
Fecal Microbiota Composition, Their Interactions, and Metagenome Function in US Adults with Type 2 Diabetes According to Enterotypes




Abstract
1. Introduction
2. Results
2.1. Collection of Fecal Bacteria and Their Enterotypes
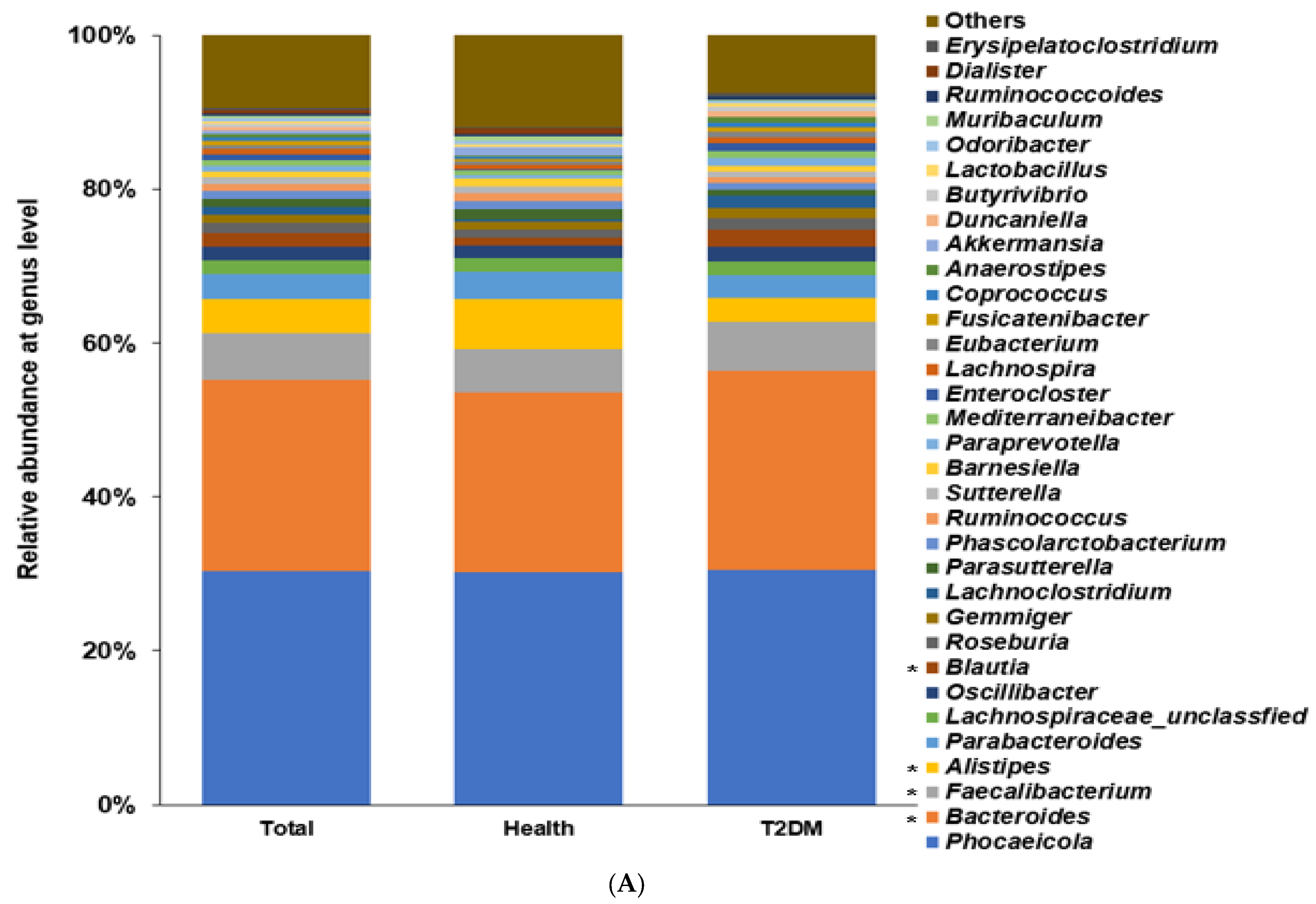
2.2. Differences in the Bacterial Composition between the T2DM and Healthy Groups in Total Participants
2.3. Differences in the Bacterial Composition between the T2DM and Healthy Groups in ET-B
2.4. Differences in the Bacterial Composition between the T2DM and Healthy Groups in ET-L
2.5. Differences in the Bacterial Composition of the T2DM and Healthy Groups in ET-P
2.6. Metagenome Function of Fecal Bacteria
3. Discussion
4. Methods
4.1. Collection and Pooling of Fecal Bacteria FASTA/Q Files for Healthy and T2DM Adults
4.2. Fecal Bacterial Composition and Community Analysis
4.3. Enterotypes
4.4. α-Diversity, β-Diversity, and Linear Discriminant Analysis (LDA) Scores of Fecal Bacteria
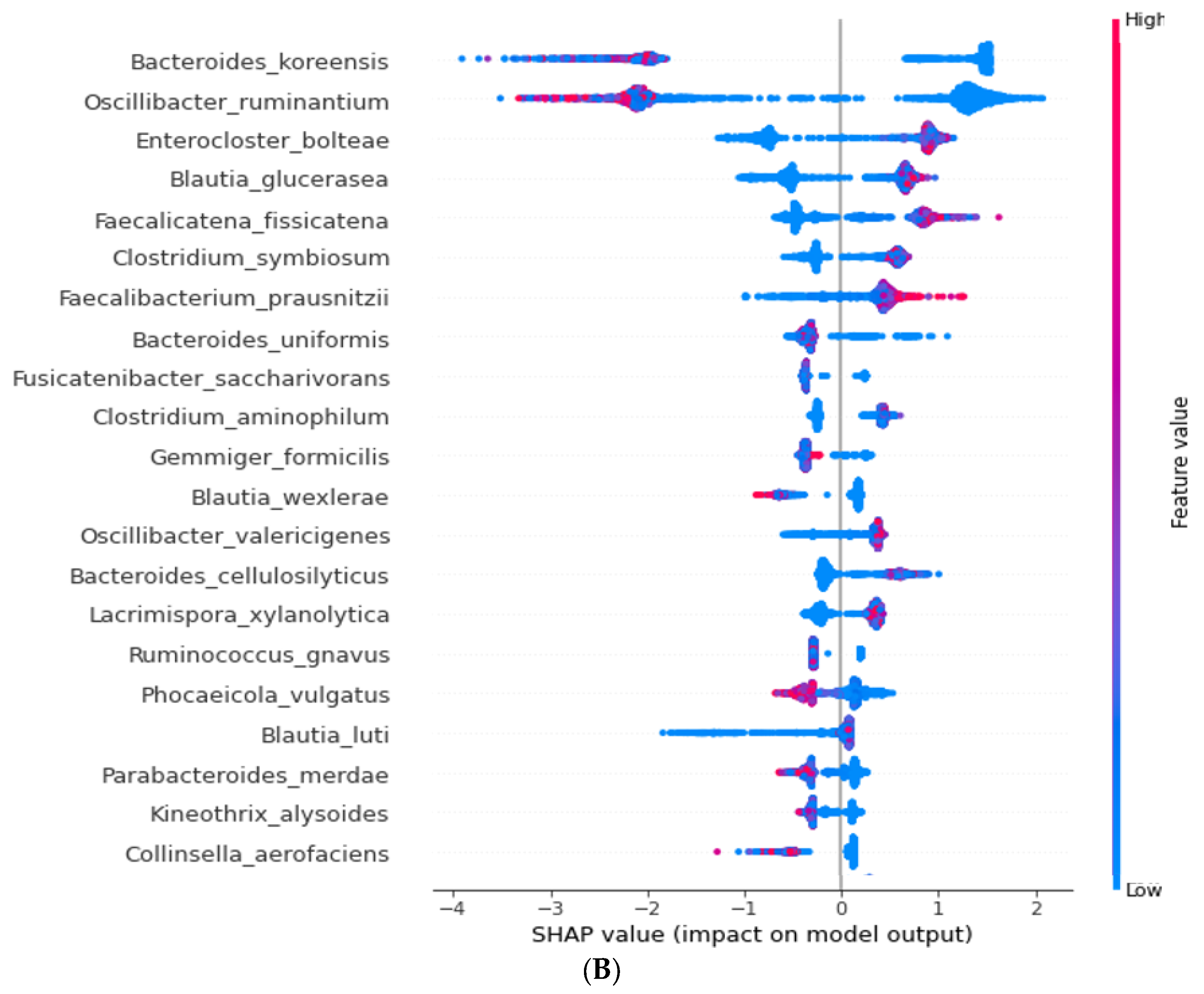
4.5. Extreme Gradient Boosting (XGBoost) Classifier Training and SHapley Additive exPlanations (SHAP) Interpreter
4.6. Metagenome Function of Fecal Bacteria by Phylogenetic Investigation of Communities by Reconstruction of Unobserved States (Picrust2)
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Abdullah, N.; Attia, J.; Oldmeadow, C.; Scott, R.J.; Holliday, E.G. The Architecture of Risk for Type 2 Diabetes: Understanding Asia in the Context of Global Findings. Int. J. Endocrinol. 2014, 2014, 593982. [Google Scholar] [CrossRef] [PubMed]
- Ma, R.C.; Chan, J.C. Type 2 diabetes in East Asians: Similarities and differences with populations in Europe and the United States. Ann. N. Y. Acad. Sci. 2013, 1281, 64–91. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.M. Characteristics of the pathophysiology of type 2 diabetes in Asians. Ann. Laparosc. Endosc. Surg. 2017, 2, 14. [Google Scholar] [CrossRef]
- Golden, S.H.; Yajnik, C.; Phatak, S.; Hanson, R.L.; Knowler, W.C. Racial/ethnic differences in the burden of type 2 diabetes over the life course: A focus on the USA and India. Diabetologia 2019, 62, 1751–1760. [Google Scholar] [CrossRef]
- Park, S.; Park, C.H.; Jang, J.S. Antecedent intake of traditional Asian-style diets exacerbates pancreatic beta-cell function, growth and survival after Western-style diet feeding in weaning male rats. J. Nutr. Biochem. 2006, 17, 307–318. [Google Scholar] [CrossRef]
- Fretts, A.M.; Follis, J.L.; Nettleton, J.A.; Lemaitre, R.N.; Ngwa, J.S.; Wojczynski, M.K.; Kalafati, I.P.; Varga, T.V.; Frazier-Wood, A.C.; Houston, D.K.; et al. Consumption of meat is associated with higher fasting glucose and insulin concentrations regardless of glucose and insulin genetic risk scores: A meta-analysis of 50,345 Caucasians. Am. J. Clin. Nutr. 2015, 102, 1266–1278. [Google Scholar] [CrossRef]
- Janssen, J.A.M.J.L. Hyperinsulinemia and Its Pivotal Role in Aging, Obesity, Type 2 Diabetes, Cardiovascular Disease and Cancer. Int. J. Mol. Sci. 2021, 22, 7797. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Zhang, J.; Cheng, Y.; Zhu, M.; Xiao, Z.; Ruan, G.; Wei, Y. Gut microbiota: A new target for T2DM prevention and treatment. Front. Endocrinol. 2022, 13, 958218. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, V.F.; Elias-Oliveira, J.; Pereira, Í.S.; Pereira, J.A.; Barbosa, S.C.; Machado, M.S.G.; Carlos, D. Akkermansia muciniphila and Gut Immune System: A Good Friendship That Attenuates Inflammatory Bowel Disease, Obesity, and Diabetes. Front. Immunol. 2022, 13, 934695. [Google Scholar] [CrossRef] [PubMed]
- Mayorga-Ramos, A.; Barba-Ostria, C.; Simancas-Racines, D.; Guamán, L.P. Protective role of butyrate in obesity and diabetes: New insights. Front. Nutr. 2022, 9, 1067647. [Google Scholar] [CrossRef]
- Wu, X.; Park, S. Fecal Bacterial Community and Metagenome Function in Asians with Type 2 Diabetes, According to Enterotypes. Biomedicines 2022, 10, 2998. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, A.L.; Stephens, J.W.; Harris, D.A. Gut microbiota influence in type 2 diabetes mellitus (T2DM). Gut Pathog. 2021, 13, 50. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Chu, J.; Hao, W.; Zhang, J.; Li, H.; Yang, C.; Yang, J.; Chen, X.; Wang, H. Gut Microbiota and Type 2 Diabetes Mellitus: Association, Mechanism, and Translational Applications. Med. Inflamm. 2021, 2021, 5110276. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Havulinna, A.S.; Liu, Y.; Jousilahti, P.; Ritchie, S.C.; Tokolyi, A.; Sanders, J.G.; Valsta, L.; Brożyńska, M.; Zhu, Q.; et al. Combined effects of host genetics and diet on human gut microbiota and incident disease in a single population cohort. Nat. Genet. 2022, 54, 134–142. [Google Scholar] [CrossRef]
- Martin, C.R.; Osadchiy, V.; Kalani, A.; Mayer, E.A. The Brain-Gut-Microbiome Axis. Cell Mol. Gastroenterol. Hepatol. 2018, 6, 133–148. [Google Scholar] [CrossRef]
- Meyers, E.E.; Kronemberger, A.; Lira, V.; Rahmouni, K.; Stauss, H.M. Contrasting effects of afferent and efferent vagal nerve stimulation on insulin secretion and blood glucose regulation. Physiol. Rep. 2016, 4, 12718. [Google Scholar] [CrossRef]
- Knauf, C.; Abot, A.; Wemelle, E.; Cani, P.D. Targeting the Enteric Nervous System to Treat Metabolic Disorders? “Enterosynes” as Therapeutic Gut Factors. Neuroendocrinology 2020, 110, 139–146. [Google Scholar] [CrossRef]
- Brooks, A.W.; Priya, S.; Blekhman, R.; Bordenstein, S.R. Gut microbiota diversity across ethnicities in the United States. PLoS Biol. 2018, 16, e2006842. [Google Scholar] [CrossRef]
- Wu, X.; Unno, T.; Kang, S.; Park, S. A Korean-Style Balanced Diet Has a Potential Connection with Ruminococcaceae Enterotype and Reduction of Metabolic Syndrome Incidence in Korean Adults. Nutrients 2021, 13, 495. [Google Scholar] [CrossRef]
- Que, Y.; Cao, M.; He, J.; Zhang, Q.; Chen, Q.; Yan, C.; Lin, A.; Yang, L.; Wu, Z.; Zhu, D.; et al. Gut Bacterial Characteristics of Patients With Type 2 Diabetes Mellitus and the Application Potential. Front. Immunol. 2021, 12, 722206. [Google Scholar] [CrossRef]
- Park, S.; Zhang, T.; Yue, Y.; Wu, X. Effects of Bile Acid Modulation by Dietary Fat, Cholecystectomy, and Bile Acid Sequestrant on Energy, Glucose, and Lipid Metabolism and Gut Microbiota in Mice. Int. J. Mol. Sci. 2022, 23, 5935. [Google Scholar] [CrossRef] [PubMed]
- Wegierska, A.E.; Charitos, I.A.; Topi, S.; Potenza, M.A.; Montagnani, M.; Santacroce, L. The Connection Between Physical Exercise and Gut Microbiota: Implications for Competitive Sports Athletes. Sports Med. 2022, 52, 2355–2369. [Google Scholar] [CrossRef] [PubMed]
- Thomas, F.; Hehemann, J.-H.; Rebuffet, E.; Czjzek, M.; Michel, G. Environmental and Gut Bacteroidetes: The Food Connection. Front. Microbiol. 2011, 2, 93. [Google Scholar] [CrossRef] [PubMed]
- Roager, H.M.; Licht, T.R.; Poulsen, S.K.; Larsen, T.M.; Bahl, M.I. Microbial enterotypes, inferred by the prevotella-to-bacteroides ratio, remained stable during a 6-month randomized controlled diet intervention with the new nordic diet. Appl. Environ. Microbiol. 2014, 80, 1142–1149. [Google Scholar] [CrossRef] [PubMed]
- Hur, H.J.; Wu, X.; Yang, H.J.; Kim, M.J.; Lee, K.-H.; Hong, M.; Park, S.; Kim, M.-S. Beneficial Effects of a Low-Glycemic Diet on Serum Metabolites and Gut Microbiota in Obese Women With Prevotella and Bacteriodes Enterotypes: A Randomized Clinical Trial. Front. Nutr. 2022, 9, 861880. [Google Scholar] [CrossRef]
- Guzior, D.V.; Quinn, R.A. Review: Microbial transformations of human bile acids. Microbiome 2021, 9, 140. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhi, F. Lower Level of Bacteroides in the Gut Microbiota Is Associated with Inflammatory Bowel Disease: A Meta-Analysis. BioMed Res. Int. 2016, 2016, 5828959. [Google Scholar] [CrossRef]
- Chen, Z.; Radjabzadeh, D.; Chen, L.; Kurilshikov, A.; Kavousi, M.; Ahmadizar, F.; Ikram, M.A.; Uitterlinden, A.G.; Zhernakova, A.; Fu, J.; et al. Association of Insulin Resistance and Type 2 Diabetes With Gut Microbial Diversity: A Microbiome-Wide Analysis From Population Studies. JAMA Netw. Open 2021, 4, e2118811. [Google Scholar] [CrossRef]
- Maioli, T.U.; Borras-Nogues, E.; Torres, L.; Barbosa, S.C.; Martins, V.D.; Langella, P.; Azevedo, V.A.; Chatel, J.-M. Possible Benefits of Faecalibacterium prausnitzii for Obesity-Associated Gut Disorders. Front. Pharmacol. 2021, 12, 740636. [Google Scholar] [CrossRef]
- Fabersani, E.; Portune, K.; Campillo, I.; López-Almela, I.; la Paz, S.M.-D.; Romaní-Pérez, M.; Benítez-Páez, A.; Sanz, Y. Bacteroides uniformis CECT 7771 alleviates inflammation within the gut-adipose tissue axis involving TLR5 signaling in obese mice. Sci. Rep. 2021, 11, 11788. [Google Scholar] [CrossRef]
- Hu, Q.; Tan, L.; Gu, S.; Xiao, Y.; Xiong, X.; Zeng, W.-A.; Feng, K.; Wei, Z.; Deng, Y. Network analysis infers the wilt pathogen invasion associated with non-detrimental bacteria. NPJ Biofilms Microbiomes 2020, 6, 8. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Collij, V.; Jaeger, M.; van den Munckhof, I.C.L.; Vich Vila, A.; Kurilshikov, A.; Gacesa, R.; Sinha, T.; Oosting, M.; Joosten, L.A.B.; et al. Gut microbial co-abundance networks show specificity in inflammatory bowel disease and obesity. Nat. Commun. 2020, 11, 4018. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.Y.; Lee, M.K. Autonomic dysfunction, diabetes and metabolic syndrome. J. Diab. Investig. 2021, 12, 2108–2111. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Wu, X. Modulation of the Gut Microbiota in Memory Impairment and Alzheimer’s Disease via the Inhibition of the Parasympathetic Nervous System. Int. J. Mol. Sci. 2022, 23, 13574. [Google Scholar] [CrossRef]
- Zhang, T.; Wu, X.; Yuan, H.; Huang, S.; Park, S. Mitigation of Memory Impairment with Fermented Fucoidan and λ-Carrageenan Supplementation through Modulating the Gut Microbiota and Their Metagenome Function in Hippocampal Amyloid-β Infused Rats. Cells 2022, 11, 2301. [Google Scholar] [CrossRef] [PubMed]
- Bergstra, J.; Bengio, Y. Random search for hyper-parameter optimization. J. Mach. Learn. Res. 2012, 13, 281–305. [Google Scholar]
- Wu, X.; Park, S. An Inverse Relation between Hyperglycemia and Skeletal Muscle Mass Predicted by Using a Machine Learning Approach in Middle-Aged and Older Adults in Large Cohorts. J. Clin. Med. 2021, 10, 2133. [Google Scholar] [CrossRef]
- Park, S.; Kim, C.; Wu, X. Development and Validation of an Insulin Resistance Predicting Model Using a Machine-Learning Approach in a Population-Based Cohort in Korea. Diagnostics 2022, 12, 212. [Google Scholar] [CrossRef]
- Lundberg, S.M.; Erion, G.; Chen, H.; DeGrave, A.; Prutkin, J.M.; Nair, B.; Katz, R.; Himmelfarb, J.; Bansal, N.; Lee, S.-I. From local explanations to global understanding with explainable AI for trees. Nat. Mach. Intell. 2020, 2, 56–67. [Google Scholar] [CrossRef]
- Yang, H.J.; Zhang, T.; Wu, X.G.; Kim, M.J.; Kim, Y.H.; Yang, E.S.; Yoon, Y.S.; Park, S. Aqueous Blackcurrant Extract Improves Insulin Sensitivity and Secretion and Modulates the Gut Microbiome in Non-Obese Type 2 Diabetic Rats. Antioxidants 2021, 10, 756. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Total Participants | AUROC | Accuracy | Sensitivity | Specificity | Precision | F1 |
|---|---|---|---|---|---|---|
| XGBoost | 1.0 ± 4.1 × 10−6 | 9.9 ± 1.7 × 10−4 | 0.99 ± 2.5 × 10−4 | 0.99 ± 2.0 × 10−4 | 1.0 ± 1.6 × 10−4 | 0.99 ± 1.52 × 10−4 |
| Random forest | 1.0 ± 0.001 | 1.0. ± 8.2 × 10−5 | 1.0 ± 0.0 | 0.99 ± 1.9 × 10−4 | 1.0 ± 1.5 × 10−4 | 1.0 ± 7.36 × 10−5 |
| Linear regress | 0.99 ± 2.0 × 10−4 | 0.97 ± 2.0 × 10−4 | 0.97 ± 3.4 × 10−4 | 0.96 ± 4.6 × 10−4 | 0.97 ± 4.6 × 10−4 | 0.97 ± 2.55 × 10−4 |
| ET-B | AUROC | Accuracy | Sensitivity | Specificity | Precision | F1 |
| XGBoost | 1.0 ± 1.1 × 10−5 | 0.97 ± 3.9 × 10−4 | 0.96 ± 5.3 × 10−4 | 0.97 ± 1.3 × 10−4 | 0.97 ± 0.0005 | 0.97 ± 0.0004 |
| Random forest | 1.0 ± 3.0 × 10−5 | 0.97 ± 3.9 × 10−4 | 0.98 ± 3.9 × 10−4 | 0.97 ± 6.4 × 10−4 | 0.97 ± 0.0005 | 0.97 ± 0.0003 |
| Linear regress | 0.98 ± 2.8 × 10−4 | 0.95 ± 5.2 × 10−4 | 0.97 ± 5.4 × 10−4 | 0.93 ± 7.7 × 10−4 | 0.94 ± 0.0007 | 0.96 ± 0.0005 |
| ET-L | AUROC | Accuracy | Sensitivity | Specificity | Precision | F1 |
| XGBoost | 1.0 ± 0.0 | 0.99 ± 3.4 × 10−6 | 0.99 ± 3.7 × 10−4 | 1.0 ± 0.0 | 1.0 ± 0.0 | 0.99 ± 1.9 × 10−4 |
| Random forest | 1.0 ± 0.0 | 0.99 ± 1.9 × 10−4 | 0.99 ± 4.3 × 10−4 | 1.0 ± 0.0 | 1.0 ± 0.0 | 0.99 ± 2.3 × 10−4 |
| Linear regress | 1.0 ± 0.0001 | 0.98 ± 3.9 × 10−4 | 0.99 ± 4.4 × 10−4 | 0.97 ± 6.8 × 10−4 | 0.98 ± 5.2 × 10−4 | 0.98 ± 3.2 × 10−4 |
| ET-P | AUROC | Accuracy | Sensitivity | Specificity | Precision | F1 |
| XGBoost | 1.0 ± 0.0 | 0.98 ± 6.9 × 10−4 | 0.954 ± 0.001 | 1.0 ± 0.0 | 1.0 ± 0.0 | 0.98 ± 8.3 × 10−5 |
| Random forest | 1.0 ± 0.0 | 0.96 ± 9.2 × 10−4 | 0.911 ± 0.002 | 1.0 ± 0.0 | 1.0 ± 0.0 | 0.95 ± 0.002 |
| Linear regress | 0.95 ± 0.001 | 0.96 ± 0.001 | 0.954 ± 0.001 | 0.957 ± 0.001 | 0.954 ± 0.001 | 0.95 ± 0.001 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, S.; Zhang, T.; Kang, S. Fecal Microbiota Composition, Their Interactions, and Metagenome Function in US Adults with Type 2 Diabetes According to Enterotypes. Int. J. Mol. Sci. 2023, 24, 9533. https://doi.org/10.3390/ijms24119533
Park S, Zhang T, Kang S. Fecal Microbiota Composition, Their Interactions, and Metagenome Function in US Adults with Type 2 Diabetes According to Enterotypes. International Journal of Molecular Sciences. 2023; 24(11):9533. https://doi.org/10.3390/ijms24119533
Chicago/Turabian StylePark, Sunmin, Ting Zhang, and Suna Kang. 2023. "Fecal Microbiota Composition, Their Interactions, and Metagenome Function in US Adults with Type 2 Diabetes According to Enterotypes" International Journal of Molecular Sciences 24, no. 11: 9533. https://doi.org/10.3390/ijms24119533
APA StylePark, S., Zhang, T., & Kang, S. (2023). Fecal Microbiota Composition, Their Interactions, and Metagenome Function in US Adults with Type 2 Diabetes According to Enterotypes. International Journal of Molecular Sciences, 24(11), 9533. https://doi.org/10.3390/ijms24119533