; Korn, W.M.; Lynce, F.; Pohlmann, P.R.; Isaacs, C.; Swain, S.M.; Vidal, G.; Schwartzberg, L.S.; et al. Actionable co-alterations in breast tumors with pathogenic mutations in the homologous recombination DNA damage repair pathway. Breast Cancer Res. Treat. 2020, 184, 265–275. [Google Scholar] [CrossRef]
- Herbst, R.S.; Soria, J.-C.; Kowanetz, M.; Fine, G.D.; Hamid, O.; Gordon, M.S.; Sosman, J.A.; McDermott, D.F.; Powderly, J.D.; Gettinger, S.N.; et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 2014, 515, 563–567. [Google Scholar] [CrossRef] [PubMed]
- Akinleye, A.; Rasool, Z. Immune checkpoint inhibitors of PD-L1 as cancer therapeutics. J. Hematol. Oncol. 2019, 12, 92. [Google Scholar] [CrossRef]
- Hoda, R.S.; Brogi, E.; Dos Anjos, C.H.; Grabenstetter, A.; Ventura, K.; Patil, S.; Selenica, P.; Weigelt, B.; Reis-Filho, J.S.; Traina, T.; et al. Clinical and pathologic features associated with PD-L1 (SP142) expression in stromal tumor-infiltrating immune cells of triple-negative breast carcinoma. Mod. Pathol. 2020, 33, 2221–2232. [Google Scholar] [CrossRef]
- Razavi, P.; Chang, M.T.; Xu, G.; Bandlamudi, C.; Ross, D.S.; Vasan, N.; Cai, Y.; Bielski, C.M.; Donoghue, M.T.A.; Jonsson, P.; et al. The Genomic Landscape of Endocrine-Resistant Advanced Breast Cancers. Cancer Cell 2018, 34, 427–438.e6. [Google Scholar] [CrossRef]
- Tan, Q.; Chi, Y.; Su, M.; Zhou, J.; Zhou, D.; Zheng, F.; Man, X.; Sun, S.; Huang, J.; Li, H. Potential predictive value of circulating tumor DNA (ctDNA) mutations for the efficacy of immune checkpoint inhibitors in advanced triple-negative breast cancer. Front. Genet. 2023, 14, 1125970. [Google Scholar] [CrossRef]
- Fallah, Y.; Brundage, J.; Allegakoen, P.; Shajahan-Haq, A.N. MYC-Driven pathways in breast cancer subtypes. Biomolecules 2017, 7, 53. [Google Scholar] [CrossRef]
- Su, M.; Huang, C.-X.; Dai, A.-P. Immune checkpoint inhibitors: Therapeutic tools for breast cancer. Asian Pac. J. Cancer Prev. 2016, 17, 905–910. [Google Scholar] [CrossRef]
- Kelly, G.L.; Strasser, A. The essential role of evasion from cell death in cancer. In Advances in Cancer Research; Academic Press Inc.: Cambridge, MA, USA, 2011; Volume 111, pp. 39–96. [Google Scholar] [CrossRef]
- Jin, X.; Zhu, L.; Cui, Z.; Tang, J.; Xie, M.; Ren, G. Elevated expression of GNAS promotes breast cancer cell proliferation and migration via the PI3K/AKT/Snail1/E-cadherin axis. Clin. Transl. Oncol. 2019, 21, 1207–1219. [Google Scholar] [CrossRef] [PubMed]
- Hung, C.-S.; Wang, S.-C.; Yen, Y.-T.; Lee, T.-H.; Wen, W.-C.; Lin, R.-K. Hypermethylation of CCND2 in lung and breast cancer is a potential biomarker and drug target. Int. J. Mol. Sci. 2018, 19, 3096. [Google Scholar] [CrossRef]
- Du, H.; Yi, Z.; Wang, L.; Li, Z.; Niu, B.; Ren, G.; Du, H.; Yi, Z.; Wang, L.; Li, Z.; et al. The co-expression characteristics of LAG3 and PD-1 on the T cells of patients with breast cancer reveal a new therapeutic strategy. Int. Immunopharmacol. 2020, 78, 106113. [Google Scholar] [CrossRef] [PubMed]
- DU, S.M. The SNHG16/miR-30a axis promotes breast cancer cell proliferation and invasion by regulating RRM2. Neoplasma 2020, 67, 567–575. [Google Scholar] [CrossRef] [PubMed]
- Marra, A.; Viale, G.; Curigliano, G. Recent advances in triple negative breast cancer: The immunotherapy era. BMC Med. 2019, 17, 90. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Sun, H.; Zhao, S.; Wang, Y.; Pu, H.; Zhang, Q. Expression of PD-L1 and prognosis in breast cancer: A meta-analysis. Oncotarget 2017, 8, 31347–31354. [Google Scholar] [CrossRef]
- Sivapiragasam, A.; Kumar, P.A.; Sokol, E.S.; Albacker, L.A.; Killian, J.K.; Ramkissoon, S.H.; Huang, R.S.P.; Severson, E.A.; Brown, C.A.; Danziger, N.; et al. Predictive Biomarkers for Immune Checkpoint Inhibitors in Metastatic Breast Cancer. Cancer Med. 2021, 10, 53–61. [Google Scholar] [CrossRef]
- Skoulidis, F.; Goldberg, M.E.; Greenawalt, D.M.; Hellmann, M.D.; Awad, M.M.; Gainor, J.F.; Schrock, A.B.; Hartmaier, R.J.; Trabucco, S.E.; Gay, L.; et al. STK11/LKB1 mutations and pd-1 inhibitor resistance in kras-mutant lung adenocarcinoma. Cancer Discov. 2018, 8, 822–835. [Google Scholar] [CrossRef]
- Xu, J.; Guo, X.; Jing, M.; Sun, T. Prediction of tumor mutation burden in breast cancer based on the expression of ER, PR, HER-2, and Ki-67. OncoTargets Ther. 2018, 11, 2269–2275. [Google Scholar] [CrossRef]
- Nakagawara, A. Trk receptor tyrosine kinases: A bridge between cancer and neural development. Cancer Lett. 2001, 169, 107–114. [Google Scholar] [CrossRef]
- Wu, S.; Shi, X.; Ren, X.; Li, K.; Pang, J.; Liang, Z. Evaluation of NTRK Gene Fusion by Five Different Platforms in Triple-Negative Breast Carcinoma. Front. Mol. Biosci. 2021, 8, 654387. [Google Scholar] [CrossRef] [PubMed]
- Anwar, T.; Rufail, M.L.; Djomehri, S.I.; Gonzalez, M.E.; de la Vega, L.L.; Tomlins, S.A.; Newman, L.A.; Kleer, C.G. Next-generation sequencing identifies recurrent copy number variations in invasive breast carcinomas from Ghana. Mod. Pathol. 2020, 33, 1537–1545. [Google Scholar] [CrossRef] [PubMed]
- Anwar, T.; Arellano-Garcia, C.; Ropa, J.; Chen, Y.-C.; Kim, H.S.; Yoon, E.; Grigsby, S.; Basrur, V.; Nesvizhskii, A.I.; Muntean, A.; et al. p38-mediated phosphorylation at T367 induces EZH2 cytoplasmic localization to promote breast cancer metastasis. Nat. Commun. 2018, 9, 2801. [Google Scholar] [CrossRef] [PubMed]
- Pang, J.; Toy, K.A.; Griffith, K.A.; Awuah, B.; Quayson, S.; Newman, L.A.; Kleer, C.G. Invasive breast carcinomas in Ghana: High frequency of high grade, basal-like histology and high EZH2 expression. Breast Cancer Res. Treat. 2012, 135, 59–66. [Google Scholar] [CrossRef]
- Ben Ayed-Guerfali, D.; Ben Kridis-Rejab, W.; Ammous-Boukhris, N.; Ayadi, W.; Charfi, S.; Khanfir, A.; Sellami-Boudawara, T.; Frikha, M.; Daoud, J.; Mokdad-Gargouri, R. Novel and recurrent BRCA1/BRCA2 germline mutations in patients with breast/ovarian cancer: A series from the south of Tunisia. J. Transl. Med. 2021, 19, 108. [Google Scholar] [CrossRef]
- Cherbal, F.; Bakour, R.; Adane, S.; Boualga, K. BRCA1 and BRCA2 germline mutation spectrum in hereditary breast/ovarian cancer families from Maghrebian countries. Breast Dis. 2012, 34, 1–8. [Google Scholar] [CrossRef]
- Mahfoudh, W.; Bouaouina, N.; Ben Ahmed, S.; Gabbouj, S.; Shan, J.; Mathew, R.; Uhrhammer, N.; Bignon, Y.-J.; Troudi, W.; Elgaaied, A.B.A.; et al. Hereditary breast cancer in Middle Eastern and North African (MENA) populations: Identification of novel, recurrent and founder BRCA1 mutations in the Tunisian population. Mol. Biol. Rep. 2012, 39, 1037–1046. [Google Scholar] [CrossRef]
- Laraqui, A.; Cavaillé, M.; Uhrhammer, N.; ElBiad, O.; Bidet, Y.; El Rhaffouli, H.; El Anaz, H.; Rahali, D.M.; Kouach, J.; Guelzim, K.; et al. Identification of a novel pathogenic variant in PALB2 and BARD1 genes by a multigene sequencing panel in triple negative breast cancer in Morocco. J. Genom. 2021, 9, 43–54. [Google Scholar] [CrossRef]
- Ricker, C.; Culver, J.O.; Lowstuter, K.; Sturgeon, D.; Sturgeon, J.D.; Chanock, C.R.; Gauderman, W.J.; McDonnell, K.J.; Idos, G.E.; Gruber, S.B. Increased yield of actionable mutations using multi-gene panels to assess hereditary cancer susceptibility in an ethnically diverse clinical cohort. Cancer Genet. 2016, 209, 130–137. [Google Scholar] [CrossRef]
- Dai, X.; Cheng, H.; Bai, Z.; Li, J. Breast cancer cell line classification and its relevance with breast tumor subtyping. J. Cancer 2017, 8, 3131–3141. [Google Scholar] [CrossRef]
- Aganezov, S.; Goodwin, S.; Sherman, R.M.; Sedlazeck, F.J.; Arun, G.; Bhatia, S.; Lee, I.; Kirsche, M.; Wappel, R.; Kramer, M.; et al. Comprehensive analysis of structural variants in breast cancer genomes using single-molecule sequencing. Genome Res. 2020, 30, 1258–1273. [Google Scholar] [CrossRef] [PubMed]
- Zaccaria, S.; Raphael, B.J. Accurate quantification of copy-number aberrations and whole-genome duplications in multi-sample tumor sequencing data. Nat. Commun. 2020, 11, 4301. [Google Scholar] [CrossRef] [PubMed]
- Leung, E.; Kim, J.E.; Askarian-Amiri, M.; Finlay, G.J.; Baguley, B.C. Evidence for the existence of triple-negative variants in the mcf-7 breast cancer cell population. BioMed Res. Int. 2014, 2014, 836769. [Google Scholar] [CrossRef] [PubMed]
- Weirather, J.L.; Afshar, P.T.; Clark, T.A.; Tseng, E.; Powers, L.S.; Underwood, J.G.; Zabner, J.; Korlach, J.; Wong, W.H.; Au, K.F. Characterization of fusion genes and the significantly expressed fusion isoforms in breast cancer by hybrid sequencing. Nucleic Acids Res. 2015, 43, e116. [Google Scholar] [CrossRef] [PubMed]
- Ferla, R.; Calò, V.; Cascio, S.; Rinaldi, G.; Badalamenti, G.; Carreca, I.; Surmacz, E.; Colucci, G.; Bazan, V.; Russo, A. Founder mutations in BRCA1 and BRCA2 genes. In Annals of Oncology; Oxford University Press: Oxford, UK, 2007; Volume 18. [Google Scholar] [CrossRef]
- Tierno, D.; Grassi, G.; Zanconati, F.; Bortul, M.; Scaggiante, B. An Overview of Circulating Cell-Free Nucleic Acids in Diagnosis and Prognosis of Triple-Negative Breast Cancer. Int. J. Mol. Sci. 2023, 24, 1799. [Google Scholar] [CrossRef]
- Chen, L.; Bode, A.M.; Dong, Z. Circulating tumor cells: Moving biological insights into detection. Theranostics 2017, 7, 2606–2619. [Google Scholar] [CrossRef]
- Lo, Y.M.D.; Han, D.S.C.; Jiang, P.; Chiu, R.W.K. Epigenetics, fragmentomics, and topology of cell-free DNA in liquid biopsies. Science 2021, 372, eaaw3616. [Google Scholar] [CrossRef]
- Alborelli, I.; Generali, D.; Jermann, P.; Cappelletti, M.R.; Ferrero, G.; Scaggiante, B.; Bortul, M.; Zanconati, F.; Nicolet, S.; Haegele, J.; et al. Cell-free DNA analysis in healthy individuals by next-generation sequencing: A proof of concept and technical validation study. Cell Death Dis. 2019, 10, 534. [Google Scholar] [CrossRef]
- Selvakumar, S.C.; Preethi, K.A.; Ross, K.; Tusubira, D.; Khan, M.W.A.; Mani, P.; Rao, T.N.; Sekar, D. CRISPR/Cas9 and next generation sequencing in the personalized treatment of Cancer. Mol. Cancer 2022, 21, 83. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tierno, D.; Grassi, G.; Scomersi, S.; Bortul, M.; Generali, D.; Zanconati, F.; Scaggiante, B. Next-Generation Sequencing and Triple-Negative Breast Cancer: Insights and Applications. Int. J. Mol. Sci. 2023, 24, 9688. https://doi.org/10.3390/ijms24119688
Tierno D, Grassi G, Scomersi S, Bortul M, Generali D, Zanconati F, Scaggiante B. Next-Generation Sequencing and Triple-Negative Breast Cancer: Insights and Applications. International Journal of Molecular Sciences. 2023; 24(11):9688. https://doi.org/10.3390/ijms24119688
Chicago/Turabian StyleTierno, Domenico, Gabriele Grassi, Serena Scomersi, Marina Bortul, Daniele Generali, Fabrizio Zanconati, and Bruna Scaggiante. 2023. "Next-Generation Sequencing and Triple-Negative Breast Cancer: Insights and Applications" International Journal of Molecular Sciences 24, no. 11: 9688. https://doi.org/10.3390/ijms24119688
APA StyleTierno, D., Grassi, G., Scomersi, S., Bortul, M., Generali, D., Zanconati, F., & Scaggiante, B. (2023). Next-Generation Sequencing and Triple-Negative Breast Cancer: Insights and Applications. International Journal of Molecular Sciences, 24(11), 9688. https://doi.org/10.3390/ijms24119688