
Untargeted Metabolomics Identifies Biomarkers for MCADD Neonates in Dried Blood Spots


Abstract
1. Introduction
2. Results
2.1. Demographic Data
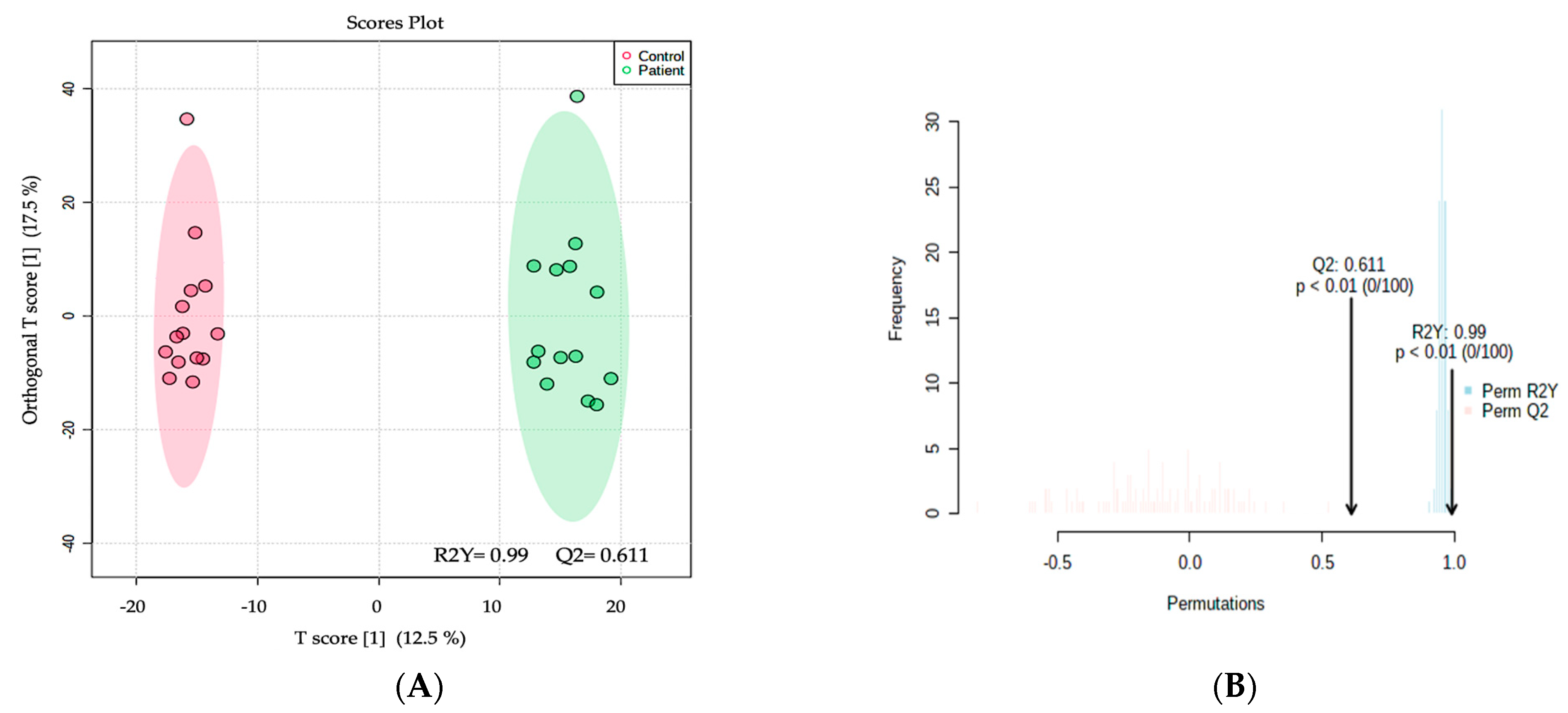
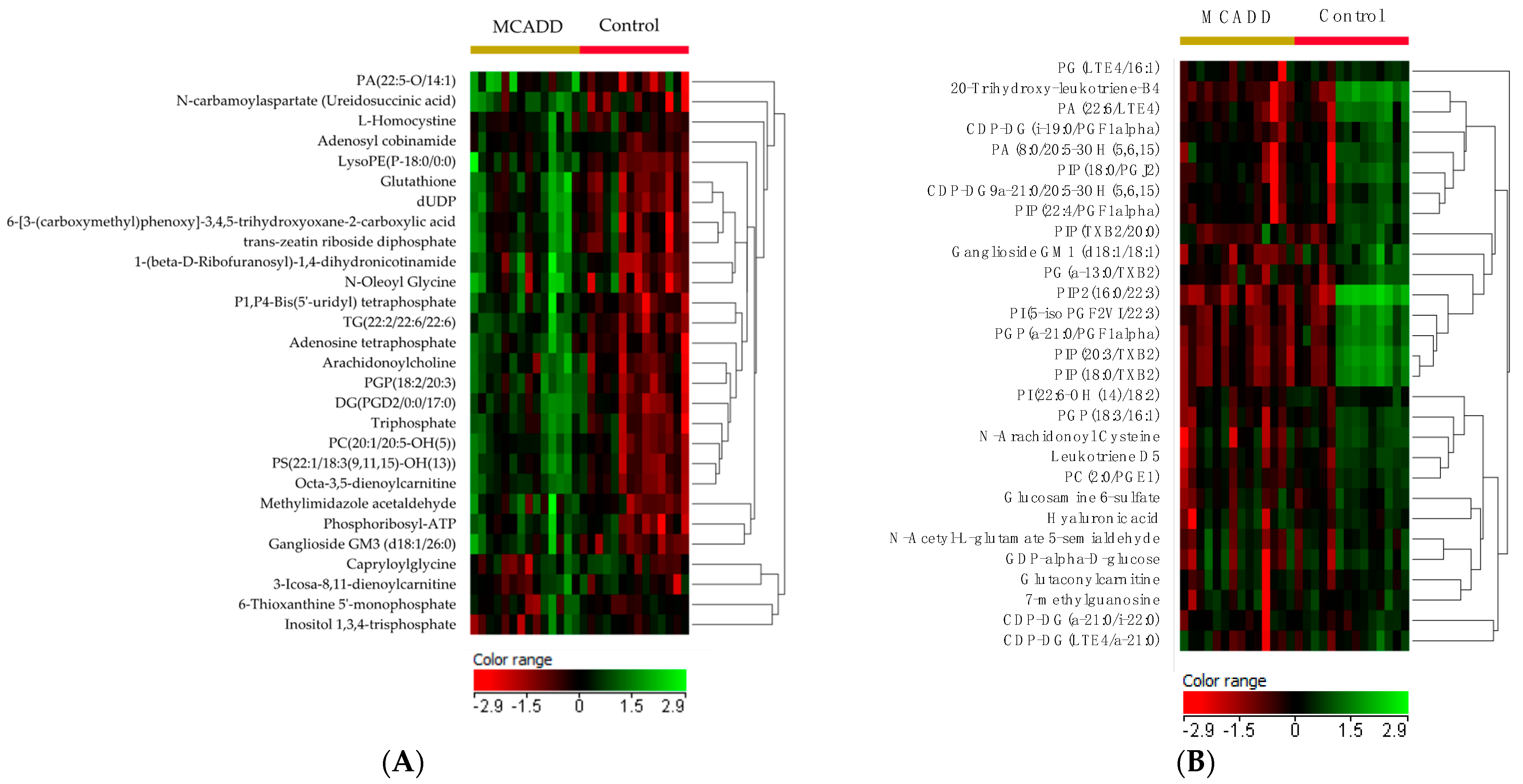
2.2. Metabolomics Profiling of MCADD Newborns
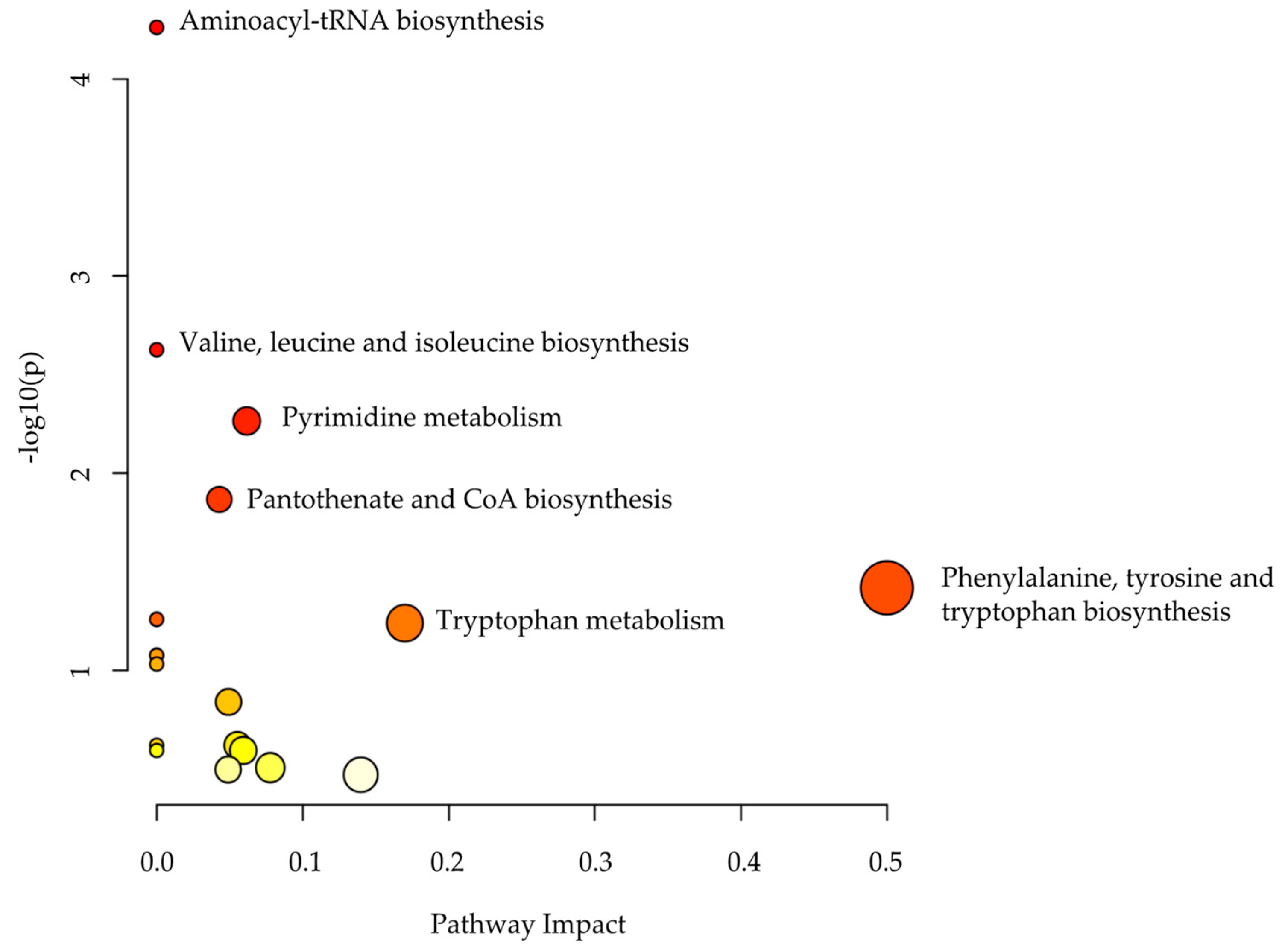
2.3. Metabolomics Pathway Analysis
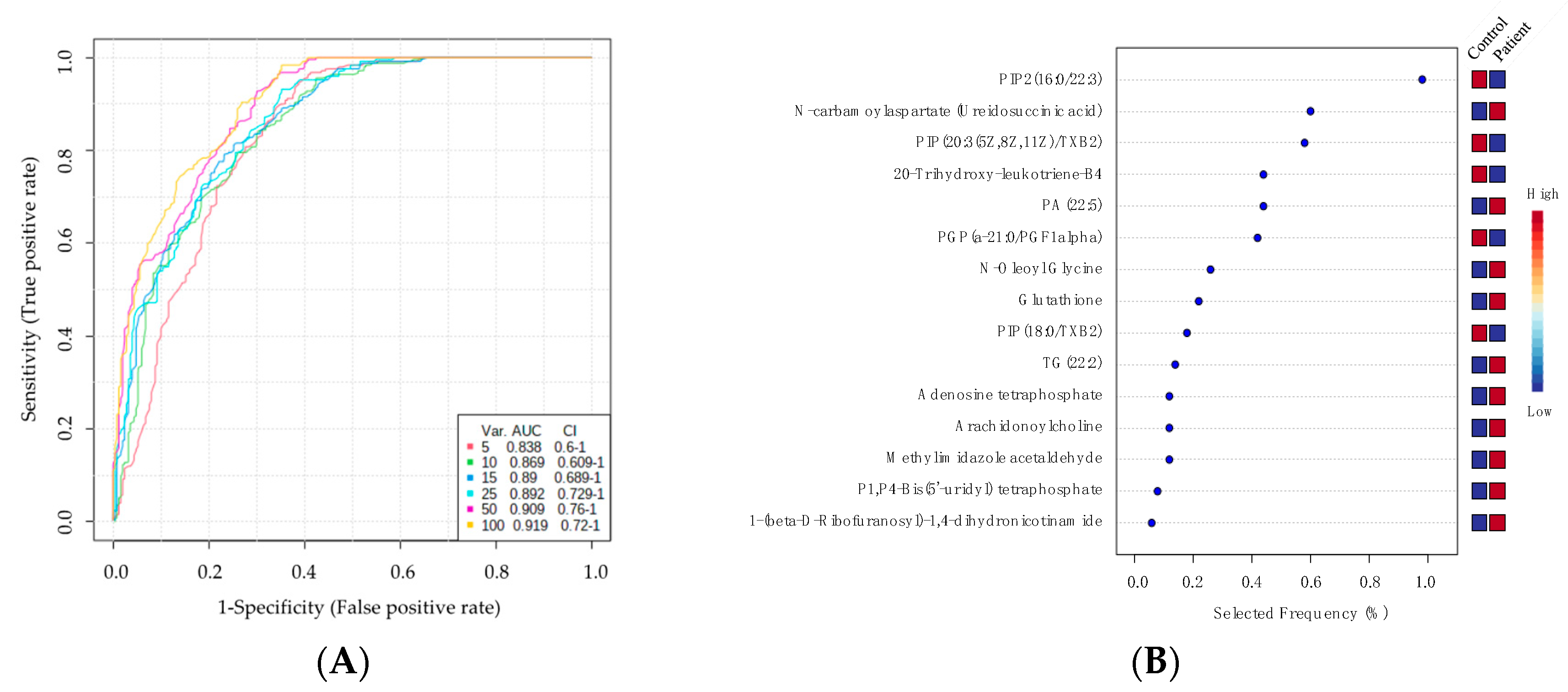
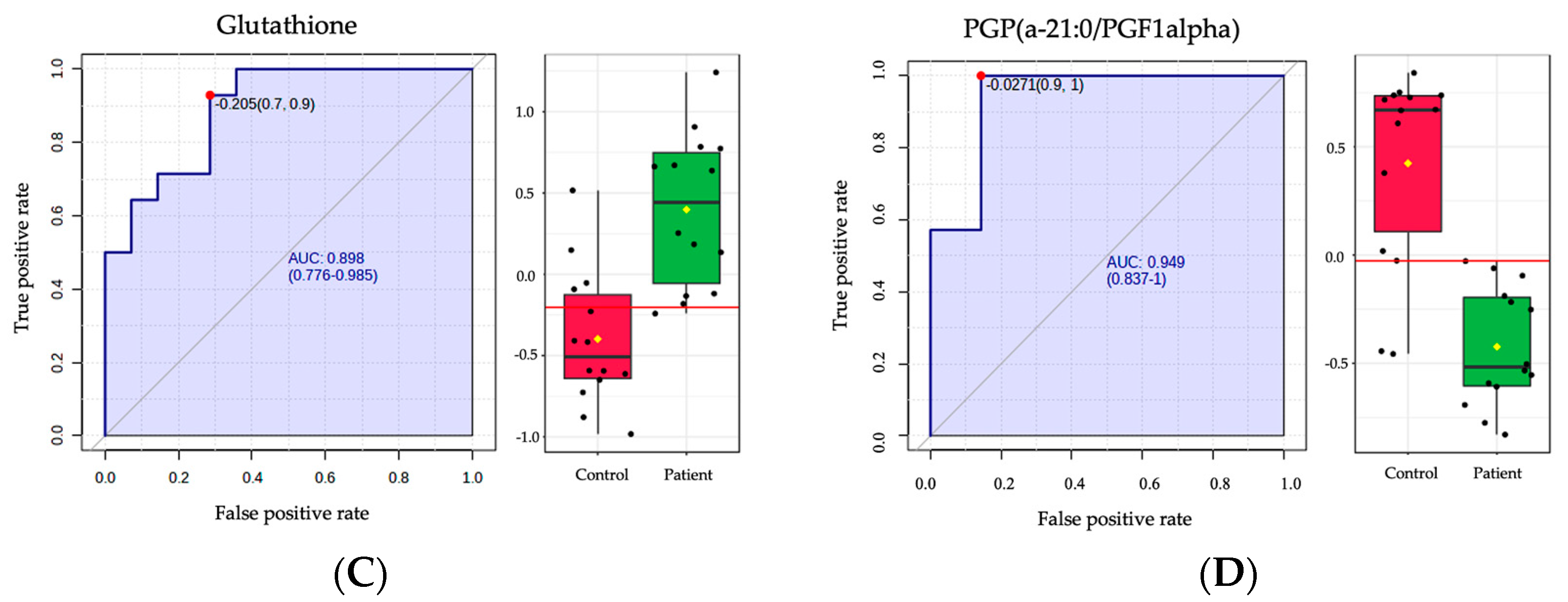
2.4. Biomarker Analysis
3. Discussion
3.1. Untargeted Metabolomics as a Complementary Diagnostic Approach for MCADD
3.2. Dysregulated Amino Acids Resulted from the Defective Mitochondrial Oxidation of Medium-Chain Fatty Acid in MCADD Newborns
3.3. Distinctive Lipid Patterns Observed in MCADD Newborns
3.4. Elevated Glutathione, a Non-Enzymatic Antioxidant Defense System, Found in MCADD Newborns
4. Materials and Methods
4.1. Ethical Approval
4.2. Biological Samples
4.3. Chemicals and Materials
4.4. Sample Preparation
4.5. LC-MS Metabolomics
4.6. Data Processing and Statistical Analyses
4.7. Peak Annotation (Metabolite Identification)
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mason, E.; Hindmarch, C.C.T.; Dunham-Snary, K.J. Medium-chain Acyl-COA Dehydrogenase Deficiency: Pathogenesis, Diagnosis, and Treatment. Endocrinol. Diabetes Metab. 2023, 6, e385. [Google Scholar] [CrossRef] [PubMed]
- Nohara, F.; Tajima, G.; Sasai, H.; Makita, Y. MCAD Deficiency Caused by Compound Heterozygous Pathogenic Variants in ACADM. Hum. Genome Var. 2022, 9, 2. [Google Scholar] [CrossRef] [PubMed]
- Afzal, R.M.; Lund, A.M.; Skovby, F. The Impact of Consanguinity on the Frequency of Inborn Errors of Metabolism. Mol. Genet. Metab. Rep. 2018, 15, 6–10. [Google Scholar] [CrossRef]
- Moammar, H.; Cheriyan, G.; Mathew, R.; Al-Sannaa, N. Incidence and Patterns of Inborn Errors of Metabolism in the Eastern Province of Saudi Arabia, 1983–2008. Ann. Saudi Med. 2010, 30, 271–277. [Google Scholar] [CrossRef] [PubMed]
- Alfadhel, M.; Al Othaim, A.; Al Saif, S.; Al Mutairi, F.; Alsayed, M.; Rahbeeni, Z.; Alzaidan, H.; Alowain, M.; Al-Hassnan, Z.; Saeedi, M.; et al. She expanded the Newborn Screening Program in Saudi Arabia: Incidence of Screened Disorders. J. Paediatr. Child Health 2017, 53, 585–591. [Google Scholar] [CrossRef] [PubMed]
- Karaceper, M.D.; Khangura, S.D.; Wilson, K.; Coyle, D.; Brownell, M.; Davies, C.; Dodds, L.; Feigenbaum, A.; Fell, D.B.; Grosse, S.D.; et al. Health Services Use among Children Diagnosed with Medium-Chain Acyl-CoA Dehydrogenase Deficiency through Newborn Screening: A Cohort Study in Ontario, Canada. Orphanet. J. Rare Dis. 2019, 14, 70. [Google Scholar] [CrossRef]
- Rhead, W.J. Newborn Screening for Medium-Chain Acyl-CoA Dehydrogenase Deficiency: A Global Perspective. J. Inherit. Metab. Dis. 2006, 29, 370–377. [Google Scholar] [CrossRef]
- Grosse, S.D.; Khoury, M.J.; Greene, C.L.; Crider, K.S.; Pollitt, R.J. The Epidemiology of Medium Chain Acyl-CoA Dehydrogenase Deficiency: An Update. Genet. Med. 2006, 8, 205–212. [Google Scholar] [CrossRef]
- Tian, Y.; Zhu, X.; Lv, S.; Jia, C.; Zhang, L.; Ni, M.; Xu, Y.; Peng, R.; Liu, S.; Zhao, D. Analysis of Gene Mutations of Medium-Chain Acyl-Coenzyme a Dehydrogenase Deficiency (MCADD) by Next-Generation Sequencing in Henan, China. Clin. Chim Acta 2022, 536, 155–161. [Google Scholar] [CrossRef]
- Al-Hassnan, Z.N.; Imtiaz, F.; Al-Amoudi, M.; Rahbeeni, Z.; Al-Sayed, M.; Al-Owain, M.; Al-Zaidan, H.; Al-Odaib, A.; Rashed, M.S. Medium-Chain Acyl-CoA Dehydrogenase Deficiency in Saudi Arabia: Incidence, Genotype, and Preventive Implications. J. Inherit. Metab. Dis. 2010, 33 (Suppl. 3), S263–S267. [Google Scholar] [CrossRef]
- El-Hattab, A.W. Systemic Primary Carnitine Deficiency. In GeneReviews®; Adam, M.P., Everman, D.B., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Longo, N.; Amat di San Filippo, C.; Pasquali, M. Disorders of Carnitine Transport and the Carnitine Cycle. Am. J. Med. Genet. Part C Semin. Med. Genet. 2006, 142C, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Schönfeld, P.; Wojtczak, L. Short- and Medium-Chain Fatty Acids in Energy Metabolism: The Cellular Perspective. J. Lipid Res. 2016, 57, 943–954. [Google Scholar] [CrossRef] [PubMed]
- Jager, E.A.; Schaafsma, M.; van der Klauw, M.M.; Heiner-Fokkema, M.R.; Derks, T.G.J. Plasma Carnitine Concentrations in Medium-Chain Acyl-CoA Dehydrogenase Deficiency: Lessons from an Observational Cohort Study. J. Inherit. Metab. Dis. 2022, 45, 1118–1129. [Google Scholar] [CrossRef] [PubMed]
- Couce, M.L.; Sánchez-Pintos, P.; Diogo, L.; Leão-Teles, E.; Martins, E.; Santos, H.; Bueno, M.A.; Delgado-Pecellín, C.; Castiñeiras, D.E.; Cocho, J.A.; et al. Newborn Screening for Medium-Chain Acyl-CoA Dehydrogenase Deficiency: Regional Experience and High Incidence of Carnitine Deficiency. Orphanet. J. Rare Dis. 2013, 8, 102. [Google Scholar] [CrossRef] [PubMed]
- Pickens, C.A.; Petritis, K. High Resolution Mass Spectrometry Newborn Screening Applications for Quantitative Analysis of Amino Acids and Acylcarnitines from Dried Blood Spots. Anal. Chim. Acta 2020, 1120, 85–96. [Google Scholar] [CrossRef]
- Neemuchwala, F.; Taki, M.; Secord, E.; Nasr, S.Z. Newborn Screening Saves Lives but Cannot Replace the Need for Clinical Vigilance. Case Rep. Pediatr. 2018, 2018, e7217326. [Google Scholar] [CrossRef]
- Dunn, C.T.; Skrypek, M.M.; Powers, A.L.R.; Laguna, T.A. The Need for Vigilance: The Case of a False-Negative Newborn Screen for Cystic Fibrosis. Pediatrics 2011, 128, e446–e449. [Google Scholar] [CrossRef]
- Crushell, E.; Chukwu, J.; Mayne, P.; Blatny, J.; Treacy, E.P. Negative Screening Tests in Classical Galactosaemia Caused by S135L Homozygosity. J. Inherit. Metab. Dis. 2009, 32, 412–415. [Google Scholar] [CrossRef]
- Kwon, C.; Farrell, P.M. The Magnitude and Challenge of False-Positive Newborn Screening Test Results. Arch. Pediatr. Adolesc. Med. 2000, 154, 714–718. [Google Scholar] [CrossRef]
- Schmidt, J.L.; Castellanos-Brown, K.; Childress, S.; Bonhomme, N.; Oktay, J.S.; Terry, S.F.; Kyler, P.; Davidoff, A.; Greene, C. The Impact of False-Positive Newborn Screening Results on Families: A Qualitative Study. Genet. Med. 2012, 14, 76–80. [Google Scholar] [CrossRef]
- Tarini, B.A.; Clark, S.J.; Pilli, S.; Dombkowski, K.J.; Korzeniewski, S.J.; Gebremariam, A.; Eisenhandler, J.; Grigorescu, V. False-Positive Newborn Screening Result and Future Health Care Use in a State Medicaid Cohort. Pediatrics 2011, 128, 715–722. [Google Scholar] [CrossRef] [PubMed]
- Karaceper, M.D.; Chakraborty, P.; Coyle, D.; Wilson, K.; Kronick, J.B.; Hawken, S.; Davies, C.; Brownell, M.; Dodds, L.; Feigenbaum, A.; et al. The Health System Impact of False Positive Newborn Screening Results for Medium-Chain Acyl-CoA Dehydrogenase Deficiency: A Cohort Study. Orphanet. J. Rare Dis. 2016, 11, 12. [Google Scholar] [CrossRef] [PubMed]
- McCandless, S.E.; Chandrasekar, R.; Linard, S.; Kikano, S.; Rice, L. Sequencing from Dried Blood Spots in Infants with “False Positive” Newborn Screen for MCAD Deficiency. Mol. Genet. Metab. 2013, 108, 51–55. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.H.; Thomas, C.; McHugh, D.; Gavrilov, D.; Raymond, K.; Rinaldo, P.; Tortorelli, S.; Matern, D.; Highsmith, W.E.; Oglesbee, D. Allelic Diversity in MCAD Deficiency: The Biochemical Classification of 54 Variants Identified during 5years of ACADM Sequencing. Mol. Genet. Metab. 2010, 100, 241–250. [Google Scholar] [CrossRef]
- Coene, K.L.M.; Kluijtmans, L.A.J.; van der Heeft, E.; Engelke, U.F.H.; de Boer, S.; Hoegen, B.; Kwast, H.J.T.; van de Vorst, M.; Huigen, M.C.D.G.; Keularts, I.M.L.W.; et al. Next-Generation Metabolic Screening: Targeted and Untargeted Metabolomics for the Diagnosis of Inborn Errors of Metabolism in Individual Patients. J. Inherit. Metab. Dis. 2018, 41, 337–353. [Google Scholar] [CrossRef]
- Liu, N.; Xiao, J.; Gijavanekar, C.; Pappan, K.L.; Glinton, K.E.; Shayota, B.J.; Kennedy, A.D.; Sun, Q.; Sutton, V.R.; Elsea, S.H. Comparison of Untargeted Metabolomic Profiling vs Traditional Metabolic Screening to Identify Inborn Errors of Metabolism. JAMA Netw. Open 2021, 4, e2114155. [Google Scholar] [CrossRef]
- Najdekr, L.; Gardlo, A.; Mádrová, L.; Friedecký, D.; Janečková, H.; Correa, E.S.; Goodacre, R.; Adam, T. Oxidized Phosphatidylcholines Suggest Oxidative Stress in Patients with Medium-Chain Acyl-CoA Dehydrogenase Deficiency. Talanta 2015, 139, 62–66. [Google Scholar] [CrossRef]
- Miller, M.J.; Kennedy, A.D.; Eckhart, A.D.; Burrage, L.C.; Wulff, J.E.; Miller, L.A.D.; Milburn, M.V.; Ryals, J.A.; Beaudet, A.L.; Sun, Q.; et al. Untargeted Metabolomic Analysis for the Clinical Screening of Inborn Errors of Metabolism. J. Inherit. Metab. Dis. 2015, 38, 1029–1039. [Google Scholar] [CrossRef]
- Vettore, L.A.; Westbrook, R.L.; Tennant, D.A. Proline Metabolism and Redox; Maintaining a Balance in Health and Disease. Amino Acids 2021, 53, 1779–1788. [Google Scholar] [CrossRef]
- Olpin, S.E. Pathophysiology of Fatty Acid Oxidation Disorders and Resultant Phenotypic Variability. J. Inherit. Metab. Dis. 2013, 36, 645–658. [Google Scholar] [CrossRef]
- Hollinshead, K.E.R.; Munford, H.; Eales, K.L.; Bardella, C.; Li, C.; Escribano-Gonzalez, C.; Thakker, A.; Nonnenmacher, Y.; Kluckova, K.; Jeeves, M.; et al. Oncogenic IDH1 Mutations Promote Enhanced Proline Synthesis through PYCR1 to Support the Maintenance of Mitochondrial Redox Homeostasis. Cell. Rep. 2018, 22, 3107–3114. [Google Scholar] [CrossRef] [PubMed]
- Lindquist, C.; Bjørndal, B.; Lund, A.; Slettom, G.; Skorve, J.; Nygård, O.; Svardal, A.; Berge, R.K. Increased Fatty Acid Oxidation and Mitochondrial Proliferation in Liver Are Associated with Increased Plasma Kynurenine Metabolites and Nicotinamide Levels in Normolipidemic and Carnitine-Depleted Rats. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2020, 1865, 158543. [Google Scholar] [CrossRef] [PubMed]
- Mileykovskaya, E.; Zhang, M.; Dowhan, W. Cardiolipin in Energy Transducing Membranes. Biochem. Mosc. 2005, 70, 154–158. [Google Scholar] [CrossRef] [PubMed]
- Vergeade, A.; Bertram, C.C.; Bikineyeva, A.T.; Zackert, W.E.; Zinkel, S.S.; May, J.M.; Dikalov, S.I.; Roberts, L.J.; Boutaud, O. Cardiolipin Fatty Acid Remodeling Regulates Mitochondrial Function by Modifying the Electron Entry Point in the Respiratory Chain. Mitochondrion 2016, 28, 88–95. [Google Scholar] [CrossRef]
- Chicco, A.J.; Sparagna, G.C. Role of Cardiolipin Alterations in Mitochondrial Dysfunction and Disease. Am. J. Physiol. Cell Physiol. 2007, 292, C33–C44. [Google Scholar] [CrossRef]
- Paradies, G.; Paradies, V.; Ruggiero, F.M.; Petrosillo, G. Cardiolipin and Mitochondrial Function in Health and Disease. Antioxid. Redox Signal. 2014, 20, 1925–1953. [Google Scholar] [CrossRef]
- Lim, S.C.; Tajika, M.; Shimura, M.; Carey, K.T.; Stroud, D.A.; Murayama, K.; Ohtake, A.; McKenzie, M. Loss of the Mitochondrial Fatty Acid β-Oxidation Protein Medium-Chain Acyl-Coenzyme A Dehydrogenase Disrupts Oxidative Phosphorylation Protein Complex Stability and Function. Sci. Rep. 2018, 8, 153. [Google Scholar] [CrossRef]
- Ribas, V.; García-Ruiz, C.; Fernández-Checa, J.C. Glutathione and Mitochondria. Front. Pharmacol. 2014, 5, 151. [Google Scholar] [CrossRef]
- Pessayre, D. Role of Mitochondria in Non-Alcoholic Fatty Liver Disease. J. Gastroenterol. Hepatol. 2007, 22, S20–S27. [Google Scholar] [CrossRef]
- Koruk, M.; Taysi, S.; Savas, M.C.; Yilmaz, O.; Akcay, F.; Karakok, M. Oxidative Stress and Enzymatic Antioxidant Status in Patients with Nonalcoholic Steatohepatitis. Ann. Clin. Lab Sci. 2004, 34, 57–62. [Google Scholar]
- Schuck, P.F.; Ferreira, G.C.; Moura, A.P.; Busanello, E.N.B.; Tonin, A.M.; Dutra-Filho, C.S.; Wajner, M. Medium-Chain Fatty Acids Accumulating in MCAD Deficiency Elicit Lipid and Protein Oxidative Damage and Decrease Non-Enzymatic Antioxidant Defenses in Rat Brain. Neurochem. Int. 2009, 54, 519–525. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Shen, L. Advances and Trends in Omics Technology Development. Front. Med. 2022, 9, 1546. [Google Scholar] [CrossRef] [PubMed]
- Micheel, C.M.; Nass, S.J.; Omenn, G.S. Omics-Based Clinical Discovery: Science, Technology, and Applications; National Academies Press: Washington, DC, USA, 2012. [Google Scholar]
- Alodaib, A.; Nimer, R.; AlMalki, R.; Alhumaidy, R.; Alhenaky, A.; Abdel Jabar, M.; Abdel Raman, A.M. Biomarker Discovery in Galactosemia: Metabolomics with UPLC/HRMS in Dried Blood Spots. Front. Mol. Biosci. 2023, 10, 1154149. [Google Scholar] [CrossRef] [PubMed]
- Jaber, M.A.; Benabdelkamel, H.; Dahabiyeh, L.A.; Masood, A.; AlMalki, R.H.; Musambil, M.; Alfadda, A.A.; Abdel Rahman, A.M. The Metabolomics Approach Revealed a Distinctive Metabolomics Pattern Associated with Hyperthyroidism Treatment. Front. Endocrinol. 2022, 13, 1050201. [Google Scholar] [CrossRef] [PubMed]
- Pang, Z.; Chong, J.; Zhou, G.; de Lima Morais, D.A.; Chang, L.; Barrette, M.; Gauthier, C.; Jacques, P.-É.; Li, S.; Xia, J. MetaboAnalyst 5.0: Narrowing the Gap between Raw Spectra and Functional Insights. Nucleic Acids Res 2021, 49, W388–W396. [Google Scholar] [CrossRef]
- Gu, X.; Al Dubayee, M.; Alshahrani, A.; Masood, A.; Benabdelkamel, H.; Zahra, M.; Li, L.; Abdel Rahman, A.M.; Aljada, A. Distinctive Metabolomics Patterns Associated With Insulin Resistance and Type 2 Diabetes Mellitus. Front. Mol. Biosci. 2020, 7, 411. [Google Scholar] [CrossRef]
- Wishart, D.S.; Guo, A.; Oler, E.; Wang, F.; Anjum, A.; Peters, H.; Dizon, R.; Sayeeda, Z.; Tian, S.; Lee, B.L.; et al. HMDB 5.0: The Human Metabolome Database for 2022. Nucleic Acids Res. 2021, 50, D622–D631. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | MCADD | Healthy Control | p-Value | |
|---|---|---|---|---|
| Number | 14 | 14 | NA | |
| Age average (Day) | 15.3 | 11 | 0.2710 | |
| Female (%) | 78% | 71% | NA | |
| Data | C8\C10-carnitine ratio (cutoff: <1.6) | 15.2 | <1.6 | 5.30 × 10−6 |
| C8-carnitine (cutoff: <0.32 µM) | 2.1 | <0.32 | 0.0007 | |
| C6-carnitine (cutoff: <0.35 µM) | 0.6 | <0.35 | 0.0004 | |
| C8\C2-carnitine ratio (cutoff: <0.1) | 0.3 | <0.1 | 0.04 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sebaa, R.; AlMogren, M.; Alseraty, W.; Abdel Rahman, A.M. Untargeted Metabolomics Identifies Biomarkers for MCADD Neonates in Dried Blood Spots. Int. J. Mol. Sci. 2023, 24, 9657. https://doi.org/10.3390/ijms24119657
Sebaa R, AlMogren M, Alseraty W, Abdel Rahman AM. Untargeted Metabolomics Identifies Biomarkers for MCADD Neonates in Dried Blood Spots. International Journal of Molecular Sciences. 2023; 24(11):9657. https://doi.org/10.3390/ijms24119657
Chicago/Turabian StyleSebaa, Rajaa, Maha AlMogren, Wafaa Alseraty, and Anas M. Abdel Rahman. 2023. "Untargeted Metabolomics Identifies Biomarkers for MCADD Neonates in Dried Blood Spots" International Journal of Molecular Sciences 24, no. 11: 9657. https://doi.org/10.3390/ijms24119657
APA StyleSebaa, R., AlMogren, M., Alseraty, W., & Abdel Rahman, A. M. (2023). Untargeted Metabolomics Identifies Biomarkers for MCADD Neonates in Dried Blood Spots. International Journal of Molecular Sciences, 24(11), 9657. https://doi.org/10.3390/ijms24119657