
Precision Editing as a Therapeutic Approach for β-Hemoglobinopathies


{kind=link}
{kind=link}
Abstract
1. Introduction
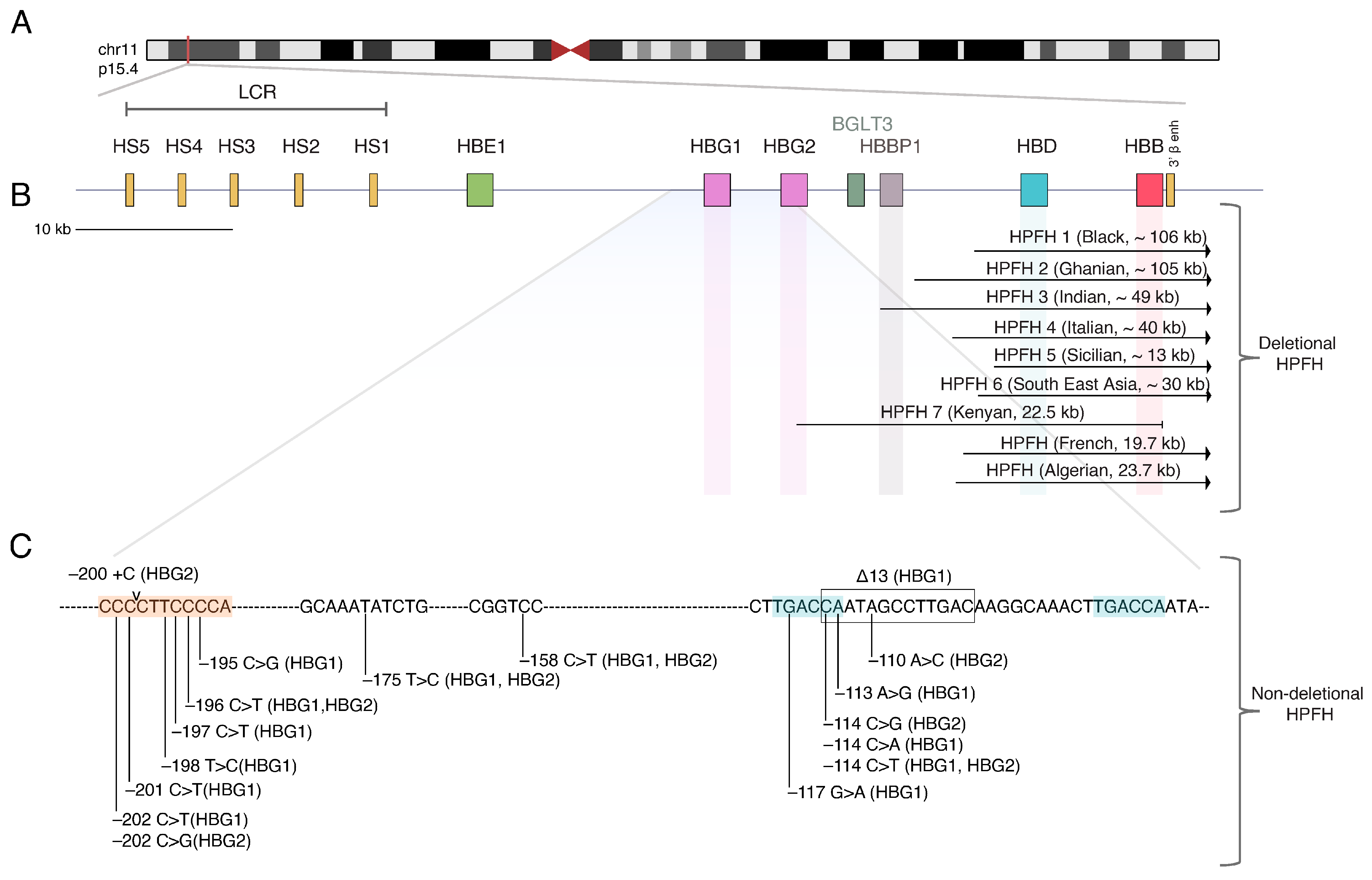
2. The Beta-Globin Locus and the Hemoglobin Switching
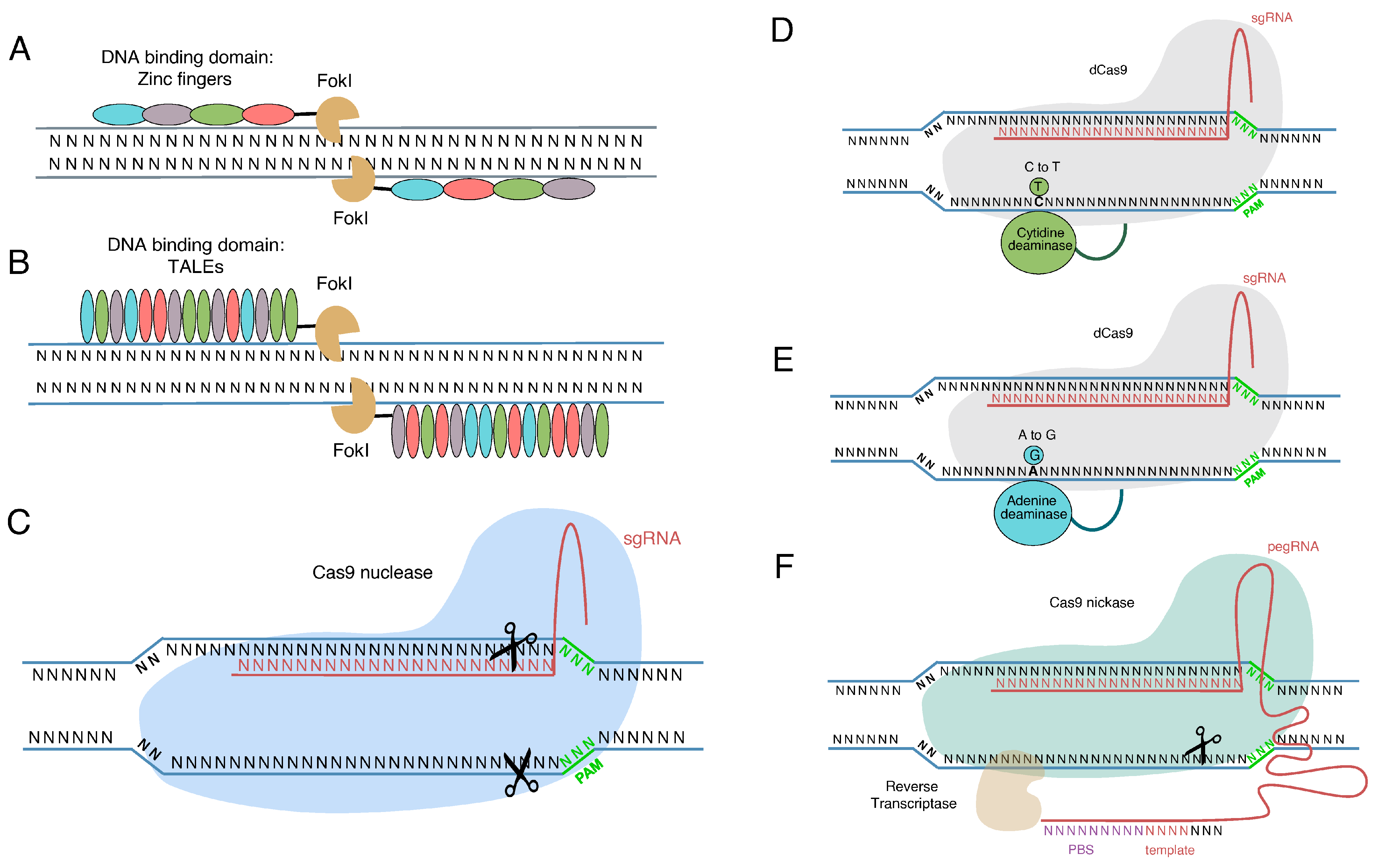
3. Genome Editing Tools
4. In Situ Mutation Correction
Sickle Cell Disease
5. Beta Thalassemia
6. HbF Reactivation by Recapitulating Natural HPFH Mutations
7. HbF Reactivation by Modulating γ-Globin Modifiers
7.1. BCL11A and Its Erythroid Enhancer
7.2. Other HbF Modifiers
8. Current Clinical Trials
9. Remaining Challenges
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Modell, B.; Darlison, M. Global Epidemiology of Haemoglobin Disorders and Derived Service Indicators. Bull. World Health Organ. 2008, 86, 480–487. [Google Scholar] [CrossRef]
- Higgs, D.R.; Engel, J.D.; Stamatoyannopoulos, G. Thalassaemia. Lancet 2012, 379, 373–383. [Google Scholar] [CrossRef]
- Rees, D.C.; Williams, T.N.; Gladwin, M.T. Sickle-Cell Disease. Lancet 2010, 376, 2018–2031. [Google Scholar] [CrossRef]
- Steinberg, M.H. Fetal Hemoglobin in Sickle Hemoglobinopathies: High Hbf Genotypes and Phenotypes. J. Clin. Med. 2020, 9, 3782. [Google Scholar] [CrossRef]
- Lettre, G.; Bauer, D.E. Fetal Haemoglobin in Sickle-Cell Disease: From Genetic Epidemiology to New Therapeutic Strategies. Lancet 2016, 387, 2554–2564. [Google Scholar] [CrossRef]
- Ferrone, F.A. Sickle Cell Disease: Its Molecular Mechanism and the One Drug That Treats It. Int. J. Biol. Macromol. 2016, 93, 1168–1173. [Google Scholar] [CrossRef]
- Hulbert, M.L.; Shenoy, S. Hematopoietic Stem Cell Transplantation for Sickle Cell Disease: Progress and Challenges. Pediatr. Blood Cancer 2018, 65, e27263. [Google Scholar] [CrossRef]
- Psatha, N.; Papayanni, P.-G.; Yannaki, E. A New Era for Hemoglobinopathies: More Than One Curative Option. Curr. Gene Ther. 2018, 17, 364–378. [Google Scholar] [CrossRef]
- Karponi, G.; Psatha, N.; Lederer, C.W.; Adair, J.E.; Zervou, F.; Zogas, N.; Kleanthous, M.; Tsatalas, C.; Anagnostopoulos, A.; Sadelain, M.; et al. Plerixafor+G-CSF-Mobilized CD34+ Cells Represent an Optimal Graft Source for Thalassemia Gene Therapy. Blood 2015, 126, 616–619. [Google Scholar] [CrossRef]
- Yannaki, E.; Karponi, G.; Zervou, F.; Constantinou, V.; Bouinta, A.; Tachynopoulou, V.; Kotta, K.; Jonlin, E.; Papayannopoulou, T.; Anagnostopoulos, A.; et al. Hematopoietic Stem Cell Mobilization for Gene Therapy: Superior Mobilization by the Combination of Granulocyte-Colony Stimulating Factor plus Plerixafor in Patients with β-Thalassemia Major. Hum. Gene Ther. 2013, 24, 852–860. [Google Scholar] [CrossRef]
- Psatha, N.; Sgouramali, E.; Gkountis, A.; Siametis, A.; Baliakas, P.; Constantinou, V.; Athanasiou, E.; Arsenakis, M.; Anagnostopoulos, A.; Papayannopoulou, T.; et al. Superior Long-Term Repopulating Capacity of G-CSF + Plerixafor-Mobilized Blood: Implications for Stem Cell Gene Therapy by Studies in the Hbbth-3 Mouse Model. Hum. Gene Ther. Methods 2014, 25, 317–327. [Google Scholar] [CrossRef]
- Esrick, E.B.; Manis, J.P.; Daley, H.; Baricordi, C.; Trébéden-Negre, H.; Pierciey, F.J.; Armant, M.; Nikiforow, S.; Heeney, M.M.; London, W.B.; et al. Successful Hematopoietic Stem Cell Mobilization and Apheresis Collection Using Plerixafor Alone in Sickle Cell Patients. Blood Adv. 2018, 2, 2505–2512. [Google Scholar] [CrossRef]
- Cavazzana, M.; Mavilio, F. Gene Therapy for Hemoglobinopathies. Hum. Gene Ther. 2018, 29, 1106–1113. [Google Scholar] [CrossRef]
- Pawliuk, R.; Westerman, K.A.; Fabry, M.E.; Payen, E.; Tighe, R.; Bouhassira, E.E.; Acharya, S.A.; Ellis, J.; London, I.M.; Eaves, C.J.; et al. Correction of Sickle Cell Disease in Transgenic Mouse Models by Gene Therapy. Science 2001, 294, 2368–2371. [Google Scholar] [CrossRef]
- Perumbeti, A.; Higashimoto, T.; Urbinati, F.; Franco, R.; Meiselman, H.J.; Witte, D.; Malik, P. A Novel Human Gamma-Globin Gene Vector for Genetic Correction of Sickle Cell Anemia in a Humanized Sickle Mouse Model: Critical Determinants for Successful Correction. Blood 2009, 114, 1174–1185. [Google Scholar] [CrossRef]
- Pestina, T.I.; Hargrove, P.W.; Jay, D.; Gray, J.T.; Boyd, K.M.; Persons, D.A. Correction of Murine Sickle Cell Disease Using γ-Globin Lentiviral Vectors to Mediate High-Level Expression of Fetal Hemoglobin. Mol. Ther. 2009, 17, 245–252. [Google Scholar] [CrossRef]
- Rivella, S.; May, C.; Chadburn, A.; Rivière, I.; Sadelain, M. A Novel Murine Model of Cooley Anemia and Its Rescue by Lentiviral-Mediated Human β-Globin Gene Transfer. Blood 2003, 101, 2932–2939. [Google Scholar] [CrossRef]
- Chad, M.; Rivella, S.; Callegari, J.; Heller, G.; Gaensler, K.M.L.; Luzzatto, L.; Sadelain, M. Therapeutic Haemoglobin Synthesis in β-Thalassaemic Mice Expressing Lentivirus-Encoded Human β-Globin. Nature 2000, 406, 82–86. [Google Scholar] [CrossRef]
- Hanawa, H.; Hargrove, P.W.; Kepes, S.; Srivastava, D.K.; Nienhuis, A.W.; Persons, D.A. Extended β-Globin Locus Control Region Elements Promote Consistent Therapeutic Expression of a γ-Globin Lentiviral Vector in Murine β-Thalassemia. Blood 2004, 104, 2281–2290. [Google Scholar] [CrossRef]
- Miccio, A.; Cesari, R.; Lotti, F.; Rossi, C.; Sanvito, F.; Ponzoni, M.; Routledge, S.J.E.; Chow, C.M.; Antoniou, M.N.; Ferrari, G. In Vivo Selection of Genetically Modified Erythroblastic Progenitors Leads to Long-Term Correction of β-Thalassemia. Proc. Natl. Acad. Sci. USA 2008, 105, 10547–10552. [Google Scholar] [CrossRef]
- Thompson, A.A.; Walters, M.C.; Kwiatkowski, J.; Rasko, J.E.J.; Ribeil, J.-A.; Hongeng, S.; Magrin, E.; Schiller, G.J.; Payen, E.; Semeraro, M.; et al. Gene Therapy in Patients with Transfusion-Dependent β-Thalassemia. N. Engl. J. Med. 2018, 378, 1479–1493. [Google Scholar] [CrossRef]
- Ribeil, J.-A.; Hacein-Bey-Abina, S.; Payen, E.; Magnani, A.; Semeraro, M.; Magrin, E.; Caccavelli, L.; Neven, B.; Bourget, P.; El Nemer, W.; et al. Gene Therapy in a Patient with Sickle Cell Disease. N. Engl. J. Med. 2017, 376, 848–855. [Google Scholar] [CrossRef]
- Locatelli, F.; Thompson, A.; Kwiatkowski, J.; Porter, J.; Thrasher, A.; Hongeng, S.; Sauer, M.; Thuret, I.; Lal, A.; Algeri, M.; et al. Betibeglogene Autotemcel Gene Therapy for Non-Β0/Β0 Genotype β-Thalassemia. N. Engl. J. Med. 2022, 356, 415–427. [Google Scholar] [CrossRef]
- Ghiaccio, V.; Chappell, M.; Rivella, S.; Breda, L. Gene Therapy for Beta-Hemoglobinopathies: Milestones, New Therapies and Challenges. Mol. Diagn. Ther. 2019, 23, 173–186. [Google Scholar] [CrossRef]
- Lux, C.T.; Pattabhi, S.; Berger, M.; Nourigat, C.; Flowers, D.A.; Negre, O.; Humbert, O.; Yang, J.G.; Lee, C.; Jacoby, K.; et al. TALEN-Mediated Gene Editing of HBG in Human Hematopoietic Stem Cells Leads to Therapeutic Fetal Hemoglobin Induction. Mol. Ther. Methods Clin. Dev. 2019, 12, 175–183. [Google Scholar] [CrossRef]
- Hoban, M.D.; Cost, G.J.; Mendel, M.C.; Romero, Z.; Kaufman, M.L.; Joglekar, A.V.; Ho, M.; Lumaquin, D.; Gray, D.; Lill, G.R.; et al. Correction of the Sickle Cell Disease Mutation in Human Hematopoietic Stem/Progenitor Cells. Blood 2015, 125, 2597–2604. [Google Scholar] [CrossRef]
- Psatha, N.; Reik, A.; Phelps, S.; Zhou, Y.; Dalas, D.; Yannaki, E.; Levasseur, D.N.; Urnov, F.D.; Holmes, M.C.; Papayannopoulou, T. Disruption of the BCL11A Erythroid Enhancer Reactivates Fetal Hemoglobin in Erythroid Cells of Patients with β-Thalassemia Major. Mol. Ther. Methods Clin. Dev. 2018, 10, 313–326. [Google Scholar] [CrossRef]
- Romero, Z.; Lomova, A.; Said, S.; Miggelbrink, A.; Kuo, C.Y.; Campo-Fernandez, B.; Hoban, M.D.; Masiuk, K.E.; Clark, D.N.; Long, J.; et al. Editing the Sickle Cell Disease Mutation in Human Hematopoietic Stem Cells: Comparison of Endonucleases and Homologous Donor Templates. Mol. Ther. 2019, 27, 1389–1406. [Google Scholar] [CrossRef] [PubMed]
- Psatha, N.; Paschoudi, K.; Papadopoulou, A.; Yannaki, E. In Vivo Hematopoietic Stem Cell Genome Editing: Perspectives and Limitations. Genes 2022, 13, 2222. [Google Scholar] [CrossRef]
- Rahimmanesh, I.; Boshtam, M.; Kouhpayeh, S.; Khanahmad, H.; Dabiri, A.; Ahangarzadeh, S.; Esmaeili, Y.; Bidram, E.; Vaseghi, G.; Javanmard, S.H.; et al. Gene Editing-Based Technologies for Beta-Hemoglobinopathies Treatment. Biology 2022, 11, 862. [Google Scholar] [CrossRef]
- Brusson, M.; Miccio, A. Genome Editing Approaches to β-Hemoglobinopathies. Prog. Mol. Biol. Transl. Sci. 2021, 182, 153–183. [Google Scholar] [CrossRef]
- Yannaki, E.; Psatha, N.; Anastasia, P.; Athanasopoulos, T.; Gravanis, A.; Roubelakis, M.G.; Tsirigotis, P.; Anagnostopoulos, A.; Anagnou, N.P.; Vassilopoulos, G. Success Stories and Challenges Ahead in Hematopoietic Stem Cell Gene Therapy: Hemoglobinopathies as Disease Models. Hum Gene Ther. 2021, 32, 1120–1137. [Google Scholar] [CrossRef]
- Saleh-Gohari, N.; Helleday, T. Conservative Homologous Recombination Preferentially Repairs DNA Double-Strand Breaks in the S Phase of the Cell Cycle in Human Cells. Nucleic Acids Res. 2004, 32, 3683–3688. [Google Scholar] [CrossRef] [PubMed]
- Branzei, D.; Foiani, M. Regulation of DNA Repair throughout the Cell Cycle. Nat. Rev. Mol. Cell Biol. 2008, 9, 297–308. [Google Scholar] [CrossRef]
- Milone, M.C.; O’Doherty, U. Clinical Use of Lentiviral Vectors. Leukemia 2018, 32, 1529–1541. [Google Scholar] [CrossRef]
- Papanikolaou, E.; Bosio, A. The Promise and the Hope of Gene Therapy. Front. Genome Ed. 2021, 3, 618346. [Google Scholar] [CrossRef]
- Li, Q.; Peterson, K.R.; Fang, X.; Stamatoyannopoulos, G. Locus Control Regions. Control 2002, 100, 3077–3086. [Google Scholar] [CrossRef] [PubMed]
- Palstra, R.J.; de Laat, W.; Grosveld, F. β-Globin Regulation and Long-Range Interactions. Adv. Genet. 2008, 61, 107–142. [Google Scholar] [CrossRef]
- Higgs, D.R.; Wood, W.G. Long-Range Regulation of α Globin Gene Expression during Erythropoiesis. Curr. Opin. Hematol. 2008, 15, 176–183. [Google Scholar] [CrossRef]
- Huang, P.; Keller, C.A.; Giardine, B.; Grevet, J.D.; Davies, J.O.J.; Hughes, J.R.; Kurita, R.; Nakamura, Y.; Hardison, R.C.; Blobel, G.A. Comparative Analysis of Three-Dimensional Chromosomal Architecture Identifies a Novel Fetal Hemoglobin Regulatory Element. Genes Dev. 2017, 31, 1704–1713. [Google Scholar] [CrossRef] [PubMed]
- Martyn, G.E.; Wienert, B.; Yang, L.; Shah, M.; Norton, L.J.; Burdach, J.; Kurita, R.; Nakamura, Y.; Pearson, R.C.M.; Funnell, A.P.W.; et al. Natural Regulatory Mutations Elevate the Fetal Globin Gene via Disruption of BCL11A or ZBTB7A Binding. Nat. Genet. 2018, 50, 498–503. [Google Scholar] [CrossRef]
- Liu, N.; Hargreaves, V.V.; Zhu, Q.; Kurland, J.V.; Hong, J.; Kim, W.; Sher, F.; Macias-Trevino, C.; Rogers, J.M.; Kurita, R.; et al. Direct Promoter Repression by BCL11A Controls the Fetal to Adult Hemoglobin Switch. Cell 2018, 173, 430–442.e17. [Google Scholar] [CrossRef]
- Fischer, K.D.; Nowock, J. The T→C Substitution at -198 of the Aγ-Globin Gene Associated with the British Form of HPFH Generates Overlapping Recognition Sites for Two DNA-Binding Proteins. Nucleic Acids Res. 1990, 18, 5685–5693. [Google Scholar] [CrossRef] [PubMed]
- Stoming, T.A.; Stoming, G.S.; Lanclos, K.D.; Fei, Y.J.; Altay, C.; Kutlar, F.; Huisman, T.H. An A gamma type of nondeletional hereditary persistence of fetal hemoglobin with a T----C mutation at position -175 to the cap site of the A gamma globin gene. Blood 1989, 329, 329–333. [Google Scholar] [CrossRef]
- Wienert, B.; Funnell, A.P.W.; Norton, L.J.; Pearson, R.C.M.; Wilkinson-White, L.E.; Lester, K.; Vadolas, J.; Porteus, M.H.; Matthews, J.M.; Quinlan, K.G.R.; et al. Editing the Genome to Introduce a Beneficial Naturally Occurring Mutation Associated with Increased Fetal Globin. Nat. Commun. 2015, 6, 7085. [Google Scholar] [CrossRef] [PubMed]
- Martyn, G.E.; Wienert, B.; Kurita, R.; Nakamura, Y.; Quinlan, K.G.R.; Crossley, M. A Natural Regulatory Mutation in the Proximal Promoter Elevates Fetal Globin Expression by Creating a de Novo GATA1 Site. Blood 2019, 133, 852–856. [Google Scholar] [CrossRef] [PubMed]
- Jane, S.M.; Cunningham, J.M. Understanding Fetal Globin Gene Expression: A Step towards Effective HbF Reactivation in Haemoglobinopathies. Br. J. Haematol. 1998, 102, 415–423. [Google Scholar] [CrossRef]
- Powars, D.R.; Weiss, J.N.; Chan, L.S.; Schroeder, W.A. Is There a Threshold Level of Fetal Hemoglobin That Ameliorates Morbidity in Sickle Cell Anemia? Blood 1984, 63, 921–926. [Google Scholar] [CrossRef]
- Cathomen, T.; Keith Joung, J. Zinc-Finger Nucleases: The next Generation Emerges. Mol. Ther. 2008, 16, 1200–1207. [Google Scholar] [CrossRef]
- Carroll, D. Genome Engineering with Zinc-Finger Nucleases. Genetics 2011, 188, 773–782. [Google Scholar] [CrossRef]
- Perez, E.E.; Wang, J.; Miller, J.C.; Jouvenot, Y.; Kim, K.A.; Wang, N.; Lee, G.; Bartsevich, V.V.; Lee, Y.; Guschin, D.Y.; et al. Editing Using Zinc-Finger Nucleases. Nat. Biotechnol. 2012, 26, 808–816. [Google Scholar] [CrossRef] [PubMed]
- Boch, J. TALEs of Genome Targeting. Nat. Biotechnol. 2011, 29, 135–136. [Google Scholar] [CrossRef] [PubMed]
- Claudio Mussolino, T.C. TALE Nucleases: Tailored Genome Engineering Made Easy. Curr. Opin. Biotechnol. 2012, 23, 644–650. [Google Scholar] [CrossRef] [PubMed]
- Becker, S.; Boch, J. TALE and TALEN Genome Editing Technologies. Gene Genome Ed. 2021, 2, 100007. [Google Scholar] [CrossRef]
- Jiang, W.; Bikard, D.; Cox, D.; Zhang, F.; Marraffini, L.A. RNA-Guided Editing of Bacterial Genomes Using CRISPR-Cas Systems. Nat. Biotechnol. 2013, 31, 233–239. [Google Scholar] [CrossRef]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A Programmable Dual-RNA-Guided DNA Endonuclease in Adaptive Bacterial Immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef]
- Doudna, J.A.; Charpentier, E. The New Frontier of Genome Engineering with CRISPR-Cas9. Science 2014, 346, 1258096. [Google Scholar] [CrossRef]
- Jiang, F.; Doudna, J.A. CRISPR—Cas9 Structures and Mechanisms. Annu. Rev. Biophys. 2017, 46, 505–531. [Google Scholar] [CrossRef]
- Bae, S.; Kweon, J.; Kim, H.S.; Kim, J.S. Microhomology-Based Choice of Cas9 Nuclease Target Sites. Nat. Methods 2014, 11, 705–706. [Google Scholar] [CrossRef]
- Symington, L.S.; Gautier, J. Double-Strand Break End Resection and Repair Pathway Choice. Annu. Rev. Genet. 2011, 45, 247–271. [Google Scholar] [CrossRef]
- Urnov, F.D.; Rebar, E.J.; Holmes, M.C.; Zhang, H.S.; Gregory, P.D. Genome Editing with Engineered Zinc Finger Nucleases. Nat. Rev. Genet. 2010, 11, 636–646. [Google Scholar] [CrossRef]
- Singh, J.K.; van Attikum, H. DNA Double-Strand Break Repair: Putting Zinc Fingers on the Sore Spot. Semin. Cell Dev. Biol. 2021, 113, 65–74. [Google Scholar] [CrossRef]
- Bhardwaj, A.; Nain, V. TALENs—An Indispensable Tool in the Era of CRISPR: A Mini Review. J. Genet. Eng. Biotechnol. 2021, 19, 125. [Google Scholar] [CrossRef]
- Deng, D.; Yan, C.; Pan, X.; Mahfouz, M.; Wang, J.; Zhu, J.K.; Shi, Y.; Yan, N. Structural Basis for Sequence-Specific Recognition of DNA by TAL Effectors. Science 2012, 335, 720–723. [Google Scholar] [CrossRef]
- Barrangou, R.; Fremaux, C.; Deveau, H.; Richardss, M.; Boyaval, P.; Moineau, S.; Romero, D.A.; Horvath, P. CRISPR Provides Against Viruses in Prokaryotes. Science 2007, 315, 1709–1712. [Google Scholar] [CrossRef]
- Gasiunas, G.; Barrangou, R.; Horvath, P.; Siksnys, V. Cas9-CrRNA Ribonucleoprotein Complex Mediates Specific DNA Cleavage for Adaptive Immunity in Bacteria. Proc. Natl. Acad. Sci. USA 2012, 109, 2579–2586. [Google Scholar] [CrossRef] [PubMed]
- Gratz, S.J.; Rubinstein, C.D.; Harrison, M.M.; Wildonger, J.; O’connor-Giles, K.M.; Bock, R.M. CRISPR-Cas9 Genome Editing in Drosophila HHS Public Access. Curr. Protoc. Mol. Biol. Curr. Protoc. Mol. Biol. 2015, 1112, 311–331. [Google Scholar] [CrossRef]
- Cornet, C.; Di Donato, V.; Terriente, J. Combining Zebrafish and CRISPR/Cas9: Toward a More Efficient Drug Discovery Pipeline. Front. Pharmacol. 2018, 9, 703. [Google Scholar] [CrossRef]
- Schwartz, M.L.; Wayne Davis, M.; Rich, M.S.; Jorgensen, E.M. High-Efficiency CRISPR Gene Editing in C. Elegans Using Cas9 Integrated into the Genome. PLoS Genet. 2021, 17, e1009755. [Google Scholar] [CrossRef]
- Platt, R.J.; Chen, S.; Zhou, Y.; Yim, M.J.; Swiech, L.; Kempton, H.R.; Dahlman, J.E.; Parnas, O.; Eisenhaure, T.M.; Jovanovic, M.; et al. CRISPR-Cas9 Knockin Mice for Genome Editing and Cancer Modeling. Cell 2014, 159, 440–455. [Google Scholar] [CrossRef] [PubMed]
- Murty, T.; Mackall, C.L. Gene Editing to Enhance the Efficacy of Cancer Cell Therapies. Mol. Ther. 2021, 29, 3153–3162. [Google Scholar] [CrossRef]
- Le Rhun, A.; Escalera-Maurer, A.; Bratovič, M.; Charpentier, E. CRISPR-Cas in Streptococcus Pyogenes. RNA Biol. 2019, 16, 380–389. [Google Scholar] [CrossRef]
- Chakraborty, C.; Teoh, S.L.; Das, S. The Smart Programmable CRISPR Technology: A Next Generation Genome Editing Tool for Investigators. Curr. Drug Targets 2016, 18, 1653–1663. [Google Scholar] [CrossRef]
- Ihry, R.J.; Worringer, K.A.; Salick, M.R.; Frias, E.; Ho, D.; Theriault, K.; Kommineni, S.; Chen, J.; Sondey, M.; Ye, C.; et al. P53 Inhibits CRISPR-Cas9 Engineering in Human Pluripotent Stem Cells. Nat. Med. 2018, 24, 939–946. [Google Scholar] [CrossRef]
- Haapaniemi, E.; Botla, S.; Persson, J.; Schmierer, B.; Taipale, J. CRISPR-Cas9 Genome Editing Induces a P53-Mediated DNA Damage Response. Nat. Med. 2018, 24, 927–930. [Google Scholar] [CrossRef]
- Schiroli, G.; Ferrari, S.; Conway, A.; Jacob, A.; Capo, V.; Albano, L.; Plati, T.; Castiello, M.C.; Sanvito, F.; Gennery, A.R.; et al. Preclinical Modeling Highlights the Therapeutic Potential of Hematopoietic Stem Cell Gene Editing for Correction of SCID-X1. Sci. Transl. Med. 2017, 9, eaan0820. [Google Scholar] [CrossRef]
- Leibowitz, M.L.; Papathanasiou, S.; Doerfler, P.A.; Blaine, L.J.; Sun, L.; Yao, Y.; Zhang, C.Z.; Weiss, M.J.; Pellman, D. Chromothripsis as an On-Target Consequence of CRISPR–Cas9 Genome Editing. Nat. Genet. 2021, 53, 895–905. [Google Scholar] [CrossRef]
- Cullot, G.; Boutin, J.; Toutain, J.; Prat, F.; Pennamen, P.; Rooryck, C.; Teichmann, M.; Rousseau, E.; Lamrissi-Garcia, I.; Guyonnet-Duperat, V.; et al. CRISPR-Cas9 Genome Editing Induces Megabase-Scale Chromosomal Truncations. Nat. Commun. 2019, 10, 1136. [Google Scholar] [CrossRef]
- Anzalone, A.V.; Randolph, P.B.; Davis, J.R.; Sousa, A.A.; Koblan, L.W.; Levy, J.M.; Chen, P.J.; Wilson, C.; Newby, G.A.; Raguram, A.; et al. Search-and-Replace Genome Editing without Double-Strand Breaks or Donor DNA. Nature 2019, 576, 149–157. [Google Scholar] [CrossRef]
- Anzalone, A.V.; Koblan, L.W.; Liu, D.R. Genome Editing with CRISPR–Cas Nucleases, Base Editors, Transposases and Prime Editors. Nat. Biotechnol. 2020, 38, 824–844. [Google Scholar] [CrossRef]
- Porto, E.M.; Komor, A.C.; Slaymaker, I.M.; Yeo, G.W. Base Editing: Advances and Therapeutic Opportunities. Nat. Rev. Drug Discov. 2020, 19, 839–859. [Google Scholar] [CrossRef]
- Holly, A.; Rees, D.R.L. Base Editing: Precision Chemistry on the Genome and Trancriptome of Living. Nat. Rev. Genet. 2018, 19, 770–788. [Google Scholar] [CrossRef]
- Gaudelli, N.M.; Komor, A.C.; Rees, H.A.; Packer, M.S.; Badran, A.H.; Bryson, D.I.; Liu, D.R. Programmable Base Editing of T to G C in Genomic DNA without DNA Cleavage. Nature 2017, 551, 464–471. [Google Scholar] [CrossRef]
- Kim, J. Precision Genome Engineering through Adenine and Cytosine Base Editing. Nat. Plants 2018, 4, 2–5. [Google Scholar] [CrossRef]
- Moon, S.B.; Kim, D.Y.; Ko, J.H.; Kim, Y.S. Recent Advances in the CRISPR Genome Editing Tool Set. Exp. Mol. Med. 2019, 51, 1–11. [Google Scholar] [CrossRef]
- Kantor, A.; Mcclements, M.E. CRISPR-Cas9 DNA Base-Editing and Prime-Editing. Int. J. Mol. Sci. 2020, 21, 6240. [Google Scholar]
- Antoniou, P.; Miccio, A.; Brusson, M. Base and Prime Editing Technologies for Blood Disorders. Front. Genome Ed. 2021, 3, 618406. [Google Scholar] [CrossRef]
- Marzec, M.; Brąszewska-Zalewska, A.; Hensel, G. Prime Editing: A New Way for Genome Editing. Trends Cell Biol. 2020, 30, 257–259. [Google Scholar] [CrossRef]
- Chen, P.J.; Hussmann, J.A.; Yan, J.; Knipping, F.; Ravisankar, P.; Chen, P.-F.; Chen, C.; Nelson, J.W.; Newby, G.A.; Sahin, M.; et al. Enhanced Prime Editing Systems by Manipulating Cellular Determinants of Editing Outcomes. Cell 2021, 184, 5635–5652.e29. [Google Scholar] [CrossRef]
- Scholefield, J.; Harrison, P.T. Prime Editing—An Update on the Field. Gene Ther. 2021, 28, 396–401. [Google Scholar] [CrossRef]
- Genovese, P.; Schiroli, G.; Escobar, G.; Di Tomaso, T.; Firrito, C.; Calabria, A.; Moi, D.; Mazzieri, R.; Bonini, C.; Holmes, M.C.; et al. Targeted Genome Editing in Human Repopulating Haematopoietic Stem Cells. Nature 2014, 510, 235–240. [Google Scholar] [CrossRef] [PubMed]
- Zou, J.; Mali, P.; Huang, X.; Dowey, S.N.; Cheng, L. Site-Specific Gene Correction of a Point Mutation in Human IPS Cells Derived from an Adult Patient with Sickle Cell Disease. Blood 2011, 118, 4599–4608. [Google Scholar] [CrossRef] [PubMed]
- Sebastiano, V.; Maeder, M.L.; Angstman, J.F.; Haddad, B.; Khayter, C.; Yeo, D.T.; Goodwin, M.J.; Hawkins, J.S.; Ramirez, C.L.; Batista, L.F.Z.; et al. In Situ Genetic Correction of the Sickle Cell Anemia Mutation in Human Induced Pluripotent Stem Cells Using Engineered Zinc Finger Nucleases. Stem Cells 2011, 29, 1717–1726. [Google Scholar] [CrossRef]
- Dever, D.P.; Bak, R.O.; Reinisch, A.; Camarena, J.; Washington, G.; Nicolas, C.E.; Pavel-Dinu, M.; Saxena, N.; Wilkens, A.B.; Mantri, S.; et al. CRISPR/Cas9 β-Globin Gene Targeting in Human Haematopoietic Stem Cells. Nature 2016, 539, 384–389. [Google Scholar] [CrossRef]
- Sun, W.; Liu, H.; Yin, W.; Qiao, J.; Zhao, X.; Liu, Y. Strategies for Enhancing the Homology-Directed Repair Efficiency of CRISPR-Cas Systems. Cris. J. 2022, 5, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Georgakopoulou, A.; Newby, G.A.; Chen, P.J.; Everette, K.A.; Paschoudi, K.; Vlachaki, E.; Gil, S.; Anderson, A.K.; Koob, T.; et al. In Vivo HSC Prime Editing Rescues Sickle Cell Disease in a Mouse Model. Blood 2023, 141, 2085–2099. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yang, Y.; Kang, X.; Lin, B.; Yu, Q.; Song, B.; Gao, G.; Chen, Y.; Sun, X.; Li, X.; et al. One-Step Biallelic and Scarless Correction of a β-Thalassemia Mutation in Patient-Specific IPSCs without Drug Selection. Mol. Ther. Nucleic Acids 2017, 6, 57–67. [Google Scholar] [CrossRef]
- Cheng, T.C.; Orkin, S.H.; Antonarakis, S.E.; Potter, M.J.; Sexton, J.P.; Markham, A.F.; Giardina, P.J.; Li, A.; Kazazian, H.H. β-Thalassemia in Chinese: Use of in Vivo RNA Analysis and Oligonucleotide Hybridization in Systematic Characterization of Molecular Defects. Proc. Natl. Acad. Sci. USA 1984, 81, 2821–2825. [Google Scholar] [CrossRef]
- Spritz, R.A.; Jagadeeswaran, P.; Choudary, P.V.; Biro, P.A.; Elder, J.T.; deRiel, J.K.; Manley, J.L.; Gefter, M.L.; Forget, B.G.; Weissman, S.M. Base Substitution in an Intervening Sequence of a β +-Thalassemic Human Globin Gene. Proc. Natl. Acad. Sci. USA 1981, 78, 2455–2459. [Google Scholar] [CrossRef]
- Xu, P.; Tong, Y.; Liu, X.Z.; Wang, T.T.; Cheng, L.; Wang, B.Y.; Lv, X.; Huang, Y.; Liu, D.P. Both TALENs and CRISPR/Cas9 Directly Target the HBB IVS2-654 (C > T) Mutation in β-Thalassemiaderived IPSCs. Sci. Rep. 2015, 5, 12065. [Google Scholar] [CrossRef]
- Fang, Y.; Cheng, Y.; Lu, D.; Gong, X.; Yang, G.; Gong, Z.; Zhu, Y.; Sang, X.; Fan, S.; Zhang, J.; et al. Treatment of Β654-Thalassaemia by TALENs in a Mouse Model. Cell Prolif. 2018, 51, e12491. [Google Scholar] [CrossRef]
- Kountouris, P.; Lederer, C.W.; Fanis, P.; Feleki, X.; Old, J.; Kleanthous, M. IthaGenes: An Interactive Database for Haemoglobin Variations and Epidemiology. PLoS ONE 2014, 9, e103020. [Google Scholar] [CrossRef]
- Xu, S.; Luk, K.; Yao, Q.; Shen, A.H.; Zeng, J.; Wu, Y.; Luo, H.Y.; Brendel, C.; Pinello, L.; Chui, D.H.K.; et al. Editing Aberrant Splice Sites Efficiently Restores B-Globin Expression in b-Thalassemia. Blood 2019, 133, 2255–2262. [Google Scholar] [CrossRef]
- Patsali, P.; Turchiano, G.; Papasavva, P.; Romito, M.; Loucari, C.C.; Stephanou, C.; Christou, S.; Sitarou, M.; Mussolino, C.; Cornu, T.I.; et al. Correction of IVS I-110(G>A) β-Thalassemia by CRISPR/Cas- And TALEN-Mediated Disruption of Aberrant Regulatory Elements in Human Hematopoietic Stem and Progenitor Cells. Haematologica 2019, 104, E497–E501. [Google Scholar] [CrossRef]
- Cosenza, L.C.; Zuccato, C.; Zurlo, M.; Gambari, R.; Finotti, A. Co-Treatment of Erythroid Cells from β-Thalassemia Patients with CRISPR-Cas9-Based Β039-Globin Gene Editing and Induction of Fetal Hemoglobin. Genes 2022, 13, 1727. [Google Scholar] [CrossRef]
- Ye, L.; Wang, J.; Tan, Y.; Beyer, A.I.; Xie, F.; Muench, M.O.; Kan, Y.W. Genome Editing Using CRISPR-Cas9 to Create the HPFH Genotype in HSPCs: An Approach for Treating Sickle Cell Disease and β-Thalassemia. Proc. Natl. Acad. Sci. USA 2016, 113, 10661–10665. [Google Scholar] [CrossRef]
- Traxler, E.A.; Yao, Y.; Wang, Y.D.; Woodard, K.J.; Kurita, R.; Nakamura, Y.; Hughes, J.R.; Hardison, R.C.; Blobel, G.A.; Li, C.; et al. A Genome-Editing Strategy to Treat β-Hemoglobinopathies That Recapitulates a Mutation Associated with a Benign Genetic Condition. Nat. Med. 2016, 22, 987–990. [Google Scholar] [CrossRef]
- Antoniani, C.; Meneghini, V.; Lattanzi, A.; Felix, T.; Romano, O.; Magrin, E.; Weber, L.; Pavani, G.; El Hoss, S.; Kurita, R.; et al. Induction of Fetal Hemoglobin Synthesis by CRISPR/Cas9-Mediated Editing of the Human b-Globin Locus. Blood 2018, 131, 1960–1973. [Google Scholar] [CrossRef]
- Métais, J.Y.; Doerfler, P.A.; Mayuranathan, T.; Bauer, D.E.; Fowler, S.C.; Hsieh, M.M.; Katta, V.; Keriwala, S.; Lazzarotto, C.R.; Luk, K.; et al. Genome Editing of HBG1 and HBG2 to Induce Fetal Hemoglobin. Blood Adv. 2019, 3, 3379–3392. [Google Scholar] [CrossRef]
- Li, C.; Psatha, N.; Sova, P.; Gil, S.; Wang, H.; Kim, J.; Kulkarni, C.; Valensisi, C.; David Hawkins, R.; Stamatoyannopoulos, G.; et al. Reactivation of γ-Globin in Adult b-YAC Mice after Ex Vivo and in Vivo Hematopoietic Stem Cell Genome Editing. Blood 2018, 131, 2915–2928. [Google Scholar] [CrossRef]
- Wang, L.; Li, L.; Ma, Y.; Hu, H.; Li, Q.; Yang, Y.; Liu, W.; Yin, S.; Li, W.; Fu, B.; et al. Reactivation of γ-Globin Expression through Cas9 or Base Editor to Treat β-Hemoglobinopathies. Cell Res. 2020, 30, 276–278. [Google Scholar] [CrossRef]
- Li, C.; Georgakopoulou, A.; Mishra, A.; Gil, S.; Hawkins, R.D.; Yannaki, E.; Lieber, A. In Vivo HSPC Gene Therapy with Base Editors Allows for Efficient Reactivation of Fetal G-Globin in b-YAC Mice. Blood Adv. 2021, 5, 1122–1135. [Google Scholar] [CrossRef]
- Hardouin, G.; Antoniou, P.; Martinucci, P.; Felix, T.; Manceau, S.; Joseph, L.; Masson, C.; Scaramuzza, S.; Ferrari, G.; Cavazzana, M.; et al. Adenine Base Editor-Mediated Correction of the Common and Severe IVS1-110 (G > A) β-Thalassemia Mutation. Blood 2022, 141, 1169–1179. [Google Scholar] [CrossRef]
- Liu, P.; Keller, J.R.; Ortiz, M.; Tessarollo, L.; Rachel, R.A.; Nakamura, T.; Jenkins, N.A.; Copeland, N.G. Bcl11a Is Essential for Normal Lymphoid Development. Nat. Immunol. 2003, 4, 525–532. [Google Scholar] [CrossRef]
- Tolve, M.; Ulusoy, A.; Patikas, N.; Islam, K.U.S.; Bodea, G.O.; Öztürk, E.; Broske, B.; Mentani, A.; Wagener, A.; van Loo, K.M.J.; et al. The Transcription Factor BCL11A Defines Distinct Subsets of Midbrain Dopaminergic Neurons. Cell Rep. 2021, 36. [Google Scholar] [CrossRef]
- Mitelman, F.; Johansson, B.; Mertens, F. Fusion Genes and Rearranged Genes as a Linear Function of Chromosome Aberrations in Cancer. Nat. Genet. 2004, 36, 331–334. [Google Scholar] [CrossRef]
- Satterwhite, E.; Sonoki, T.; Willis, T.G.; Harder, L.; Nowak, R.; Arriola, E.L.; Liu, H.; Price, H.P.; Gesk, S.; Steinemann, D.; et al. The BCL11 Gene Family: Involvement of BCL11A in Lymphoid Malignancies. Blood 2001, 98, 3413–3420. [Google Scholar] [CrossRef]
- Yu, Y.; Wang, J.; Khaled, W.; Burke, S.; Li, P.; Chen, X.; Yang, W.; Jenkins, N.A.; Copeland, N.G.; Zhang, S.; et al. Bcl11a Is Essential for Lymphoid Development and Negatively Regulates P53. J. Exp. Med. 2012, 209, 2467–2483. [Google Scholar] [CrossRef]
- Sankaran, V.G.; Xu, J.; Ragoczy, T.; Ippolito, G.C.; Walkley, C.R.; Maika, S.D.; Fujiwara, Y.; Ito, M.; Groudine, M.; Bender, M.A.; et al. Developmental and Species-Divergent Globin Switching Are Driven by BCL11A. Nature 2009, 460, 1093–1097. [Google Scholar] [CrossRef]
- Luc, S.; Huang, J.; McEldoon, J.L.; Somuncular, E.; Li, D.; Rhodes, C.; Mamoor, S.; Hou, S.; Xu, J.; Orkin, S.H. Bcl11a Deficiency Leads to Hematopoietic Stem Cell Defects with an Aging-like Phenotype. Cell Rep. 2016, 16, 3181–3194. [Google Scholar] [CrossRef]
- Menzel, S.; Garner, C.; Gut, I.; Matsuda, F.; Yamaguchi, M.; Heath, S.; Foglio, M.; Zelenika, D.; Boland, A.; Rooks, H.; et al. A QTL Influencing F Cell Production Maps to a Gene Encoding a Zinc-Finger Protein on Chromosome 2p15. Nat. Genet. 2007, 39, 1197–1199. [Google Scholar] [CrossRef]
- Lettre, G.; Sankaran, V.G.; Bezerra, M.C.; Araujo, A.S.; Uda, M.; Sanna, S.; Cao, A.; Schlessinger, D.; Costa, F.F.; Hirschhorn, J.N.; et al. DNA polymorphisms at the BCL11A, HBS1L-MYB, and β-globin loci associate with fetal hemoglobin levels and pain crises in sickle cell disease. Proc. Natl. Acad. Sci. USA 2008, 105, 11869–11874. [Google Scholar] [CrossRef]
- Bhatnagar, P.; Purvis, S.; Barron-casella, E.; Debaun, M.R.; Arking, D.E.; Keefer, J.R.; Hopkins, J.; Louis, S. Genome-Wide Association Study Identifies Genetic Variants Influencing F-Cell Levels in Sickle-Cell Patients. J. Hum. Genet. 2011, 56, 316–323. [Google Scholar] [CrossRef]
- Nuinoon, M.; Makarasara, W.; Mushiroda, T.; Setianingsih, I.; Wahidiyat, P.A.; Sripichai, O.; Kumasaka, N.; Takahashi, A.; Svasti, S.; Munkongdee, T.; et al. A Genome-Wide Association Identified the Common Genetic Variants Influence Disease Severity in Β0-Thalassemia/Hemoglobin, E. Hum. Genet. 2010, 127, 303–314. [Google Scholar] [CrossRef]
- Solovieff, N.; Milton, J.N.; Hartley, S.W.; Sherva, R.; Sebastiani, P.; Dworkis, D.A.; Klings, E.S.; Farrer, L.A.; Garrett, M.E.; Ashley-Koch, A.; et al. Fetal Hemoglobin in Sickle Cell Anemia: Genome-Wide Association Studies Suggest a Regulatory Region in the 5′ Olfactory Receptor Gene Cluster. Blood 2010, 115, 1815–1822. [Google Scholar] [CrossRef]
- Xu, J.; Bauer, D.E.; Kerenyi, M.A.; Vo, T.D.; Hou, S.; Hsu, Y.J.; Yao, H.; Trowbridge, J.J.; Mandel, G.; Orkin, S.H. Corepressor-Dependent Silencing of Fetal Hemoglobin Expression by BCL11A. Proc. Natl. Acad. Sci. USA 2013, 110, 6518–6523. [Google Scholar] [CrossRef]
- Sankaran, V.G.; Menne, T.F.; Xu, J.; Akie, T.E.; Lettre, G.; Van Handel, B.; Mikkola, H.K.; Hirschhorn, J.N.; Cantor, A.B.; Orkin, S.H. Human Fetal Hemoglobin Expression Is Regulated by the Developmental Stage-Specific Repressor BCL11A. Science 2008, 322, 1839–1842. [Google Scholar] [CrossRef]
- Xu, J.; Peng, C.; Sankaran, V.G.; Shao, Z.; Esrick, E.B.; Chong, B.G.; Ippolito, G.C.; Fujiwara, Y.; Ebert, B.L.; Tucker, P.W.; et al. Correction of Sickle Cell Disease in Adult Mice by Interference with Fetal Hemoglobin Silencing. Science 2011, 334, 993–996. [Google Scholar] [CrossRef]
- Psatha, N.; Sova, P.; Georgolopoulos, G.; Iwata, M.; Paschoudi, K.; Kirtsou, A.; Ulyanova, T.; Stamatoyannopoulos, J.A.; Yannaki, E.; Papayannopoulou, T.; et al. P1437: Novel Erythroid Enhancers Improve Gene Therapy Vectors for Beta-Hemoglobinopathies. In Proceedings of the EHA Congress 2022, Vienna, Austria, 9–12 June 2022; Volume 6, pp. 1320–1321. [Google Scholar]
- Bauer, D.E.; Kamran, S.C.; Lessard, S.; Xu, J.; Fujiwara, Y.; Lin, C.; Shao, Z.; Canver, M.C.; Smith, E.C.; Pinello, L.; et al. An Erythroid Enhancer of BCL11A Subject to Genetic Variation Determines Fetal Hemoglobin Level. Science 2013, 342, 253–257. [Google Scholar] [CrossRef]
- Canver, M.C.; Smith, E.C.; Sher, F.; Pinello, L.; Sanjana, N.E.; Shalem, O.; Chen, D.D.; Schupp, P.G.; Vinjamur, D.S.; Garcia, S.P.; et al. BCL11A Enhancer Dissection by Cas9-Mediated in Situ Saturating Mutagenesis. Nature 2015, 527, 192–197. [Google Scholar] [CrossRef]
- Vierstra, J.; Reik, A.; Chang, K.H.; Stehling-Sun, S.; Zhou, Y.; Hinkley, S.J.; Paschon, D.E.; Zhang, L.; Psatha, N.; Bendana, Y.R.; et al. Functional Footprinting of Regulatory DNA. Nat. Methods 2015, 12, 927–930. [Google Scholar] [CrossRef]
- Georgolopoulos, G.; Psatha, N.; Iwata, M.; Nishida, A.; Som, T.; Yiangou, M.; Stamatoyannopoulos, J.A.; Vierstra, J. Discrete Regulatory Modules Instruct Hematopoietic Lineage Commitment and Differentiation. Nat. Commun. 2021, 12, 6790. [Google Scholar] [CrossRef]
- Humbert, O.; Peterson, C.W.; Norgaard, Z.K.; Radtke, S.; Kiem, H.P. A Nonhuman Primate Transplantation Model to Evaluate Hematopoietic Stem Cell Gene Editing Strategies for β-Hemoglobinopathies. Mol. Ther. Methods Clin. Dev. 2018, 8, 75–86. [Google Scholar] [CrossRef]
- Wu, Y.; Zeng, J.; Roscoe, B.P.; Liu, P.; Yao, Q.; Lazzarotto, C.R.; Clement, K.; Cole, M.A.; Luk, K.; Baricordi, C.; et al. Highly Efficient Therapeutic Gene Editing of Human Hematopoietic Stem Cells. Nat. Med. 2019, 25, 776–783. [Google Scholar] [CrossRef]
- Fu, B.; Liao, J.; Chen, S.; Li, W.; Wang, Q.; Hu, J.; Yang, F.; Hsiao, S.; Jiang, Y.; Wang, L.; et al. CRISPR–Cas9-Mediated Gene Editing of the BCL11A Enhancer for Pediatric Β0/Β0 Transfusion-Dependent β-Thalassemia. Nat. Med. 2022, 28, 1573–1580. [Google Scholar] [CrossRef]
- Demirci, S.; Zeng, J.; Wu, Y.; Uchida, N.; Shen, A.H.; Pellin, D.; Gamer, J.; Yapundich, M.; Drysdale, C.; Bonanno, J.; et al. BCL11A Enhancer–Edited Hematopoietic Stem Cells Persist in Rhesus Monkeys without Toxicity. J. Clin. Investig. 2020, 130, 6677–6687. [Google Scholar] [CrossRef]
- Psatha, N.; Georgakopoulou, A.; Li, C.; Nandakumar, V.; Georgolopoulos, G.; Acosta, R.; Paschoudi, K.; Nelson, J.; Chee, D.; Athanasiadou, A.; et al. Enhanced HbF Reactivation by Multiplex Mutagenesis of Thalassemic CD34+ Cells in Vitro and in Vivo. Blood 2021, 138, 1540–1553. [Google Scholar] [CrossRef]
- Zeng, J.; Wu, Y.; Ren, C.; Bonanno, J.; Shen, A.H.; Shea, D.; Gehrke, J.M.; Clement, K.; Luk, K.; Yao, Q.; et al. Therapeutic Base Editing of Human Hematopoietic Stem Cells. Nat. Med. 2020, 26, 535–541. [Google Scholar] [CrossRef]
- Antoniou, P.; Hardouin, G.; Martinucci, P.; Frati, G.; Felix, T.; Chalumeau, A.; Fontana, L.; Martin, J.; Masson, C.; Brusson, M.; et al. Base-Editing-Mediated Dissection of a γ-Globin Cis -Regulatory Element for the Therapeutic Reactivation of Fetal Hemoglobin Expression. Nat. Commun. 2022, 13, 6618. [Google Scholar] [CrossRef]
- Maeda, T. Regulation of Hematopoietic Development by ZBTB Transcription Factors. Int. J. Hematol. 2016, 104, 310–323. [Google Scholar] [CrossRef]
- Maeda, T.; Ito, K.; Merghoub, T.; Poliseno, L.; Hobbs, R.M.; Wang, G.; Dong, L.; Maeda, M.; Dore, L.C.; Zelent, A.; et al. LRF Is an Essential Downstream Target of GATA1 in Erythroid Development and Regulates BIM-Dependent Apoptosis. Dev. Cell 2009, 17, 527–540. [Google Scholar] [CrossRef]
- Masuda, T.; Wang, X.; Maeda, M.; Canver, M.C.; Sher, F.; Funnell, A.P.W.; Fisher, C.; Suciu, M.; Martyn, G.E.; Norton, L.J.; et al. Gene Regulation: Transcription Factors LRF and BCL11A Independently Repress Expression of Fetal Hemoglobin. Science 2016, 351, 285–289. [Google Scholar] [CrossRef]
- Weber, L.; Frati, G.; Felix, T.; Hardouin, G.; Casini, A.; Wollenschlaeger, C.; Meneghini, V.; Masson, C.; de Cian, A.; Chalumeau, A.; et al. Editing a γ-Globin Repressor Binding Site Restores Fetal Hemoglobin Synthesis and Corrects the Sickle Cell Disease Phenotype. Sci. Adv. 2020, 6, eaay9392. [Google Scholar] [CrossRef]
- Tallack, M.R.; Whitington, T.; Yuen, W.S.; Wainwright, E.N.; Keys, J.R.; Gardiner, B.B.; Nourbakhsh, E.; Cloonan, N.; Grimmond, S.M.; Bailey, T.L.; et al. A Global Role for KLF1 in Erythropoiesis Revealed by ChIP-Seq in Primary Erythroid Cells. Genome Res. 2010, 20, 1052–1063. [Google Scholar] [CrossRef]
- Hodge, D.; Coghill, E.; Keys, J.; Maguire, T.; Hartmann, B.; McDowall, A.; Weiss, M.; Grimmond, S.; Perkins, A. A Global Role for EKLF in Definitive and Primitive Erythropoiesis. Blood 2006, 107, 3359–3370. [Google Scholar] [CrossRef]
- Miller, I.J.; Bieker, J.J. A Novel, Erythroid Cell-Specific Murine Transcription Factor That Binds to the CACCC Element and Is Related to the Krüppel Family of Nuclear Proteins. Mol. Cell. Biol. 1993, 13, 2776–2786. [Google Scholar] [CrossRef]
- Borg, J.; Papadopoulos, P.; Georgitsi, M.; Gutiérrez, L.; Grech, G.; Fanis, P.; Phylactides, M.; Verkerk, A.J.M.H.; van der Spek, P.J.; Scerri, C.A.; et al. Haploinsufficiency for the Erythroid Transcription Factor KLF1 Causes Hereditary Persistence of Fetal Hemoglobin. Nat. Genet. 2010, 42, 801–807. [Google Scholar] [CrossRef]
- Gillinder, K.R.; Reed, C.L.; Malelang, S.; Mitchell, H.L.; Hoskin, E.; Magor, G.W.; Kaplan, Z.; Perkins, A.C. Gene Editing of KLF1 to Cure Sickle Cell Disease. Blood 2020, 136, 30–31. [Google Scholar] [CrossRef]
- Shariati, L.; Khanahmad, H.; Salehi, M.; Hejazi, Z.; Rahimmanesh, I.; Tabatabaiefar, M.A.; Modarressi, M.H. Genetic Disruption of the KLF1 Gene to Overexpress the γ-Globin Gene Using the CRISPR/Cas9 System. J. Gene Med. 2016, 18, 294–301. [Google Scholar] [CrossRef] [PubMed]
- Lamsfus-Calle, A.; Daniel-Moreno, A.; Antony, J.S.; Epting, T.; Heumos, L.; Baskaran, P.; Admard, J.; Casadei, N.; Latifi, N.; Siegmund, D.M.; et al. Comparative Targeting Analysis of KLF1, BCL11A, and HBG1/2 in CD34+ HSPCs by CRISPR/Cas9 for the Induction of Fetal Hemoglobin. Sci. Rep. 2020, 10, 10133. [Google Scholar] [CrossRef]
- Gaudelli, N.M.; Lam, D.K.; Rees, H.A.; Solá-Esteves, N.M.; Barrera, L.A.; Born, D.A.; Edwards, A.; Gehrke, J.M.; Lee, S.J.; Liquori, A.J.; et al. Directed Evolution of Adenine Base Editors with Increased Activity and Therapeutic Application. Nat. Biotechnol. 2020, 38, 892–900. [Google Scholar] [CrossRef]
- Connor, F.; Cary, P.D.; Read, C.M.; Preston, N.S.; Driscoll, P.C.; Denny, P.; Crane-robinson, C.; Ashworth, A. DNA Binding and Bending Properties of the Postmeiotically Expressed Sry-Related Protein Sox-5. Nucleic Acids Res. 1994, 22, 3339–3346. [Google Scholar] [CrossRef]
- Ferrari, S.; Harley, V.R.; Pontiggia, A.; Goodfellow, P.N.; Lovell-Badge, R.; Bianchi, M.E. SRY, like HMG1, Recognizes Sharp Angles in DNA. EMBO J. 1992, 11, 4497–4506. [Google Scholar] [CrossRef]
- Dumitriu, B.; Dy, P.; Smits, P.; Lefebvre, V. Generation of Mice Harboring a Sox6 Conditional Null Allele. Genesis 2006, 44, 219–224. [Google Scholar] [CrossRef] [PubMed]
- Yi, Z.; Cohen-Barak, O.; Hagiwara, N.; Kingsley, P.D.; Fuchs, D.A.; Erickson, D.T.; Epner, E.M.; Palis, J.; Brilliant, M.H. Sox6 Directly Silences Epsilon Globin Expression in Definitive Erythropoiesis. PLoS Genet. 2006, 2, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Sankaran, V.G.; Ni, M.; Menne, T.F.; Puram, R.V.; Kim, W.; Orkin, S.H. Transcriptional Silencing of γ-Globin by BCL11A Involves Long-Range Interactions and Cooperation with SOX6. Genes Dev. 2010, 24, 783–789. [Google Scholar] [CrossRef] [PubMed]
- Modares Sadeghi, M.; Shariati, L.; Hejazi, Z.; Shahbazi, M.; Tabatabaiefar, M.A.; Khanahmad, H. Inducing Indel Mutation in the SOX6 Gene by Zinc Finger Nuclease for Gamma Reactivation: An Approach towards Gene Therapy of Beta Thalassemia. J. Cell. Biochem. 2018, 119, 2512–2519. [Google Scholar] [CrossRef]
- Shariati, L.; Rohani, F.; Heidari Hafshejani, N.; Kouhpayeh, S.; Boshtam, M.; Mirian, M.; Rahimmanesh, I.; Hejazi, Z.; Modarres, M.; Pieper, I.L.; et al. Disruption of SOX6 Gene Using CRISPR/Cas9 Technology for Gamma-Globin Reactivation: An Approach towards Gene Therapy of β-Thalassemia. J. Cell. Biochem. 2018, 119, 9357–9363. [Google Scholar] [CrossRef]
- Vinjamur, D.S.; Yao, Q.; Cole, M.A.; McGuckin, C.; Ren, C.; Zeng, J.; Hossain, M.; Luk, K.; Wolfe, S.A.; Pinello, L.; et al. ZNF410 Represses Fetal Globin by Singular Control of CHD4. Nat. Genet. 2021, 53, 719–728. [Google Scholar] [CrossRef] [PubMed]
- Lan, X.; Ren, R.; Feng, R.; Ly, L.C.; Lan, Y.; Zhang, Z.; Aboreden, N.; Qin, K.; Horton, J.R.; Grevet, J.D.; et al. ZNF410 Uniquely Activates the NuRD Component CHD4 to Silence Fetal Hemoglobin Expression. Mol. Cell 2021, 81, 239–254.e8. [Google Scholar] [CrossRef] [PubMed]
- Tumburu, L.; Thein, S.L. Targeting ZNF410 as a Potential β-Hemoglobinopathy Therapy. Nat. Genet. 2021, 53, 589–590. [Google Scholar] [CrossRef] [PubMed]
- Chefalo, P.J.; Oh, J.; Rafie-Kolpin, M.; Kan, B.; Chen, J.J. Heme-Regulated EIF-2α Kinase Purifies as a Hemoprotein. Eur. J. Biochem. 1998, 258, 820–830. [Google Scholar] [CrossRef] [PubMed]
- Crosby, J.S.; Lee, K.; London, I.M.; Chen, J.J. Erythroid Expression of the Heme-Regulated EIF-2 Alpha Kinase. Mol. Cell. Biol. 1994, 14, 3906–3914. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.; Peslak, S.A.; Lan, X.; Khandros, E.; Yano, J.A.; Sharma, M.; Keller, C.A.; Giardine, B.; Qin, K.; Abdulmalik, O.; et al. The HRI-Regulated Transcription Factor ATF4 Activates BCL11A Transcription to Silence Fetal Hemoglobin Expression. Blood 2020, 135, 2121–2132. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.J.; Zhang, S. Heme-Regulated EIF2α Kinase in Erythropoiesis and Hemoglobinopathies. Blood 2019, 134, 1697–1707. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.; Peslak, S.A.; Khandros, E.; Lan, X.; Qin, K.; Sharma, M.; Keller, C.A.; Giardine, B.; Abdulmalik, O.; Chou, S.T.; et al. HIC2 Controls Developmental Hemoglobin Switching By Repressing BCL11A Transcription. Blood 2021, 138, 571. [Google Scholar] [CrossRef]
- Huang, P.; Peslak, S.A.; Ren, R.; Khandros, E.; Qin, K.; Keller, C.A.; Giardine, B.; Bell, H.W.; Lan, X.; Sharma, M.; et al. HIC2 Controls Developmental Hemoglobin Switching by Repressing BCL11A Transcription. Nat. Genet. 2022, 54, 1417–1426. [Google Scholar] [CrossRef]
- Peslak, S.A.; Khandros, E.; Huang, P.; Wakabayashi, A.; Abbas, T.; Abdulmalik, O.Y.; Keller, C.A.; Hardison, R.C.; Shi, J.; Blobel, G.A. Regulation of Fetal Hemoglobin Production in Adult Erythroid Cells By Protein Phosphatase 6C (PPP6C). Blood 2022, 140, 2502–2503. [Google Scholar] [CrossRef]
- Sangamo, Sanofi Show Positive Early Data for SCD Gene-Edited Cell Therapy, Genetic Engineering & Biotechnology News. Available online: https://www.genengnews.com/news/sangamo-sanofi-show-positive-early-data-for-scd-gene-edited-cell-therapy/ (accessed on 3 April 2023).
- Frangoul, H.; Altshuler, D.; Cappellini, M.D.; Chen, Y.-S.; Domm, J.; Eustace, B.K.; Foell, J.; de la Fuente, J.; Grupp, S.; Handgretinger, R.; et al. CRISPR-Cas9 Gene Editing for Sickle Cell Disease and β-Thalassemia. N. Engl. J. Med. 2021, 384, 252–260. [Google Scholar] [CrossRef]
- Locatelli, F.; Frangoul, H.; Corbacioglu, S.; de la Fuente, J.; Wall, D.; Capellini, M.D.; de Montalembert, M.; Kattamis, A.; Lobitz, S.; Rondelli, D.; et al. Efficacy and Safety of A Single Dose Of Ctx001 For Transfusion-Dependent Βeta-Thalassemia And Severe Sickle Cell Disease. In Proceedings of the Conference of European Hematology Association, Vienna, Austria, 9–12 June 2022. [Google Scholar]
- ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/home (accessed on 25 February 2023).
- Maddalo, D.; Manchado, E.; Concepcion, C.P.; Bonetti, C.; Vidigal, J.A.; Han, Y.C.; Ogrodowski, P.; Crippa, A.; Rekhtman, N.; De Stanchina, E.; et al. In Vivo Engineering of Oncogenic Chromosomal Rearrangements with the CRISPR/Cas9 System. Nature 2014, 516, 423–428. [Google Scholar] [CrossRef]
- Brunet, E.; Simsek, D.; Tomishima, M.; DeKelver, R.; Choi, V.M.; Gregory, P.; Urnov, F.; Weinstock, D.M.; Jasin, M. Chromosomal Translocations Induced at Specified Loci in Human Stem Cells. Proc. Natl. Acad. Sci. USA 2009, 106, 10620–10625. [Google Scholar] [CrossRef] [PubMed]
- Tsai, S.Q.; Zheng, Z.; Nguyen, N.T.; Liebers, M.; Topkar, V.V.; Thapar, V.; Wyvekens, N.; Khayter, C.; Iafrate, A.J.; Le, L.P.; et al. GUIDE-Seq Enables Genome-Wide Profiling of off-Target Cleavage by CRISPR-Cas Nucleases. Nat. Biotechnol. 2015, 33, 187–198. [Google Scholar] [CrossRef]
- Park, S.H.; Lee, C.M.; Dever, D.P.; Davis, T.H.; Camarena, J.; Srifa, W.; Zhang, Y.; Paikari, A.; Chang, A.K.; Porteus, M.H.; et al. Highly Efficient Editing of the β-Globin Gene in Patient-Derived Hematopoietic Stem and Progenitor Cells to Treat Sickle Cell Disease. Nucleic Acids Res. 2019, 47, 7955–7972. [Google Scholar] [CrossRef]
- Cradick, T.J.; Qiu, P.; Lee, C.M.; Fine, E.J.; Bao, G. COSMID: A Web-Based Tool for Identifying and Validating CRISPR/Cas off-Target Sites. Mol. Ther. Nucleic Acids 2014, 3, e214. [Google Scholar] [CrossRef]
- Turchiano, G.; Andrieux, G.; Klermund, J.; Blattner, G.; Pennucci, V.; El Gaz, M.; Monaco, G.; Poddar, S.; Mussolino, C.; Cornu, T.I.; et al. Quantitative Evaluation of Chromosomal Rearrangements in Gene-Edited Human Stem Cells by CAST-Seq. Cell Stem Cell 2021, 28, 1136–1147.e5. [Google Scholar] [CrossRef] [PubMed]
- Wienert, B.; Wyman, S.K.; Richardson, C.D.; Yeh, C.D.; Akcakaya, P.; Porritt, M.J.; Morlock, M.; Vu, J.T.; Kazane, K.R.; Watry, H.L.; et al. Unbiased Detection of CRISPR Off-Targets in Vivo Using DISCOVER-Seq. Science 2019, 364, 286–289. [Google Scholar] [CrossRef]
- Papathanasiou, S.; Markoulaki, S.; Blaine, L.J.; Leibowitz, M.L.; Zhang, C.Z.; Jaenisch, R.; Pellman, D. Whole Chromosome Loss and Genomic Instability in Mouse Embryos after CRISPR-Cas9 Genome Editing. Nat. Commun. 2021, 12, 5855. [Google Scholar] [CrossRef] [PubMed]
- Boutin, J.; Cappellen, D.; Rosier, J.; Amintas, S.; Dabernat, S.; Bedel, A.; Moreau-Gaudry, F. ON-Target Adverse Events of CRISPR-Cas9 Nuclease: More Chaotic than Expected. Cris. J. 2022, 5, 19–30. [Google Scholar] [CrossRef]
- FDA Approves First Cell-Based Gene Therapy to Treat Adult and Pediatric Patients with Beta-Thalassemia Who Require Regular Blood Transfusions. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-first-cell-based-gene-therapy-treat-adult-and-pediatric-patients-beta-thalassemia-who (accessed on 20 September 2022).
- Richter, M.; Saydaminova, K.; Yumul, R.; Krishnan, R.; Liu, J.; Nagy, E.E.; Singh, M.; Izsvák, Z.; Cattaneo, R.; Uckert, W.; et al. In Vivo Transduction of Primitive Mobilized Hematopoietic Stem Cells after Intravenous Injection of Integrating Adenovirus Vectors. Blood 2016, 128, 2206–2217. [Google Scholar] [CrossRef]
- Wang, H.; Georgakopoulou, A.; Psatha, N.; Li, C.; Capsali, C.; Samal, H.B.; Anagnostopoulos, A.; Ehrhardt, A.; Izsvák, Z.; Papayannopoulou, T.; et al. In Vivo Hematopoietic Stem Cell Gene Therapy Ameliorates Murine Thalassemia Intermedia. J. Clin. Investig. 2019, 129, 598–615. [Google Scholar] [CrossRef]
- Li, C.; Wang, H.; Gil, S.; Germond, A.; Fountain, C.; Baldessari, A.; Kim, J.; Liu, Z.; Georgakopoulou, A.; Radtke, S.; et al. Safe and Efficient in Vivo Hematopoietic Stem Cell Transduction in Nonhuman Primates Using HDAd5/35++ Vectors. Mol. Ther. Methods Clin. Dev. 2022, 24, 127–141. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paschoudi, K.; Yannaki, E.; Psatha, N. Precision Editing as a Therapeutic Approach for β-Hemoglobinopathies. Int. J. Mol. Sci. 2023, 24, 9527. https://doi.org/10.3390/ijms24119527
Paschoudi K, Yannaki E, Psatha N. Precision Editing as a Therapeutic Approach for β-Hemoglobinopathies. International Journal of Molecular Sciences. 2023; 24(11):9527. https://doi.org/10.3390/ijms24119527
Chicago/Turabian StylePaschoudi, Kiriaki, Evangelia Yannaki, and Nikoletta Psatha. 2023. "Precision Editing as a Therapeutic Approach for β-Hemoglobinopathies" International Journal of Molecular Sciences 24, no. 11: 9527. https://doi.org/10.3390/ijms24119527
APA StylePaschoudi, K., Yannaki, E., & Psatha, N. (2023). Precision Editing as a Therapeutic Approach for β-Hemoglobinopathies. International Journal of Molecular Sciences, 24(11), 9527. https://doi.org/10.3390/ijms24119527