
Towards Novel Potential Molecular Targets for Antidepressant and Antipsychotic Pharmacotherapies

 , ,
, ,  , , and
, , and
Abstract
1. Introduction
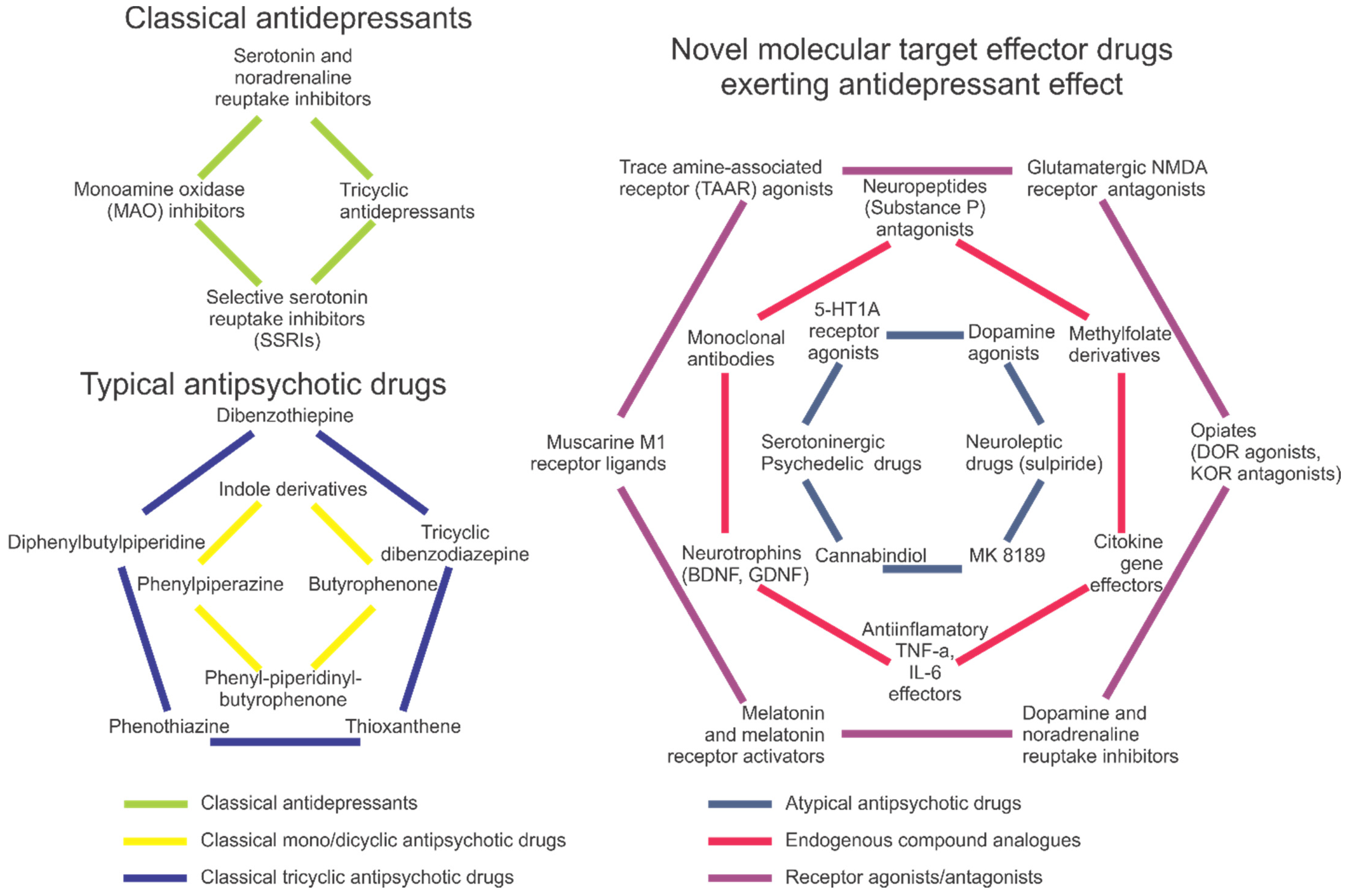
2. Pharmacotherapy of Depression
3. Approaches to Antipsychotic Therapy
4. In Silico-Driven Search for Novel Therapeutic Agents
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Depression, W. Other Common Mental Disorders: Global Health Estimates; World Health Organization: Geneva, Switzerland, 2017; Volume 24. [Google Scholar]
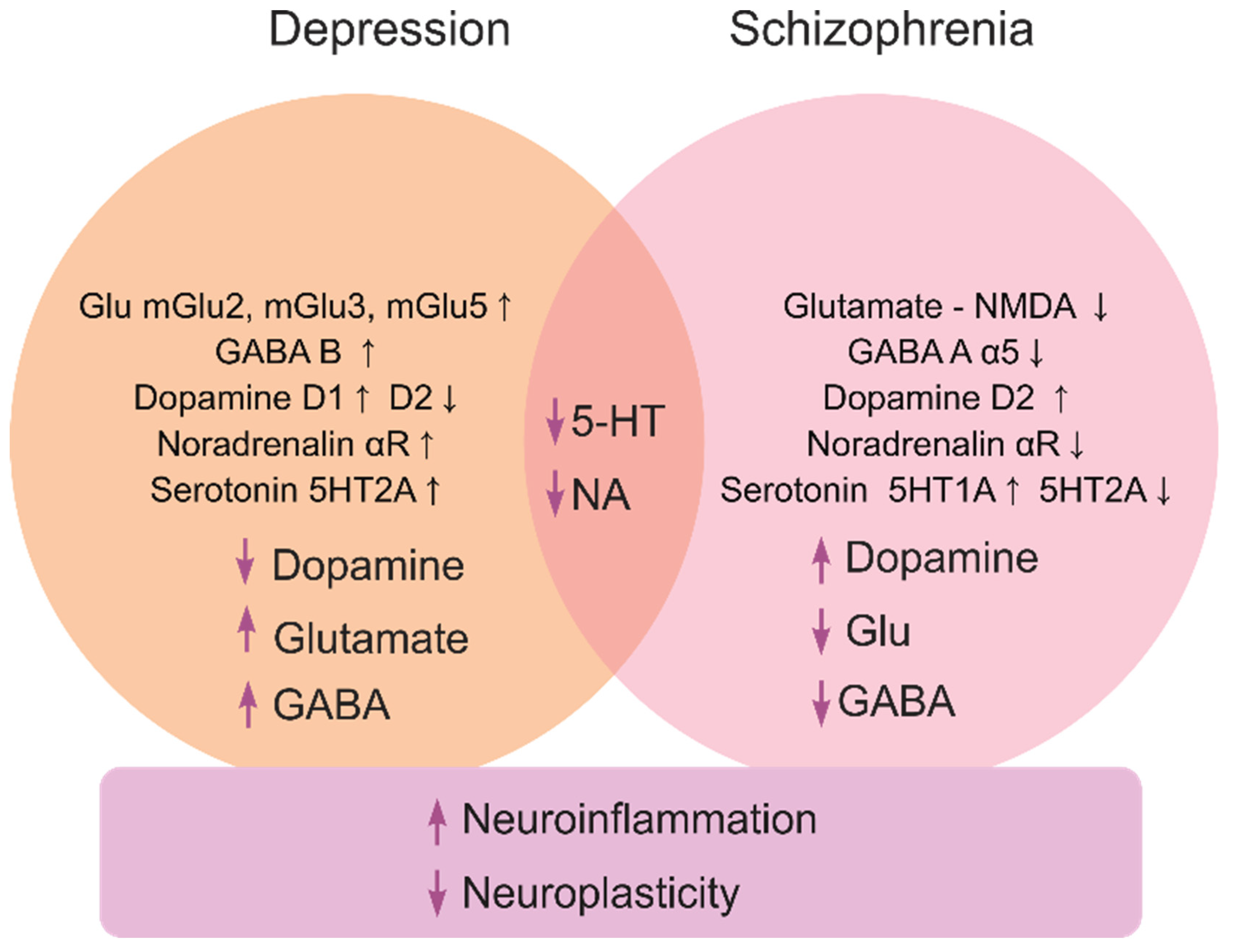
- Wierońska, J.M.; Pilc, A. Depression and schizophrenia viewed from the perspective of amino acidergic neurotransmission: Antipodes of psychiatric disorders. Pharmacol. Ther. 2019, 193, 75–82. [Google Scholar] [CrossRef]
- Pandarakalam, J.P. Challenges of treatment-resistant depression. Psychiatr. Danub. 2018, 30, 273–284. [Google Scholar] [CrossRef] [PubMed]
- Nucifora, F.C., Jr.; Woznica, E.; Lee, B.J.; Cascella, N.; Sawa, A. Treatment resistant schizophrenia: Clinical, biological, and therapeutic perspectives. Neurobiol. Dis. 2019, 131, 104257. [Google Scholar] [CrossRef]
- de Abreu, M.S.; Friend, A.J.; Demin, K.A.; Amstislavskaya, T.G.; Bao, W.; Kalueff, A.V. Zebrafish models: Do we have valid paradigms for depression? J. Pharmacol. Toxicol. Methods 2018, 94, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Demin, K.A.; Kolesnikova, T.O.; Khatsko, S.L.; Zhu, X.; Yuan, X.; Song, C.; Meshalkina, D.A.; Leonard, B.E.; Tian, L.; et al. Animal inflammation-based models of depression and their application to drug discovery. Expert Opin. Drug Discov. 2017, 12, 995–1009. [Google Scholar] [CrossRef]
- Venzala, E.; Garcia-Garcia, A.L.; Elizalde, N.; Tordera, R.M. Social vs. environmental stress models of depression from a behavioural and neurochemical approach. Eur. Neuropsychopharmacol. 2013, 23, 697–708. [Google Scholar] [CrossRef]
- Rutter, M. Commentary: Nature-nurture interplay in emotional disorders. J. Child Psychol. Psychiatry 2003, 44, 934–944. [Google Scholar] [CrossRef]
- Cryan, J.F.; Mombereau, C. In search of a depressed mouse: Utility of models for studying depression-related behavior in genetically modified mice. Mol. Psychiatry 2004, 9, 326–357. [Google Scholar] [CrossRef] [PubMed]
- Huynh, N.N.; McIntyre, R.S. What are the implications of the STAR* D trial for primary care? A review and synthesis. Prim. Care Companion J. Clin. Psychiatry 2008, 10, 91. [Google Scholar] [CrossRef]
- Wong, M.-L.; Licinio, J. From monoamines to genomic targets: A paradigm shift for drug discovery in depression. Nat. Rev. Drug Discov. 2004, 3, 136–151. [Google Scholar] [CrossRef]
- Haeffel, G.J.; Getchell, M.; Koposov, R.A.; Yrigollen, C.M.; Deyoung, C.G.; Klinteberg, B.A.; Oreland, L.; Ruchkin, V.V.; Grigorenko, E.L. Association between polymorphisms in the dopamine transporter gene and depression: Evidence for a gene-environment interaction in a sample of juvenile detainees. Psychol. Sci. 2008, 19, 62–69. [Google Scholar] [CrossRef]
- Risch, N.; Herrell, R.; Lehner, T.; Liang, K.Y.; Eaves, L.; Hoh, J.; Griem, A.; Kovacs, M.; Ott, J.; Merikangas, K.R. Interaction between the serotonin transporter gene (5-HTTLPR), stressful life events, and risk of depression: A meta-analysis. JAMA 2009, 301, 2462–2471. [Google Scholar] [CrossRef] [PubMed]
- Edition, F. Diagnostic and statistical manual of mental disorders. Am. Psychiatr. Assoc. 2013, 21, 591–643. [Google Scholar]
- Walther, S. Psychomotor symptoms of schizophrenia map on the cerebral motor circuit. Psychiatry Res. 2015, 233, 293–298. [Google Scholar] [CrossRef]
- Gawel, K.; Banono, N.S.; Michalak, A.; Esguerra, C.V. A critical review of zebrafish schizophrenia models: Time for validation? Neurosci. Biobehav. Rev. 2019, 107, 6–22. [Google Scholar] [CrossRef] [PubMed]
- Coyle, J.T. Glutamate and schizophrenia: Beyond the dopamine hypothesis. Cell. Mol. Neurobiol. 2006, 26, 363–382. [Google Scholar] [CrossRef] [PubMed]
- Ross, C.A.; Margolis, R.L.; Reading, S.A.; Pletnikov, M.; Coyle, J.T. Neurobiology of schizophrenia. Neuron 2006, 52, 139–153. [Google Scholar] [CrossRef]
- Dean, B. Neurochemistry of schizophrenia: The contribution of neuroimaging postmortem pathology and neurochemistry in schizophrenia. Curr. Top. Med. Chem. 2012, 12, 2375–2392. [Google Scholar] [CrossRef]
- Salavati, B.; Rajji, T.K.; Price, R.; Sun, Y.; Graff-Guerrero, A.; Daskalakis, Z.J. Imaging-based neurochemistry in schizophrenia: A systematic review and implications for dysfunctional long-term potentiation. Schizophr. Bull. 2015, 41, 44–56. [Google Scholar] [CrossRef]
- Girgis, R.R.; Slifstein, M.; Brucato, G.; Kegeles, L.S.; Colibazzi, T.; Lieberman, J.A.; Abi-Dargham, A. Imaging synaptic dopamine availability in individuals at clinical high-risk for psychosis: A [(11)C]-(+)-PHNO PET with methylphenidate challenge study. Mol. Psychiatry 2021, 26, 2504–2513. [Google Scholar] [CrossRef]
- Howes, O.D.; Kapur, S. The dopamine hypothesis of schizophrenia: Version III--the final common pathway. Schizophr. Bull. 2009, 35, 549–562. [Google Scholar] [CrossRef]
- Howes, O.; McCutcheon, R.; Stone, J. Glutamate and dopamine in schizophrenia: An update for the 21st century. J. Psychopharmacol. 2015, 29, 97–115. [Google Scholar] [CrossRef]
- Stahl, S.M. Beyond the dopamine hypothesis of schizophrenia to three neural networks of psychosis: Dopamine, serotonin, and glutamate. CNS Spectr. 2018, 23, 187–191. [Google Scholar] [CrossRef] [PubMed]
- Lewis, D.A.; Moghaddam, B. Cognitive dysfunction in schizophrenia: Convergence of γ-aminobutyric acid and glutamate alterations. Arch. Neurol. 2006, 63, 1372–1376. [Google Scholar] [CrossRef]
- Dell’Osso, L.; Carmassi, C.; Mucci, F.; Marazziti, D. Depression, serotonin and tryptophan. Curr. Pharm. Des. 2016, 22, 949–954. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, K.; Miyazaki, K.W.; Doya, K. The role of serotonin in the regulation of patience and impulsivity. Mol. Neurobiol. 2012, 45, 213–224. [Google Scholar] [CrossRef]
- Cowen, P.J. Serotonin and depression: Pathophysiological mechanism or marketing myth? Trends Pharmacol. Sci. 2008, 29, 433–436. [Google Scholar] [CrossRef] [PubMed]
- Slifirski, G.; Krol, M.; Turlo, J. 5-HT receptors and the development of new antidepressants. Int. J. Mol. Sci. 2021, 22, 9015. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; MacDonald, M.L.; Elswick, D.E.; Sweet, R.A. The glutamate hypothesis of schizophrenia: Evidence from human brain tissue studies. Ann. N. Y. Acad. Sci. 2015, 1338, 38–57. [Google Scholar] [CrossRef] [PubMed]
- Trullas, R.; Skolnick, P. Functional antagonists at the NMDA receptor complex exhibit antidepressant actions. Eur. J. Pharmacol. 1990, 185, 1–10. [Google Scholar] [CrossRef]
- Rodrigues, S.M.; Bauer, E.P.; Farb, C.R.; Schafe, G.E.; LeDoux, J.E. The group I metabotropic glutamate receptor mGluR5 is required for fear memory formation and long-term potentiation in the lateral amygdala. J. Neurosci. 2002, 22, 5219–5229. [Google Scholar] [CrossRef]
- Schoepp, D.D. Unveiling the functions of presynaptic metabotropic glutamate receptors in the central nervous system. J. Pharmacol. Exp. Ther. 2001, 299, 12–20. [Google Scholar]
- Khoodoruth, M.A.S.; Estudillo-Guerra, M.A.; Pacheco-Barrios, K.; Nyundo, A.; Chapa-Koloffon, G.; Ouanes, S. Glutamatergic System in Depression and Its Role in Neuromodulatory Techniques Optimization. Front. Psychiatry 2022, 13, 886918. [Google Scholar] [CrossRef]
- Verrotti, A.; Striano, P.; Iapadre, G.; Zagaroli, L.; Bonanni, P.; Coppola, G.; Elia, M.; Mecarelli, O.; Franzoni, E.; De Liso, P. The pharmacological management of Lennox-Gastaut syndrome and critical literature review. Seizure 2018, 63, 17–25. [Google Scholar] [CrossRef]
- Gorman, J.M.; Docherty, J.P. A hypothesized role for dendritic remodeling in the etiology of mood and anxiety disorders. J. Neuropsychiatry Clin. Neurosci. 2010, 22, 256–264. [Google Scholar] [CrossRef]
- Altamura, C.A.; Mauri, M.C.; Ferrara, A.; Moro, A.R.; D’Andrea, G.; Zamberlan, F. Plasma and platelet excitatory amino acids in psychiatric disorders. Am. J. Psychiatry 1993, 150, 1731–1733. [Google Scholar]
- Altamura, C.; Maes, M.; Dai, J.; Meltzer, H.Y. Plasma concentrations of excitatory amino acids, serine, glycine, taurine and histidine in major depression. Eur. Neuropsychopharmacol. 1995, 5 (Suppl. S1), 71–75. [Google Scholar] [CrossRef]
- Mauri, M.C.; Ferrara, A.; Boscati, L.; Bravin, S.; Zamberlan, F.; Alecci, M.; Invernizzi, G. Plasma and platelet amino acid concentrations in patients affected by major depression and under fluvoxamine treatment. Neuropsychobiology 1998, 37, 124–129. [Google Scholar] [CrossRef]
- Mitani, H.; Shirayama, Y.; Yamada, T.; Maeda, K.; Ashby, C.R., Jr.; Kawahara, R. Correlation between plasma levels of glutamate, alanine and serine with severity of depression. Prog. Neuropsychopharmacol. Biol. Psychiatry 2006, 30, 1155–1158. [Google Scholar] [CrossRef]
- Kucukibrahimoglu, E.; Saygin, M.Z.; Caliskan, M.; Kaplan, O.K.; Unsal, C.; Goren, M.Z. The change in plasma GABA, glutamine and glutamate levels in fluoxetine- or S-citalopram-treated female patients with major depression. Eur. J. Clin. Pharmacol. 2009, 65, 571–577. [Google Scholar] [CrossRef]
- Racagni, G.; Popoli, M. Cellular and molecular mechanisms in the long-term action of antidepressants. Dialogues Clin. Neurosci. 2008, 10, 385–400. [Google Scholar] [CrossRef] [PubMed]
- Musazzi, L.; Racagni, G.; Popoli, M. Stress, glucocorticoids and glutamate release: Effects of antidepressant drugs. Neurochem. Int. 2011, 59, 138–149. [Google Scholar] [CrossRef] [PubMed]
- Skolnick, P.; Layer, R.T.; Popik, P.; Nowak, G.; Paul, I.A.; Trullas, R. Adaptation of N-methyl-D-aspartate (NMDA) receptors following antidepressant treatment: Implications for the pharmacotherapy of depression. Pharmacopsychiatry 1996, 29, 23–26. [Google Scholar] [CrossRef]
- Nowak, G.; Trullas, R.; Layer, R.T.; Skolnick, P.; Paul, I.A. Adaptive changes in the N-methyl-D-aspartate receptor complex after chronic treatment with imipramine and 1-aminocyclopropanecarboxylic acid. J. Pharmacol. Exp. Ther. 1993, 265, 1380–1386. [Google Scholar]
- Li, C.T.; Yang, K.C.; Lin, W.C. Glutamatergic Dysfunction and Glutamatergic Compounds for Major Psychiatric Disorders: Evidence From Clinical Neuroimaging Studies. Front. Psychiatry 2018, 9, 767. [Google Scholar] [CrossRef]
- Ohgi, Y.; Futamura, T.; Hashimoto, K. Glutamate Signaling in Synaptogenesis and NMDA Receptors as Potential Therapeutic Targets for Psychiatric Disorders. Curr. Mol. Med. 2015, 15, 206–221. [Google Scholar] [CrossRef]
- Martin, J.L.; Finsterwald, C. Cooperation between BDNF and glutamate in the regulation of synaptic transmission and neuronal development. Commun. Integr. Biol. 2011, 4, 14–16. [Google Scholar] [CrossRef]
- Kornmeier, J.; Sosic-Vasic, Z. Parallels between spacing effects during behavioral and cellular learning. Front. Hum. Neurosci. 2012, 6, 203. [Google Scholar] [CrossRef]
- Javitt, D.C.; Schoepp, D.; Kalivas, P.W.; Volkow, N.D.; Zarate, C.; Merchant, K.; Bear, M.F.; Umbricht, D.; Hajos, M.; Potter, W.Z.; et al. Translating glutamate: From pathophysiology to treatment. Sci. Transl. Med. 2011, 3, 102mr2. [Google Scholar] [CrossRef]
- McDonald, J.W.; Johnston, M.V. Physiological and pathophysiological roles of excitatory amino acids during central nervous system development. Brain Res. Rev. 1990, 15, 41–70. [Google Scholar] [CrossRef]
- Jansson, L.C.; Åkerman, K.E. The role of glutamate and its receptors in the proliferation, migration, differentiation and survival of neural progenitor cells. J. Neural Transm. 2014, 121, 819–836. [Google Scholar] [CrossRef]
- Peyrovian, B.; Rosenblat, J.D.; Pan, Z.; Iacobucci, M.; Brietzke, E.; McIntyre, R.S. The glycine site of NMDA receptors: A target for cognitive enhancement in psychiatric disorders. Prog. Neuropsychopharmacol. Biol. Psychiatry 2019, 92, 387–404. [Google Scholar] [CrossRef] [PubMed]
- Krystal, J.H.; Sanacora, G.; Duman, R.S. Rapid-acting glutamatergic antidepressants: The path to ketamine and beyond. Biol. Psychiatry 2013, 73, 1133–1141. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Lee, B.; Liu, R.J.; Banasr, M.; Dwyer, J.M.; Iwata, M.; Li, X.Y.; Aghajanian, G.; Duman, R.S. mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science 2010, 329, 959–964. [Google Scholar] [CrossRef] [PubMed]
- Diazgranados, N.; Ibrahim, L.; Brutsche, N.E.; Newberg, A.; Kronstein, P.; Khalife, S.; Kammerer, W.A.; Quezado, Z.; Luckenbaugh, D.A.; Salvadore, G.; et al. A randomized add-on trial of an N-methyl-D-aspartate antagonist in treatment-resistant bipolar depression. Arch. Gen. Psychiatry 2010, 67, 793–802. [Google Scholar] [CrossRef]
- Murrough, J.W.; Iosifescu, D.V.; Chang, L.C.; Al Jurdi, R.K.; Green, C.E.; Perez, A.M.; Iqbal, S.; Pillemer, S.; Foulkes, A.; Shah, A.; et al. Antidepressant efficacy of ketamine in treatment-resistant major depression: A two-site randomized controlled trial. Am. J. Psychiatry 2013, 170, 1134–1142. [Google Scholar] [CrossRef]
- Walker, A.K.; Budac, D.P.; Bisulco, S.; Lee, A.W.; Smith, R.A.; Beenders, B.; Kelley, K.W.; Dantzer, R. NMDA receptor blockade by ketamine abrogates lipopolysaccharide-induced depressive-like behavior in C57BL/6J mice. Neuropsychopharmacology 2013, 38, 1609–1616. [Google Scholar] [CrossRef]
- Tan, S.; Wang, Y.; Chen, K.; Long, Z.; Zou, J. Ketamine Alleviates Depressive-Like Behaviors via Down-Regulating Inflammatory Cytokines Induced by Chronic Restraint Stress in Mice. Biol. Pharm. Bull. 2017, 40, 1260–1267. [Google Scholar] [CrossRef]
- Shibakawa, Y.S.; Sasaki, Y.; Goshima, Y.; Echigo, N.; Kamiya, Y.; Kurahashi, K.; Yamada, Y.; Andoh, T. Effects of ketamine and propofol on inflammatory responses of primary glial cell cultures stimulated with lipopolysaccharide. Br. J. Anaesth. 2005, 95, 803–810. [Google Scholar] [CrossRef]
- Zhang, K.; Yang, C.; Chang, L.; Sakamoto, A.; Suzuki, T.; Fujita, Y.; Qu, Y.; Wang, S.; Pu, Y.; Tan, Y.; et al. Essential role of microglial transforming growth factor-beta1 in antidepressant actions of (R)-ketamine and the novel antidepressant TGF-beta1. Transl. Psychiatry 2020, 10, 32. [Google Scholar] [CrossRef]
- Chang, Y.; Lee, J.J.; Hsieh, C.Y.; Hsiao, G.; Chou, D.S.; Sheu, J.R. Inhibitory effects of ketamine on lipopolysaccharide-induced microglial activation. Mediat. Inflamm. 2009, 2009, 705379. [Google Scholar] [CrossRef] [PubMed]
- Mei, X.P.; Zhou, Y.; Wang, W.; Tang, J.; Wang, W.; Zhang, H.; Xu, L.X.; Li, Y.Q. Ketamine depresses toll-like receptor 3 signaling in spinal microglia in a rat model of neuropathic pain. Neurosignals 2011, 19, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Ho, M.F.; Zhang, C.; Zhang, L.; Li, H.; Weinshilboum, R.M. Ketamine and Active Ketamine Metabolites Regulate STAT3 and the Type I Interferon Pathway in Human Microglia: Molecular Mechanisms Linked to the Antidepressant Effects of Ketamine. Front. Pharmacol. 2019, 10, 1302. [Google Scholar] [CrossRef] [PubMed]
- Haile, C.N.; Murrough, J.W.; Iosifescu, D.V.; Chang, L.C.; Al Jurdi, R.K.; Foulkes, A.; Iqbal, S.; Mahoney, J.J., 3rd; De La Garza, R., 2nd; Charney, D.S.; et al. Plasma brain derived neurotrophic factor (BDNF) and response to ketamine in treatment-resistant depression. Int. J. Neuropsychopharmacol. 2014, 17, 331–336. [Google Scholar] [CrossRef]
- Cui, W.; Ning, Y.; Hong, W.; Wang, J.; Liu, Z.; Li, M.D. Crosstalk Between Inflammation and Glutamate System in Depression: Signaling Pathway and Molecular Biomarkers for Ketamine’s Antidepressant Effect. Mol. Neurobiol. 2019, 56, 3484–3500. [Google Scholar] [CrossRef]
- Sanacora, G.; Treccani, G.; Popoli, M. Towards a glutamate hypothesis of depression: An emerging frontier of neuropsychopharmacology for mood disorders. Neuropharmacology 2012, 62, 63–77. [Google Scholar] [CrossRef]
- Vargas, R. The GABAergic system: An overview of physiology, physiopathology and therapeutics. Int. J. Clin. Pharmacol. Pharmacother. 2018, 3, 142. [Google Scholar] [CrossRef]
- Rudolph, U.; Crestani, F.; Möhler, H. GABAA receptor subtypes: Dissecting their pharmacological functions. Trends Pharmacol. Sci. 2001, 22, 188–194. [Google Scholar] [CrossRef]
- Marques, T.R.; Ashok, A.H.; Angelescu, I.; Borgan, F.; Myers, J.; Lingford-Hughes, A.; Nutt, D.J.; Veronese, M.; Turkheimer, F.E.; Howes, O.D. GABA-A receptor differences in schizophrenia: A positron emission tomography study using [11C] Ro154513. Mol. Psychiatry 2021, 26, 2616–2625. [Google Scholar] [CrossRef]
- Hoftman, G.D.; Volk, D.W.; Bazmi, H.H.; Li, S.; Sampson, A.R.; Lewis, D.A. Altered cortical expression of GABA-related genes in schizophrenia: Illness progression vs developmental disturbance. Schizophr. Bull. 2015, 41, 180–191. [Google Scholar] [CrossRef]
- Guilloux, J.-P.; Douillard-Guilloux, G.; Kota, R.; Wang, X.; Gardier, A.; Martinowich, K.; Tseng, G.C.; Lewis, D.A.; Sibille, E. Molecular evidence for BDNF-and GABA-related dysfunctions in the amygdala of female subjects with major depression. Mol. Psychiatry 2012, 17, 1130–1142. [Google Scholar] [CrossRef]
- Oh, H.; Piantadosi, S.C.; Rocco, B.R.; Lewis, D.A.; Watkins, S.C.; Sibille, E. The Role of Dendritic Brain-Derived Neurotrophic Factor Transcripts on Altered Inhibitory Circuitry in Depression. Biol. Psychiatry 2019, 85, 517–526. [Google Scholar] [CrossRef] [PubMed]
- Black, C.; Miller, B.J. Meta-Analysis of Cytokines and Chemokines in Suicidality: Distinguishing Suicidal Versus Nonsuicidal Patients. Biol. Psychiatry 2015, 78, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Engler, H.; Brendt, P.; Wischermann, J.; Wegner, A.; Rohling, R.; Schoemberg, T.; Meyer, U.; Gold, R.; Peters, J.; Benson, S.; et al. Selective increase of cerebrospinal fluid IL-6 during experimental systemic inflammation in humans: Association with depressive symptoms. Mol. Psychiatry 2017, 22, 1448–1454. [Google Scholar] [CrossRef] [PubMed]
- Schwieler, L.; Larsson, M.K.; Skogh, E.; Kegel, M.E.; Orhan, F.; Abdelmoaty, S.; Finn, A.; Bhat, M.; Samuelsson, M.; Lundberg, K.; et al. Increased levels of IL-6 in the cerebrospinal fluid of patients with chronic schizophrenia--significance for activation of the kynurenine pathway. J. Psychiatry Neurosci. 2015, 40, 126–133. [Google Scholar] [CrossRef]
- Dowlati, Y.; Herrmann, N.; Swardfager, W.; Liu, H.; Sham, L.; Reim, E.K.; Lanctot, K.L. A meta-analysis of cytokines in major depression. Biol. Psychiatry 2010, 67, 446–457. [Google Scholar] [CrossRef]
- Enache, D.; Pariante, C.M.; Mondelli, V. Markers of central inflammation in major depressive disorder: A systematic review and meta-analysis of studies examining cerebrospinal fluid, positron emission tomography and post-mortem brain tissue. Brain Behav. Immun. 2019, 81, 24–40. [Google Scholar] [CrossRef]
- Kubera, M.; Obuchowicz, E.; Goehler, L.; Brzeszcz, J.; Maes, M. In animal models, psychosocial stress-induced (neuro)inflammation, apoptosis and reduced neurogenesis are associated to the onset of depression. Prog. Neuropsychopharmacol. Biol. Psychiatry 2011, 35, 744–759. [Google Scholar] [CrossRef]
- Dantzer, R.; O’Connor, J.C.; Freund, G.G.; Johnson, R.W.; Kelley, K.W. From inflammation to sickness and depression: When the immune system subjugates the brain. Nat. Rev. Neurosci. 2008, 9, 46–56. [Google Scholar] [CrossRef]
- Couch, Y.; Anthony, D.C.; Dolgov, O.; Revischin, A.; Festoff, B.; Santos, A.I.; Steinbusch, H.W.; Strekalova, T. Microglial activation, increased TNF and SERT expression in the prefrontal cortex define stress-altered behaviour in mice susceptible to anhedonia. Brain Behav. Immun. 2013, 29, 136–146. [Google Scholar] [CrossRef]
- Raison, C.L.; Rutherford, R.E.; Woolwine, B.J.; Shuo, C.; Schettler, P.; Drake, D.F.; Haroon, E.; Miller, A.H. A randomized controlled trial of the tumor necrosis factor antagonist infliximab for treatment-resistant depression: The role of baseline inflammatory biomarkers. JAMA Psychiatry 2013, 70, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Fazzino, F.; Urbina, M.; Cedeno, N.; Lima, L. Fluoxetine treatment to rats modifies serotonin transporter and cAMP in lymphocytes, CD4+ and CD8+ subpopulations and interleukins 2 and 4. Int. Immunopharmacol. 2009, 9, 463–467. [Google Scholar] [CrossRef]
- Nguyen, K.T.; Deak, T.; Owens, S.M.; Kohno, T.; Fleshner, M.; Watkins, L.R.; Maier, S.F. Exposure to acute stress induces brain interleukin-1beta protein in the rat. J. Neurosci. 1998, 18, 2239–2246. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Chen, Y.; Liu, H.; Zhou, Z.; Zhai, Y.; Yang, J. Chronic unpredictable stress accelerates atherosclerosis through promoting inflammation in apolipoprotein E knockout mice. Thromb. Res. 2010, 126, 386–392. [Google Scholar] [CrossRef] [PubMed]
- Iwata, M.; Ishida, H.; Kaneko, K.; Shirayama, Y. Learned helplessness activates hippocampal microglia in rats: A potential target for the antidepressant imipramine. Pharmacol. Biochem. Behav. 2016, 150–151, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, K.; Sheridan, J.F. Antidepressant imipramine diminishes stress-induced inflammation in the periphery and central nervous system and related anxiety- and depressive- like behaviors. Brain Behav. Immun. 2016, 57, 293–303. [Google Scholar] [CrossRef] [PubMed]
- Alboni, S.; Benatti, C.; Montanari, C.; Tascedda, F.; Brunello, N. Chronic antidepressant treatments resulted in altered expression of genes involved in inflammation in the rat hypothalamus. Eur. J. Pharmacol. 2013, 721, 158–167. [Google Scholar] [CrossRef]
- O’Brien, S.M.; Scully, P.; Fitzgerald, P.; Scott, L.V.; Dinan, T.G. Plasma cytokine profiles in depressed patients who fail to respond to selective serotonin reuptake inhibitor therapy. J. Psychiatr. Res. 2007, 41, 326–331. [Google Scholar] [CrossRef] [PubMed]
- Warner-Schmidt, J.L.; Vanover, K.E.; Chen, E.Y.; Marshall, J.J.; Greengard, P. Antidepressant effects of selective serotonin reuptake inhibitors (SSRIs) are attenuated by antiinflammatory drugs in mice and humans. Proc. Natl. Acad. Sci. USA 2011, 108, 9262–9267. [Google Scholar] [CrossRef]
- Chung, H.S.; Kim, H.; Bae, H. Phenelzine (monoamine oxidase inhibitor) increases production of nitric oxide and proinflammatory cytokines via the NF-kappaB pathway in lipopolysaccharide-activated microglia cells. Neurochem. Res. 2012, 37, 2117–2124. [Google Scholar] [CrossRef]
- Felger, J.C.; Lotrich, F.E. Inflammatory cytokines in depression: Neurobiological mechanisms and therapeutic implications. Neuroscience 2013, 246, 199–229. [Google Scholar] [CrossRef]
- Duman, R.S.; Monteggia, L.M. A neurotrophic model for stress-related mood disorders. Biol. Psychiatry 2006, 59, 1116–1127. [Google Scholar] [CrossRef]
- Nibuya, M.; Morinobu, S.; Duman, R.S. Regulation of BDNF and trkB mRNA in rat brain by chronic electroconvulsive seizure and antidepressant drug treatments. J. Neurosci. 1995, 15, 7539–7547. [Google Scholar] [CrossRef]
- Duman, R.S.; Heninger, G.R.; Nestler, E.J. A molecular and cellular theory of depression. Arch. Gen. Psychiatry 1997, 54, 597–606. [Google Scholar] [CrossRef]
- Kavalali, E.T.; Monteggia, L.M. Targeting Homeostatic Synaptic Plasticity for Treatment of Mood Disorders. Neuron 2020, 106, 715–726. [Google Scholar] [CrossRef]
- Maya Vetencourt, J.F.; Sale, A.; Viegi, A.; Baroncelli, L.; De Pasquale, R.; O’Leary, O.F.; Castren, E.; Maffei, L. The antidepressant fluoxetine restores plasticity in the adult visual cortex. Science 2008, 320, 385–388. [Google Scholar] [CrossRef]
- Castren, E. Neuronal network plasticity and recovery from depression. JAMA Psychiatry 2013, 70, 983–989. [Google Scholar] [CrossRef]
- Martinowich, K.; Manji, H.; Lu, B. New insights into BDNF function in depression and anxiety. Nat. Neurosci. 2007, 10, 1089–1093. [Google Scholar] [CrossRef]
- Zhang, J.-C.; Wu, J.; Fujita, Y.; Yao, W.; Ren, Q.; Yang, C.; Li, S.-X.; Shirayama, Y.; Hashimoto, K. Antidepressant effects of TrkB ligands on depression-like behavior and dendritic changes in mice after inflammation. Int. J. Neuropsychopharmacol. 2015, 18, 1–12. [Google Scholar] [CrossRef]
- Lapchak, P.; Araujo, D.; Hefti, F. Systemic interleukin-1 beta decreases brain-derived neurotrophic factor messenger RNA expression in the rat hippocampal formation. Neuroscience 1993, 53, 297–301. [Google Scholar] [CrossRef]
- Guan, Z.; Fang, J. Peripheral immune activation by lipopolysaccharide decreases neurotrophins in the cortex and hippocampus in rats. Brain Behav. Immun. 2006, 20, 64–71. [Google Scholar] [CrossRef]
- Levy, M.J.F.; Boulle, F.; Steinbusch, H.W.; van den Hove, D.L.A.; Kenis, G.; Lanfumey, L. Neurotrophic factors and neuroplasticity pathways in the pathophysiology and treatment of depression. Psychopharmacology 2018, 235, 2195–2220. [Google Scholar] [CrossRef]
- Mahmoudi Asl, A.; Mehdizadeh, M.; Kulisevsky, J.; Sabet, A.; Taghavi Azar Sharabiani, P.; Mehdizadeh, H.; Ashayeri, H.; Taghizadeh, G. Reliability, Validity, and Diagnostic Accuracy of Parkinson’s Disease-Cognitive Rating Scale in Iranian Patients with Idiopathic Parkinson’s Disease. Disabil. Rehabil. 2022, 44, 2091–2098. [Google Scholar] [CrossRef]
- Ratti, E.; Bettica, P.; Alexander, R.; Archer, G.; Carpenter, D.; Evoniuk, G.; Gomeni, R.; Lawson, E.; Lopez, M.; Millns, H.; et al. Full central neurokinin-1 receptor blockade is required for efficacy in depression: Evidence from orvepitant clinical studies. J. Psychopharmacol. 2013, 27, 424–434. [Google Scholar] [CrossRef]
- Tchekalarova, J.; Kortenska, L.; Ivanova, N.; Atanasova, M.; Marinov, P. Agomelatine treatment corrects impaired sleep-wake cycle and sleep architecture and increases MT 1 receptor as well as BDNF expression in the hippocampus during the subjective light phase of rats exposed to chronic constant light. Psychopharmacology 2020, 237, 503–518. [Google Scholar] [CrossRef]
- Li, N.-X.; Hu, Y.-R.; Chen, W.-N.; Zhang, B. Dose effect of psilocybin on primary and secondary depression: A preliminary systematic review and meta-analysis. J. Affect. Disord. 2022, 296, 26–34. [Google Scholar] [CrossRef]
- Gonda, X.; Dome, P.; Neill, J.C.; Tarazi, F.I. Novel antidepressant drugs: Beyond monoamine targets. CNS Spectr. 2021, 28, 6–15. [Google Scholar] [CrossRef]
- Demin, K.A.; Kupriyanova, O.V.; Shevyrin, V.A.; Derzhavina, K.A.; Krotova, N.A.; Ilyin, N.P.; Kolesnikova, T.O.; Galstyan, D.S.; Kositsyn, Y.M.; Khaybaev, A.-A.S. Acute behavioral and Neurochemical Effects of Novel N-Benzyl-2-Phenylethylamine Derivatives in Adult Zebrafish. ACS Chem. Neurosci. 2022, 13, 1902–1922. [Google Scholar] [CrossRef]
- Goodwin, G.M.; Aaronson, S.T.; Alvarez, O.; Arden, P.C.; Baker, A.; Bennett, J.C.; Bird, C.; Blom, R.E.; Brennan, C.; Brusch, D.; et al. Single-Dose Psilocybin for a Treatment-Resistant Episode of Major Depression. N. Engl. J. Med. 2022, 387, 1637–1648. [Google Scholar] [CrossRef]
- Fawcett, J.; Rush, A.J.; Vukelich, J.; Diaz, S.H.; Dunklee, L.; Romo, P.; Yarns, B.C.; Escalona, R. Clinical experience with high-dosage pramipexole in patients with treatment-resistant depressive episodes in unipolar and bipolar depression. Am. J. Psychiatry 2016, 173, 107–111. [Google Scholar] [CrossRef]
- Papakostas, G.I.; Shelton, R.C.; Zajecka, J.M.; Bottiglieri, T.; Roffman, J.; Cassiello, C.; Stahl, S.M.; Fava, M. Effect of adjunctive L-methylfolate 15 mg among inadequate responders to SSRIs in depressed patients who were stratified by biomarker levels and genotype: Results from a randomized clinical trial. J. Clin. Psychiatry 2014, 75, 5464. [Google Scholar] [CrossRef]
- Macaluso, M. L-Methylfolate in Antidepressant Non-responders: The Impact of Body Weight and Inflammation. Front. Psychiatry 2022, 13, 840116. [Google Scholar] [CrossRef]
- Lai, C.Y.; Scarr, E.; Udawela, M.; Everall, I.; Chen, W.J.; Dean, B. Biomarkers in schizophrenia: A focus on blood based diagnostics and theranostics. World J. Psychiatry 2016, 6, 102–117. [Google Scholar] [CrossRef]
- Dold, M.; Samara, M.T.; Li, C.; Tardy, M.; Leucht, S. Haloperidol versus first-generation antipsychotics for the treatment of schizophrenia and other psychotic disorders. Cochrane Database Syst. Rev. 2015, 1, CD009831. [Google Scholar] [CrossRef]
- Brasseur, R. Clinical trial with bromperidol in psychotic states. Acta Psychiatr. Belg. 1978, 78, 110–117. [Google Scholar]
- Nestoros, J.N.; Suranyl-Cadotte, B.E.; Spees, R.C.; Schwartz, G.; Vasavan Nair, N. Diazepam in high doses is effective in schizophrenia. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 1982, 6, 513–516. [Google Scholar] [CrossRef]
- Currier, G.W.; Chou, J.C.-Y.; Feifel, D.; Bossie, C.A.; Turkoz, I.; Mahmoud, R.A.; Gharabawi, G.M. Acute treatment of psychotic agitation: A randomized comparison of oral treatment with risperidone and lorazepam versus intramuscular treatment with haloperidol and lorazepam. J. Clin. Psychiatry 2004, 65, 387. [Google Scholar] [CrossRef]
- Kousgaard, S.J.; Licht, R.W.; Nielsen, R.E. Effects of intramuscular midazolam and lorazepam on acute agitation in non-Elderly Subjects—A systematic review. Pharmacopsychiatry 2017, 50, 129–135. [Google Scholar] [CrossRef]
- Madras, B.K. History of the discovery of the antipsychotic dopamine D2 receptor: A basis for the dopamine hypothesis of schizophrenia. J. Hist. Neurosci. 2013, 22, 62–78. [Google Scholar] [CrossRef]
- Borroto-Escuela, D.O.; Rodriguez, D.; Romero-Fernandez, W.; Kapla, J.; Jaiteh, M.; Ranganathan, A.; Lazarova, T.; Fuxe, K.; Carlsson, J. Mapping the Interface of a GPCR Dimer: A Structural Model of the A(2A) Adenosine and D(2) Dopamine Receptor Heteromer. Front. Pharmacol. 2018, 9, 829. [Google Scholar] [CrossRef]
- Meltzer, H.Y.; Stahl, S.M. The dopamine hypothesis of schizophrenia: A review. Schizophr. Bull. 1976, 2, 19–76. [Google Scholar] [CrossRef] [PubMed]
- Carlsson, A. The current status of the dopamine hypothesis of schizophrenia. Neuropsychopharmacology 1988, 1, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Bach, M.-E.; Simpson, E.H.; Kahn, L.; Marshall, J.J.; Kandel, E.R.; Kellendonk, C. Transient and selective overexpression of D2 receptors in the striatum causes persistent deficits in conditional associative learning. Proc. Natl. Acad. Sci. USA 2008, 105, 16027–16032. [Google Scholar] [CrossRef] [PubMed]
- Drew, M.; Simpson, E.; Kellendonk, C.; Herzberg, W.; Lipatova, O.; Fairhurst, S.; Kandel, E.R.; Malapani, C.; Balsam, P.D. Transient overexpression of striatal D2 receptors impairs operant motivation and interval timing. J. Neurosci. 2007, 27, 7731–7739. [Google Scholar] [CrossRef] [PubMed]
- Masri, B.; Salahpour, A.; Didriksen, M.; Ghisi, V.; Beaulieu, J.M.; Gainetdinov, R.R.; Caron, M.G. Antagonism of dopamine D2 receptor/beta-arrestin 2 interaction is a common property of clinically effective antipsychotics. Proc. Natl. Acad. Sci. USA 2008, 105, 13656–13661. [Google Scholar] [CrossRef]
- Urs, N.M.; Peterson, S.M.; Caron, M.G. New Concepts in Dopamine D(2) Receptor Biased Signaling and Implications for Schizophrenia Therapy. Biol. Psychiatry 2017, 81, 78–85. [Google Scholar] [CrossRef]
- De Vries, L.; Finana, F.; Cathala, C.; Ronsin, B.; Cussac, D. Innovative Bioluminescence Resonance Energy Transfer Assay Reveals Differential Agonist-Induced D2 Receptor Intracellular Trafficking and Arrestin-3 Recruitment. Mol. Pharmacol. 2019, 96, 308–319. [Google Scholar] [CrossRef]
- Simpson, E.H.; Gallo, E.F.; Balsam, P.D.; Javitch, J.A.; Kellendonk, C. How changes in dopamine D2 receptor levels alter striatal circuit function and motivation. Mol. Psychiatry 2022, 27, 436–444. [Google Scholar] [CrossRef]
- Mohn, A.R.; Gainetdinov, R.R.; Caron, M.G.; Koller, B.H. Mice with reduced NMDA receptor expression display behaviors related to schizophrenia. Cell 1999, 98, 427–436. [Google Scholar] [CrossRef]
- Abdolmaleky, H.M.; Cheng, K.H.; Russo, A.; Smith, C.L.; Faraone, S.V.; Wilcox, M.; Shafa, R.; Glatt, S.J.; Nguyen, G.; Ponte, J.F. Hypermethylation of the reelin (RELN) promoter in the brain of schizophrenic patients: A preliminary report. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2005, 134, 60–66. [Google Scholar] [CrossRef]
- Ruzicka, W.; Zhubi, A.; Veldic, M.; Grayson, D.; Costa, E.; Guidotti, A. Selective epigenetic alteration of layer I GABAergic neurons isolated from prefrontal cortex of schizophrenia patients using laser-assisted microdissection. Mol. Psychiatry 2007, 12, 385–397. [Google Scholar] [CrossRef] [PubMed]
- Mill, J.; Tang, T.; Kaminsky, Z.; Khare, T.; Yazdanpanah, S.; Bouchard, L.; Jia, P.; Assadzadeh, A.; Flanagan, J.; Schumacher, A. Epigenomic profiling reveals DNA-methylation changes associated with major psychosis. Am. J. Hum. Genet. 2008, 82, 696–711. [Google Scholar] [CrossRef]
- Veldic, M.; Guidotti, A.; Maloku, E.; Davis, J.M.; Costa, E. In psychosis, cortical interneurons overexpress DNA-methyltransferase 1. Proc. Natl. Acad. Sci. USA 2005, 102, 2152–2157. [Google Scholar] [CrossRef] [PubMed]
- Dean, B.; Gibbons, A.S.; Boer, S.; Uezato, A.; Meador-Woodruff, J.; Scarr, E.; McCullumsmith, R.E. Changes in cortical N-methyl-D-aspartate receptors and post-synaptic density protein 95 in schizophrenia, mood disorders and suicide. Aust. N. Z. J. Psychiatry 2016, 50, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Kantrowitz, J.T. Targeting serotonin 5-HT2A receptors to better treat schizophrenia: Rationale and current approaches. CNS Drugs 2020, 34, 947–959. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.; Zhou, Y. NMDAR Hypofunction Animal Models of Schizophrenia. Front. Mol. Neurosci. 2019, 12, 185. [Google Scholar] [CrossRef]
- Farber, N.B. The NMDA receptor hypofunction model of psychosis. Ann. N. Y. Acad. Sci. 2003, 1003, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Pei, J.C.; Luo, D.Z.; Gau, S.S.; Chang, C.Y.; Lai, W.S. Directly and Indirectly Targeting the Glycine Modulatory Site to Modulate NMDA Receptor Function to Address Unmet Medical Needs of Patients With Schizophrenia. Front. Psychiatry 2021, 12, 742058. [Google Scholar] [CrossRef]
- Greenwood, J.; Acharya, R.B.; Marcellus, V.; Rey, J.A. Lumateperone: A Novel Antipsychotic for Schizophrenia. Ann. Pharmacother. 2021, 55, 98–104. [Google Scholar] [CrossRef]
- Davis, R.E.; Correll, C.U. ITI-007 in the treatment of schizophrenia: From novel pharmacology to clinical outcomes. Expert Rev. Neurother. 2016, 16, 601–614. [Google Scholar] [CrossRef]
- Dedic, N.; Dworak, H.; Zeni, C.; Rutigliano, G.; Howes, O.D. Therapeutic Potential of TAAR1 Agonists in Schizophrenia: Evidence from Preclinical Models and Clinical Studies. Int. J. Mol. Sci. 2021, 22, 13185. [Google Scholar] [CrossRef] [PubMed]
- Halff, E.F.; Rutigliano, G.; Garcia-Hidalgo, A.; Howes, O.D. Trace amine-associated receptor 1 (TAAR1) agonism as a new treatment strategy for schizophrenia and related disorders. Trends Neurosci. 2022, 46, 60–74. [Google Scholar] [CrossRef] [PubMed]
- Koblan, K.S.; Kent, J.; Hopkins, S.C.; Krystal, J.H.; Cheng, H.; Goldman, R.; Loebel, A. A Non-D2-Receptor-Binding Drug for the Treatment of Schizophrenia. N. Engl. J. Med. 2020, 382, 1497–1506. [Google Scholar] [CrossRef]
- Scarr, E.; Cowie, T.; Kanellakis, S.; Sundram, S.; Pantelis, C.; Dean, B. Decreased cortical muscarinic receptors define a subgroup of subjects with schizophrenia. Mol. Psychiatry 2009, 14, 1017–1023. [Google Scholar] [CrossRef]
- Dean, B.; Scarr, E. Muscarinic M1 and M4 receptors: Hypothesis driven drug development for schizophrenia. Psychiatry Res. 2020, 288, 112989. [Google Scholar] [CrossRef]
- Eggers, A.E. A serotonin hypothesis of schizophrenia. Med. Hypotheses 2013, 80, 791–794. [Google Scholar] [CrossRef]
- Srinivasan, S.; Tampi, R.R.; Balaram, K.; Kapoor, A. Pimavanserin for the treatment of psychosis in Alzheimer’s disease: A literature review. World J. Psychiatry 2020, 10, 162–174. [Google Scholar] [CrossRef]
- Di Cristo, G. Development of cortical GABAergic circuits and its implications for neurodevelopmental disorders. Clin. Genet. 2007, 72, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, T.; Volk, D.W.; Eggan, S.M.; Mirnics, K.; Pierri, J.N.; Sun, Z.; Sampson, A.R.; Lewis, D.A. Gene expression deficits in a subclass of GABA neurons in the prefrontal cortex of subjects with schizophrenia. J. Neurosci. 2003, 23, 6315–6326. [Google Scholar] [CrossRef]
- Racki, V.; Petric, D.; Kucic, N.; Grzeta, N.; Jurdana, K.; Roncevic-Grzeta, I. Cortical gray matter loss in schizophrenia: Could microglia be the culprit? Med. Hypotheses 2016, 88, 18–21. [Google Scholar] [CrossRef]
- Réus, G.Z.; Fries, G.R.; Stertz, L.; Badawy, M.; Passos, I.; Barichello, T.; Kapczinski, F.; Quevedo, J. The role of inflammation and microglial activation in the pathophysiology of psychiatric disorders. Neuroscience 2015, 300, 141–154. [Google Scholar] [CrossRef]
- Steiner, J.; Walter, M.; Gos, T.; Guillemin, G.J.; Bernstein, H.-G.; Sarnyai, Z.; Mawrin, C.; Brisch, R.; Bielau, H.; zu Schwabedissen, L.M. Severe depression is associated with increased microglial quinolinic acid in subregions of the anterior cingulate gyrus: Evidence for an immune-modulated glutamatergic neurotransmission? J. Neuroinflamm. 2011, 8, 94. [Google Scholar] [CrossRef] [PubMed]
- Di Rosa, E.; Crow, T.J.; Walker, M.A.; Black, G.; Chance, S.A. Reduced neuron density, enlarged minicolumn spacing and altered ageing effects in fusiform cortex in schizophrenia. Psychiatry Res. 2009, 166, 102–115. [Google Scholar] [CrossRef] [PubMed]
- Orihuela, R.; McPherson, C.A.; Harry, G.J. Microglial M1/M2 polarization and metabolic states. Br. J. Pharmacol. 2016, 173, 649–665. [Google Scholar] [CrossRef]
- Inamdar, A.; Merlo-Pich, E.; Gee, M.; Makumi, C.; Mistry, P.; Robertson, J.; Steinberg, E.; Zamuner, S.; Learned, S.; Alexander, R.; et al. Evaluation of antidepressant properties of the p38 MAP kinase inhibitor losmapimod (GW856553) in Major Depressive Disorder: Results from two randomised, placebo-controlled, double-blind, multicentre studies using a Bayesian approach. J. Psychopharmacol. 2014, 28, 570–581. [Google Scholar] [CrossRef]
- Muller, N.; Schwarz, M.J.; Dehning, S.; Douhe, A.; Cerovecki, A.; Goldstein-Muller, B.; Spellmann, I.; Hetzel, G.; Maino, K.; Kleindienst, N.; et al. The cyclooxygenase-2 inhibitor celecoxib has therapeutic effects in major depression: Results of a double-blind, randomized, placebo controlled, add-on pilot study to reboxetine. Mol. Psychiatry 2006, 11, 680–684. [Google Scholar] [CrossRef] [PubMed]
- Howes, O.D.; McCutcheon, R. Inflammation and the neural diathesis-stress hypothesis of schizophrenia: A reconceptualization. Transl. Psychiatry 2017, 7, e1024. [Google Scholar] [CrossRef]
- Walder, D.J.; Walker, E.F.; Lewine, R.J. Cognitive functioning, cortisol release, and symptom severity in patients with schizophrenia. Biol. Psychiatry 2000, 48, 1121–1132. [Google Scholar] [CrossRef]
- Mondelli, V.; Dazzan, P.; Hepgul, N.; Di Forti, M.; Aas, M.; D’Albenzio, A.; Di Nicola, M.; Fisher, H.; Handley, R.; Marques, T.R. Abnormal cortisol levels during the day and cortisol awakening response in first-episode psychosis: The role of stress and of antipsychotic treatment. Schizophr. Res. 2010, 116, 234–242. [Google Scholar] [CrossRef]
- Chiappelli, J.; Shi, Q.; Kodi, P.; Savransky, A.; Kochunov, P.; Rowland, L.M.; Nugent, K.L.; Hong, L.E. Disrupted glucocorticoid—Immune interactions during stress response in schizophrenia. Psychoneuroendocrinology 2016, 63, 86–93. [Google Scholar] [CrossRef]
- Ryan, M.C.; Sharifi, N.; Condren, R.; Thakore, J.H. Evidence of basal pituitary–adrenal overactivity in first episode, drug naive patients with schizophrenia. Psychoneuroendocrinology 2004, 29, 1065–1070. [Google Scholar] [CrossRef] [PubMed]
- Mondelli, V.; Pariante, C.M.; Navari, S.; Aas, M.; D’Albenzio, A.; Di Forti, M.; Handley, R.; Hepgul, N.; Marques, T.R.; Taylor, H. Higher cortisol levels are associated with smaller left hippocampal volume in first-episode psychosis. Schizophr. Res. 2010, 119, 75–78. [Google Scholar] [CrossRef] [PubMed]
- Foster, R.; Kandanearatchi, A.; Beasley, C.; Williams, B.; Khan, N.; Fagerhol, M.K.; Everall, I.P. Calprotectin in microglia from frontal cortex is up-regulated in schizophrenia: Evidence for an inflammatory process? Eur. J. Neurosci. 2006, 24, 3561–3566. [Google Scholar] [CrossRef] [PubMed]
- Bender, H.-U.; Almashanu, S.; Steel, G.; Hu, C.-A.; Lin, W.-W.; Willis, A.; Pulver, A.; Valle, D. Functional consequences of PRODH missense mutations. Am. J. Hum. Genet. 2005, 76, 409–420. [Google Scholar] [CrossRef] [PubMed]
- Savio, L.E.B.; Vuaden, F.C.; Piato, A.L.; Bonan, C.D.; Wyse, A.T. Behavioral changes induced by long-term proline exposure are reversed by antipsychotics in zebrafish. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2012, 36, 258–263. [Google Scholar] [CrossRef] [PubMed]
- Layton, M.E.; Kern, J.C.; Hartingh, T.J.; Shipe, W.D.; Raheem, I.; Kandebo, M.; Hayes, R.P.; Huszar, S.; Eddins, D.; Ma, B. Discovery of MK-8189, a Highly Potent and Selective PDE10A Inhibitor for the Treatment of Schizophrenia. J. Med. Chem. 2023, 66, 1157–1171. [Google Scholar] [CrossRef]
- McGuire, P.; Robson, P.; Cubala, W.J.; Vasile, D.; Morrison, P.D.; Barron, R.; Taylor, A.; Wright, S. Cannabidiol (CBD) as an adjunctive therapy in schizophrenia: A multicenter randomized controlled trial. Am. J. Psychiatry 2018, 175, 225–231. [Google Scholar] [CrossRef]
- Lagunin, A.A.; Goel, R.K.; Gawande, D.Y.; Pahwa, P.; Gloriozova, T.A.; Dmitriev, A.V.; Ivanov, S.M.; Rudik, A.V.; Konova, V.I.; Pogodin, P.V. Chemo-and bioinformatics resources for in silico drug discovery from medicinal plants beyond their traditional use: A critical review. Nat. Prod. Rep. 2014, 31, 1585–1611. [Google Scholar] [CrossRef]
- Van den Broeck, W.M. Drug Targets, Target Identification, Validation, and Screening. In The Practice of Medicinal Chemistry; Elsevier: Amsterdam, The Netherlands, 2015; pp. 45–70. [Google Scholar]
- Ayipo, Y.O.; Alananzeh, W.A.; Ahmad, I.; Patel, H.; Mordi, M.N. Structural modelling and in silico pharmacology of β-carboline alkaloids as potent 5-HT1A receptor antagonists and reuptake inhibitors. J. Biomol. Struct. Dyn. 2022, 26, 1–17. [Google Scholar] [CrossRef]
- Jaramillo, D.N.; Millán, D.; Guevara-Pulido, J. Design, synthesis and cytotoxic evaluation of a selective serotonin reuptake inhibitor (SSRI) by virtual screening. Eur. J. Pharm. Sci. 2023, 183, 106403. [Google Scholar] [CrossRef]
- Javid, N.; Jalil, S.; Munir, R.; Zia-ur-Rehman, M.; Sahar, A.; Arshad, S.; Iqbal, J. 2, 1-Benzothiazine–(quinolin/thiophen) yl hydrazone frameworks as new monoamine oxidase inhibitory agents; synthesis, in vitro and in silico investigation. RSC Adv. 2023, 13, 1701–1710. [Google Scholar] [CrossRef]
- Seong, S.H.; Kim, B.-R.; Cho, M.L.; Kim, T.-S.; Im, S.; Han, S.; Jeong, J.-W.; Jung, H.A.; Choi, J.S. Phytoestrogen Coumestrol Selectively Inhibits Monoamine Oxidase-A and Amyloid β Self-Aggregation. Nutrients 2022, 14, 3822. [Google Scholar] [CrossRef] [PubMed]
- El-Damasy, A.K.; Park, J.E.; Kim, H.J.; Lee, J.; Bang, E.-K.; Kim, H.; Keum, G. Identification of New N-methyl-piperazine Chalcones as Dual MAO-B/AChE Inhibitors. Pharmaceuticals 2023, 16, 83. [Google Scholar] [CrossRef] [PubMed]
- Cabezas, D.; Mellado, G.; Espinoza, N.; Gárate, J.A.; Morales, C.; Castro-Alvarez, A.; Matos, M.J.; Mellado, M.; Mella, J. In silico approaches to develop new phenyl-pyrimidines as glycogen synthase kinase 3 (GSK-3) inhibitors with halogen-bonding capabilities: 3D-QSAR CoMFA/CoMSIA, molecular docking and molecular dynamics studies. J. Biomol. Struct. Dyn. 2023, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.-N.; Yu, M.-C. Inhibition of Voltage-Gated Na+ Currents Exerted by KB-R7943 (2-[2-[4-(4-nitrobenzyloxy) phenyl] ethyl] isothiourea), an Inhibitor of Na+-Ca2+ Exchanging Process. Int. J. Mol. Sci. 2023, 24, 1805. [Google Scholar] [CrossRef] [PubMed]
- Amin, F.; Ibrahim, M.A.; Rizwan-ul-Hasan, S.; Khaliq, S.; Gabr, G.A.; Khan, A.; Sidhom, P.A.; Tikmani, P.; Shawky, A.M.; Ahmad, S. Interactions of Apigenin and Safranal with the 5HT1A and 5HT2A Receptors and Behavioral Effects in Depression and Anxiety: A Molecular Docking, Lipid-Mediated Molecular Dynamics, and In Vivo Analysis. Molecules 2022, 27, 8658. [Google Scholar] [CrossRef]
- da Rocha, M.J.; Pires, C.S.; Presa, M.H.; Besckow, E.M.; Nunes, G.D.A.; Gomes, C.S.; Penteado, F.; Lenardão, E.J.; Bortolatto, C.F.; Brüning, C.A. Involvement of the serotonergic system in the antidepressant-like effect of 1-(phenylselanyl)-2-(p-tolyl) indolizine in mice. Psychopharmacology 2023, 240, 373–389. [Google Scholar] [CrossRef]
- Ayipo, Y.O.; Alananzeh, W.A.; Yahayaa, S.N.; Mordi, M.N. Molecular Modelling and Virtual Screening to Identify New Piperazine Derivatives as Potent Human 5-HT1A Antagonists and Reuptake Inhibitors. Comb. Chem. High Throughput Screen. 2022. [Google Scholar] [CrossRef]
- Jha, P.; Chaturvedi, S.; Bhat, R.; Jain, N.; Mishra, A.K. Insights of ligand binding in modeled h5-HT1A receptor: Homology modeling, docking, MM-GBSA, screening and molecular dynamics. J. Biomol. Struct. Dyn. 2022, 40, 11625–11637. [Google Scholar] [CrossRef]
- Camargo, A.; Bettio, L.E.; Rosa, P.B.; Rosa, J.M.; Altê, G.A.; Rodrigues, A.L.S. The antidepressant-like effect of guanosine involves the modulation of adenosine A1 and A2A receptors. Purinergic Signal. 2022. [Google Scholar] [CrossRef]
- Rangel-Galván, M.; Castro, M.E.; Perez-Aguilar, J.M.; Caballero, N.A.; Melendez, F.J. Conceptual DFT, QTAIM, and Molecular Docking Approaches to Characterize the T-Type Calcium Channel Blocker Anandamide. Front. Chem. 2022, 10, 920661. [Google Scholar] [CrossRef] [PubMed]
- Dawood, S.; Bano, S.; Badawy, A.A.-B. Inflammation and serotonin deficiency in major depressive disorder: Molecular docking of antidepressant and anti-inflammatory drugs to tryptophan and indoleamine 2, 3-dioxygenases. Biosci. Rep. 2022, 42, BSR20220426. [Google Scholar] [CrossRef] [PubMed]
- Elkholy, N.; Abdelwaly, A.; Mohamed, K.; Amata, E.; Lombino, J.; Cosentino, G.; Intagliata, S.; Helal, M.A. Discovery of 3-(2-aminoethyl)-thiazolidine-2, 4-diones as a novel chemotype of sigma-1 receptor ligands. Chem. Biol. Drug Des. 2022, 100, 25–40. [Google Scholar] [CrossRef] [PubMed]
- Gawai, A.A.; Biyani, K.; Das, S.; Tapadiya, G.; Mokale, S.; Dhawale, S.A. Design, Synthesis, Molecular Docking, and Preliminary pharmacological screening of some new benzo [d] thiazol-2-ylamino containing chromen-2-one derivatives with atypical antipsychotic profile. Curr. Comput. Aided Drug Des. 2023, 19, 465–475. [Google Scholar]
- Batista, V.S.; Gonçalves, A.M.; Nascimento-Júnior, N.M. Pharmacophore Mapping Combined with dbCICA Reveal New Structural Features for the Development of Novel Ligands Targeting α4β2 and α7 Nicotinic Acetylcholine Receptors. Molecules 2022, 27, 8236. [Google Scholar] [CrossRef]
- Nag, S.; Miranda-Azpiazu, P.; Jia, Z.; Datta, P.; Arakawa, R.; Moein, M.M.; Yang, Z.; Tu, Y.; Lemoine, L.; Ågren9(6):, H. Development of 11C-Labeled ASEM Analogues for the Detection of Neuronal Nicotinic Acetylcholine Receptors (α7-nAChR). ACS Chem. Neurosci. 2022, 13, 352–362. [Google Scholar] [CrossRef]
- Sharma, B.; Bhattacherjee, D.; Zyryanov, G.V.; Purohit, R. An insight from computational approach to explore novel, high-affinity phosphodiesterase 10A inhibitors for neurological disorders. J. Biomol. Struct. Dyn. 2022, 11, 1–13. [Google Scholar] [CrossRef]
- Hitge, R.; Petzer, J.P.; Petzer, A. The inhibition of monoamine oxidase by 2H-1, 4-benzothiazin-3 (4H)-ones. Bioorganic Med. Chem. Lett. 2022, 77, 129038. [Google Scholar] [CrossRef]
- Alharbi, A.H. Bio-Computational Evaluation of Compounds of Bacopa Monnieri as a Potential Treatment for Schizophrenia. Molecules 2022, 27, 7050. [Google Scholar] [CrossRef]
- Bhardwaj, T.; Ahmad, I.; Somvanshi, P. Systematic analysis to identify novel disease indications and plausible potential chemical leads of glutamate ionotropic receptor NMDA type subunit 1, GRIN1. J. Mol. Recognit. 2023, 36, e2997. [Google Scholar] [CrossRef]
- Wang, H.; Liu, J.; He, J.; Huang, D.; Xi, Y.; Xiao, T.; Ouyang, Q.; Zhang, S.; Wan, S.; Chen, X. Potential mechanisms underlying the therapeutic roles of sinisan formula in depression: Based on network pharmacology and molecular docking study. Front. Psychiatry 2022, 13, 1063489. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Taouil, A.; Awwa, M.; Clement, T.; Zhu, C.; Kim, J.; Rendina, D.; Jayanetti, K.; Maharaj, A.; Wang, L. SAR study on Novel truxillic acid monoester-Based inhibitors of fatty acid binding proteins as Next-Generation antinociceptive agents. Bioorganic Chem. 2022, 129, 106184. [Google Scholar] [CrossRef] [PubMed]
- Uba, A.I.; Chea, J.; Hoag, H.; Hryb, M.; Bui-Linh, C.; Wu, C. Binding of a positive allosteric modulator CDPPB to metabotropic glutamate receptor type 5 (mGluR5) probed by all-atom molecular dynamics simulations. Life Sci. 2022, 309, 121014. [Google Scholar] [CrossRef] [PubMed]
- Đorđević, V.; Petković, M.; Živković, J.; Nikolić, G.M.; Veselinović, A.M. Development of Novel Therapeutics for Schizophrenia Treatment Based on a Selective Positive Allosteric Modulation of α1-Containing GABAARs—In Silico Approach. Curr. Issues Mol. Biol. 2022, 44, 3398–3412. [Google Scholar] [CrossRef]
- El Fadili, M.; Er-Rajy, M.; Kara, M.; Assouguem, A.; Belhassan, A.; Alotaibi, A.; Mrabti, N.N.; Fidan, H.; Ullah, R.; Ercisli, S. QSAR, ADMET In silico pharmacokinetics, molecular docking and molecular dynamics studies of novel bicyclo (aryl methyl) benzamides as potent GlyT1 inhibitors for the treatment of schizophrenia. Pharmaceuticals 2022, 15, 670. [Google Scholar] [CrossRef]
- Noorbakhsh, A.; Hosseininezhadian Koushki, E.; Farshadfar, C.; Ardalan, N. Designing a natural inhibitor against human kynurenine aminotransferase type II and a comparison with PF-04859989: A computational effort against schizophrenia. J. Biomol. Struct. Dyn. 2022, 40, 7038–7051. [Google Scholar] [CrossRef]
- Hopkins, A.L. Network pharmacology: The next paradigm in drug discovery. Nat. Chem. Biol. 2008, 4, 682–690. [Google Scholar] [CrossRef]
- Wen, L.; Fan, Y.; Xiong, W.; Liu, Y.; Zhang, T.; Wei, G.; Altamirano, A.; Zhang, T.-E.; Yan, Z. Exploring the Mechanism of Action of Trachelospermi Caulis et Folium for Depression Based on Experiments: Combining Network Pharmacology and Molecular Docking. Comput. Math. Methods Med. 2022, 2022, 3945063. [Google Scholar] [CrossRef]
- Nguyen, L.T.H.; Nguyen, N.P.K.; Tran, K.N.; Shin, H.-M.; Yang, I.-J. Network Pharmacology and Experimental Validation to Investigate the Antidepressant Potential of Atractylodes lancea (Thunb.) DC. Life 2022, 12, 1925. [Google Scholar] [CrossRef]
- Zhao, Q.; Pan, W.; Shi, H.; Qi, F.; Liu, Y.; Yang, T.; Si, H.; Si, G. Network pharmacology and molecular docking analysis on the mechanism of Baihe Zhimu decoction in the treatment of postpartum depression. Medicine 2022, 101, e29323. [Google Scholar] [CrossRef]
- Podder, A.; Latha, N. New insights into schizophrenia disease genes interactome in the human brain: Emerging targets and therapeutic implications in the postgenomics era. OMICS J. Integr. Biol. 2014, 18, 754–766. [Google Scholar] [CrossRef] [PubMed]
- Paudel, S.; Min, X.; Acharya, S.; Khadka, D.B.; Yoon, G.; Kim, K.-M.; Cheon, S.H. Design, synthesis, and systematic evaluation of 4-arylpiperazine-and 4-benzylpiperidine napthyl ethers as inhibitors of monoamine neurotransmitters reuptake. Bioorg. Med. Chem. 2018, 26, 5538–5546. [Google Scholar] [CrossRef] [PubMed]
- Singla, R.K.; Ashraf, G.M.; Ganash, M.; Shen, B. Physicochemical, Interaction & Topological Descriptors vs. hMAO-A Inhibition of Aplysinopsin Analogs: A Boulevard to the Discovery of Semi-synthetic Antidepression Agents. Curr. Drug Metab. 2021, 22, 905–915. [Google Scholar] [PubMed]
- Santos, G.R.; Chiari, L.P.; da Silva, A.P.; Lipinski, C.F.; Oliveira, A.A.; Honorio, K.M.; de Sousa, A.G.; da Silva, A.B. A partial least squares and artificial neural network study for a series of arylpiperazines as antidepressant agents. J. Mol. Model. 2021, 27, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Jing, C.; Yu, P.; Lu, M.; Xu, X.; Pei, Q.; Yan, F. Profiling the structural determinants of aminoketone derivatives as hNET and hDAT reuptake inhibitors by field-based QSAR based on molecular docking. Technol. Health Care 2021, 29, 257–273. [Google Scholar] [CrossRef]
- Bukhari, S.N.A.; Elsherif, M.A.; Junaid, K.; Ejaz, H.; Alam, P.; Samad, A.; Jawarkar, R.D.; Masand, V.H. Perceiving the Concealed and Unreported Pharmacophoric Features of the 5-Hydroxytryptamine Receptor Using Balanced QSAR Analysis. Pharmaceuticals 2022, 15, 834. [Google Scholar] [CrossRef]
- Rathore, A.; Asati, V.; Mishra, M.; Das, R.; Kashaw, V.; Kashaw, S.K. Computational approaches for the design of novel dopamine D2 and serotonin 5-HT2A receptor dual antagonist towards schizophrenia. Silico Pharmacol. 2022, 10, 7. [Google Scholar] [CrossRef]
- Yu, Y.; Dong, H.; Peng, Y.; Welsh, W.J. QSAR-Based Computational Approaches to Accelerate the Discovery of Sigma-2 Receptor (S2R) Ligands as Therapeutic Drugs. Molecules 2021, 26, 5270. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissTargetPrediction: Updated data and new features for efficient prediction of protein targets of small molecules. Nucleic Acids Res. 2019, 47, W357–W364. [Google Scholar] [CrossRef]
- Poroikov, V.; Filimonov, D.; Gloriozova, T.; Lagunin, A.; Druzhilovskiy, D.; Rudik, A.; Stolbov, L.; Dmitriev, A.; Tarasova, O.; Ivanov, S. Computer-aided prediction of biological activity spectra for organic compounds: The possibilities and limitations. Russ. Chem. Bull. 2019, 68, 2143–2154. [Google Scholar] [CrossRef]
- Filimonov, D.; Lagunin, A.; Gloriozova, T.; Rudik, A.; Druzhilovskii, D.; Pogodin, P.; Poroikov, V. Prediction of the biological activity spectra of organic compounds using the PASS online web resource. Chem. Heterocycl. Compd. 2014, 50, 444–457. [Google Scholar] [CrossRef]
- Nickel, J.; Gohlke, B.-O.; Erehman, J.; Banerjee, P.; Rong, W.W.; Goede, A.; Dunkel, M.; Preissner, R. SuperPred: Update on drug classification and target prediction. Nucleic Acids Res. 2014, 42, W26–W31. [Google Scholar] [CrossRef]
- Grosdidier, A.; Zoete, V.; Michielin, O. SwissDock, a protein-small molecule docking web service based on EADock DSS. Nucleic Acids Res. 2011, 39, W270–W277. [Google Scholar] [CrossRef]
- Potemkin, V.; Potemkin, A.; Grishina, M. Internet resources for drug discovery and design. Curr. Top. Med. Chem. 2018, 18, 1955–1975. [Google Scholar] [CrossRef]
- Murtazalieva, K.; Druzhilovskiy, D.; Goel, R.; Sastry, G.; Poroikov, V. How good are publicly available web services that predict bioactivity profiles for drug repurposing? SAR QSAR Environ. Res. 2017, 28, 843–862. [Google Scholar] [CrossRef]
- Lagunin, A.; Filimonov, D.; Poroikov, V. Multi-targeted natural products evaluation based on biological activity prediction with PASS. Curr. Pharm. Des. 2010, 16, 1703–1717. [Google Scholar] [CrossRef] [PubMed]
- Goel, R.; Gawande, D.; Lagunin, A.; Poroikov, V. Pharmacological repositioning of Achyranthes aspera as an antidepressant using pharmacoinformatic tools PASS and PharmaExpert: A case study with wet lab validation. SAR QSAR Environ. Res. 2018, 29, 69–81. [Google Scholar] [CrossRef] [PubMed]
- Jha, S.; Dong, B.; Sakata, K. Enriched environment treatment reverses depression-like behavior and restores reduced hippocampal neurogenesis and protein levels of brain-derived neurotrophic factor in mice lacking its expression through promoter IV. Transl. Psychiatry 2011, 1, e40. [Google Scholar] [CrossRef]
- Marcon, M.; Mocelin, R.; Benvenutti, R.; Costa, T.; Herrmann, A.P.; de Oliveira, D.L.; Koakoski, G.; Barcellos, L.J.G.; Piato, A. Environmental enrichment modulates the response to chronic stress in zebrafish. J. Exp. Biol. 2018, 221, jeb176735. [Google Scholar] [CrossRef]
- Zhu, X.; Grace, A.A. Prepubertal Environmental Enrichment Prevents Dopamine Dysregulation and Hippocampal Hyperactivity in MAM Schizophrenia Model Rats. Biol. Psychiatry 2021, 89, 298–307. [Google Scholar] [CrossRef]
- Sramek, J.J.; Murphy, M.F.; Cutler, N.R. Sex differences in the psychopharmacological treatment of depression. Dialogues Clin. Neurosci. 2016, 18, 447–457. [Google Scholar] [CrossRef] [PubMed]
- Hoekstra, S.; Bartz-Johannessen, C.; Sinkeviciute, I.; Reitan, S.K.; Kroken, R.A.; Løberg, E.-M.; Larsen, T.K.; Rettenbacher, M.; Johnsen, E.; Sommer, I.E. Sex differences in antipsychotic efficacy and side effects in schizophrenia spectrum disorder: Results from the BeSt InTro study. NPJ Schizophr. 2021, 7, 39. [Google Scholar] [CrossRef] [PubMed]
- Furey, M.L.; Drevets, W.C. Antidepressant efficacy of the antimuscarinic drug scopolamine: A randomized, placebo-controlled clinical trial. Arch. Gen. Psychiatry 2006, 63, 1121–1129. [Google Scholar] [CrossRef] [PubMed]
- Lanquillon, S.; Krieg, J.C.; Bening-Abu-Shach, U.; Vedder, H. Cytokine production and treatment response in major depressive disorder. Neuropsychopharmacology 2000, 22, 370–379. [Google Scholar] [CrossRef]
- Kopschina Feltes, P.; Doorduin, J.; Klein, H.C.; Juarez-Orozco, L.E.; Dierckx, R.A.; Moriguchi-Jeckel, C.M.; de Vries, E.F. Anti-inflammatory treatment for major depressive disorder: Implications for patients with an elevated immune profile and non-responders to standard antidepressant therapy. J. Psychopharmacol. 2017, 31, 1149–1165. [Google Scholar] [CrossRef]
- Furtado, M.; Katzman, M.A. Examining the role of neuroinflammation in major depression. Psychiatry Res. 2015, 229, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Lotrich, F.E. Inflammatory cytokine-associated depression. Brain Res. 2015, 1617, 113–125. [Google Scholar] [CrossRef]
- Miller, A.H.; Raison, C.L. The role of inflammation in depression: From evolutionary imperative to modern treatment target. Nat. Rev. Immunol. 2016, 16, 22–34. [Google Scholar] [CrossRef]
- Yirmiya, R. Endotoxin produces a depressive-like episode in rats. Brain Res. 1996, 711, 163–174. [Google Scholar] [CrossRef]
- Capuron, L.; Ravaud, A.; Neveu, P.; Miller, A.; Maes, M.; Dantzer, R. Association between decreased serum tryptophan concentrations and depressive symptoms in cancer patients undergoing cytokine therapy. Mol. Psychiatry 2002, 7, 468–473. [Google Scholar] [CrossRef]
- Vollmer-Conna, U.A.; Fazou, C.; Cameron, B.; Li, H.; Brennan, C.; Luck, L.; Davenport, T.; Wakefield, D.; Hickie, I.; Lloyd, A. Production of pro-inflammatory cytokines correlates with the symptoms of acute sickness behaviour in humans. Psychol. Med. 2004, 34, 1289–1297. [Google Scholar] [CrossRef] [PubMed]
- Pigeon, W.R.; Hegel, M.; Unützer, J.; Fan, M.-Y.; Sateia, M.J.; Lyness, J.M.; Phillips, C.; Perlis, M.L. Is insomnia a perpetuating factor for late-life depression in the IMPACT cohort? Sleep 2008, 31, 481–488. [Google Scholar] [CrossRef] [PubMed]
- Demyttenaere, K.; De Fruyt, J.; Stahl, S.M. The many faces of fatigue in major depressive disorder. Int. J. Neuropsychopharmacol. 2005, 8, 93–105. [Google Scholar] [CrossRef] [PubMed]
- George, L.K.; Blazer, D.G.; Hughes, D.C.; Fowler, N. Social support and the outcome of major depression. Br. J. Psychiatry 1989, 154, 478–485. [Google Scholar] [CrossRef]
- Bluthé, R.-M.; Beaudu, C.; Kelley, K.W.; Dantzer, R. Differential effects of IL-1ra on sickness behavior and weight loss induced by IL-1 in rats. Brain Res. 1995, 677, 171–176. [Google Scholar] [CrossRef]
- Remus, J.L.; Dantzer, R. Inflammation models of depression in rodents: Relevance to psychotropic drug discovery. Int. J. Neuropsychopharmacol. 2016, 19, pyw028. [Google Scholar] [CrossRef] [PubMed]
- Ben-Shachar, D. The bimodal mechanism of interaction between dopamine and mitochondria as reflected in Parkinson’s disease and in schizophrenia. J. Neural Transm. 2020, 127, 159–168. [Google Scholar] [CrossRef]
- Jarskog, L.F. Apoptosis in schizophrenia: Pathophysiologic and therapeutic considerations. Curr. Opin. Psychiatry 2006, 19, 307–312. [Google Scholar] [CrossRef]
- Łukasiewicz, S. Development of a New Polymeric Nanocarrier Dedicated to Controlled Clozapine Delivery at the Dopamine D2-Serotonin 5-HT1A Heteromers. Polymers 2021, 13, 1000. [Google Scholar] [CrossRef]
- Kokras, N.; Dalla, C. Sex differences in animal models of psychiatric disorders. Br. J. Pharmacol. 2014, 171, 4595–4619. [Google Scholar] [CrossRef]
- Kokras, N.; Dalla, C. Preclinical sex differences in depression and antidepressant response: Implications for clinical research. J. Neurosci. Res. 2017, 95, 731–736. [Google Scholar] [CrossRef] [PubMed]
- Kokras, N.; Dalla, C.; Papadopoulou-Daifoti, Z. Sex differences in pharmacokinetics of antidepressants. Expert Opin. Drug Metab. Toxicol. 2011, 7, 213–226. [Google Scholar] [CrossRef] [PubMed]
- Puga-Olguín, A.; Rodríguez-Landa, J.F.; de Jesús Rovirosa-Hernández, M.; Germán-Ponciano, L.J.; Caba, M.; Meza, E.; Guillén-Ruiz, G.; Olmos-Vázquez, O.J. Long-term ovariectomy increases anxiety-and despair-like behaviors associated with lower Fos immunoreactivity in the lateral septal nucleus in rats. Behav. Brain Res. 2019, 360, 185–195. [Google Scholar] [CrossRef]
- Kokras, N.; Pastromas, N.; Papasava, D.; de Bournonville, C.; Cornil, C.; Dalla, C. Sex differences in behavioral and neurochemical effects of gonadectomy and aromatase inhibition in rats. Psychoneuroendocrinology 2018, 87, 93–107. [Google Scholar] [CrossRef] [PubMed]
- Stewart, A.; Wong, K.; Cachat, J.; Elegante, M.; Gilder, T.; Mohnot, S.; Wu, N.; Minasyan, A.; Tuohimaa, P.; Kalueff, A.V. Neurosteroid vitamin D system as a nontraditional drug target in neuropsychopharmacology. Behav. Pharmacol. 2010, 21, 420–426. [Google Scholar] [CrossRef] [PubMed]
- Kalueff, A.V.; Eremin, K.O.; Tuohimaa, P. Mechanisms of neuroprotective action of vitamin D(3). Biochemistry 2004, 69, 738–741. [Google Scholar] [CrossRef]
- Cui, X.; Eyles, D.W. Vitamin D and the Central Nervous System: Causative and Preventative Mechanisms in Brain Disorders. Nutrients 2022, 14, 4353. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| No | The Number of Active Compounds with the Respective Activity | IAP Based on Leave-One-Out Cross-Validation * | Predicted Activity Profile |
|---|---|---|---|
| 1 | 19,174 | 0.897 | Antidepressant |
| 2 | 3101 | 0.989 | Serotonin (5 Hydroxytryptamine) 1 agonist |
| 3 | 1701 | 0.991 | 5 Hydroxytryptamine 1A agonist |
| 4 | 5764 | 0.984 | 5 Hydroxytryptamine 1A antagonist |
| 5 | 135 | 0.989 | 5 Hydroxytryptamine 1B agonist |
| 6 | 7461 | 0.968 | 5 Hydroxytryptamine 2 antagonist |
| 7 | 5262 | 0.979 | 5 Hydroxytryptamine 2A antagonist |
| 8 | 2548 | 0.988 | 5 Hydroxytryptamine 6 antagonist |
| 9 | 1272 | 0.985 | 5 Hydroxytryptamine 7 antagonist |
| 10 | 6367 | 0.984 | 5 Hydroxytryptamine agonist |
| 11 | 18,747 | 0.967 | 5 Hydroxytryptamine antagonist |
| 12 | 7398 | 0.985 | 5 Hydroxytryptamine uptake inhibitor |
| 13 | 244 | 0.997 | AMPA receptor agonist |
| 14 | 4131 | 0.983 | Adrenaline uptake inhibitor |
| 15 | 2759 | 0.973 | Alpha 2 adrenoreceptor antagonist |
| 16 | 2896 | 0.983 | Dopamine agonist |
| 17 | 3932 | 0.985 | Dopamine uptake inhibitor |
| 18 | 1112 | 0.966 | GABA receptor agonist |
| 19 | 1212 | 0.996 | Glutamate (mGluR2) antagonist |
| 20 | 312 | 0.993 | Glutamate (mGluR3) antagonist |
| 21 | 2623 | 0.972 | MAO A inhibitor |
| 22 | 3993 | 0.977 | MAO B inhibitor |
| 23 | 5366 | 0.964 | MAO inhibitor |
| 24 | 593 | 0.994 | Melatonin agonist |
| 25 | 929 | 0.987 | NMDA 2B receptor antagonist |
| 26 | 27 | 0.999 | NMDA receptor glycine site B antagonist |
| 27 | 731 | 0.997 | NMDA receptor glycine site antagonist |
| 28 | 434 | 0.983 | Nicotinic alpha4beta2 receptor antagonist |
| 29 | 3884 | 0.970 | Opioid kappa receptor antagonist |
| No | The Number of Active Compounds with the Respective Activity | IAP Based on Leave-One-Out Cross-Validation * | Predicted Activity Profile |
|---|---|---|---|
| 1 | 48 | 0.910 | Antischizophrenic |
| 2 | 7461 | 0.968 | Serotonin (5 Hydroxytryptamine) 2 antagonist |
| 3 | 5262 | 0.979 | 5 Hydroxytryptamine 2A antagonist |
| 4 | 2432 | 0.986 | 5 Hydroxytryptamine 3 antagonist |
| 5 | 2548 | 0.988 | 5 Hydroxytryptamine 6 antagonist |
| 6 | 1272 | 0.985 | 5 Hydroxytryptamine 7 antagonist |
| 7 | 1835 | 0.992 | Acetylcholine M1 receptor agonist |
| 8 | 411 | 0.997 | Acetylcholine M4 receptor agonist |
| 9 | 1604 | 0.979 | Acetylcholine nicotinic agonist |
| 10 | 591 | 0.997 | Dopamine D1 agonist |
| 11 | 8375 | 0.983 | Dopamine D2 antagonist |
| 12 | 3789 | 0.984 | Dopamine D3 antagonist |
| 13 | 2387 | 0.986 | Dopamine D4 antagonist |
| 14 | 10,756 | 0.980 | Dopamine antagonist |
| 15 | 588 | 0.996 | Estrogen receptor beta agonist |
| 16 | 885 | 0.994 | Glutamate (mGluR2) agonist |
| 17 | 114 | 0.993 | Glutamate (mGluR3) agonist |
| 18 | 1999 | 0.996 | Glycine transporter 1 inhibitor |
| 19 | 6006 | 0.977 | Glutamate NMDA receptor antagonist |
| 20 | 975 | 0.988 | Nicotinic alpha7 receptor agonist |
| 21 | 5287 | 0.992 | Phosphodiesterase 10A inhibitor |
| 22 | 629 | 0.970 | Trace amine-associated receptor 1 agonist |
General conceptual questions:
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kositsyn, Y.M.; de Abreu, M.S.; Kolesnikova, T.O.; Lagunin, A.A.; Poroikov, V.V.; Harutyunyan, H.S.; Yenkoyan, K.B.; Kalueff, A.V. Towards Novel Potential Molecular Targets for Antidepressant and Antipsychotic Pharmacotherapies. Int. J. Mol. Sci. 2023, 24, 9482. https://doi.org/10.3390/ijms24119482
Kositsyn YM, de Abreu MS, Kolesnikova TO, Lagunin AA, Poroikov VV, Harutyunyan HS, Yenkoyan KB, Kalueff AV. Towards Novel Potential Molecular Targets for Antidepressant and Antipsychotic Pharmacotherapies. International Journal of Molecular Sciences. 2023; 24(11):9482. https://doi.org/10.3390/ijms24119482
Chicago/Turabian StyleKositsyn, Yuriy M., Murilo S. de Abreu, Tatiana O. Kolesnikova, Alexey A. Lagunin, Vladimir V. Poroikov, Hasmik S. Harutyunyan, Konstantin B. Yenkoyan, and Allan V. Kalueff. 2023. "Towards Novel Potential Molecular Targets for Antidepressant and Antipsychotic Pharmacotherapies" International Journal of Molecular Sciences 24, no. 11: 9482. https://doi.org/10.3390/ijms24119482
APA StyleKositsyn, Y. M., de Abreu, M. S., Kolesnikova, T. O., Lagunin, A. A., Poroikov, V. V., Harutyunyan, H. S., Yenkoyan, K. B., & Kalueff, A. V. (2023). Towards Novel Potential Molecular Targets for Antidepressant and Antipsychotic Pharmacotherapies. International Journal of Molecular Sciences, 24(11), 9482. https://doi.org/10.3390/ijms24119482