
The Role of Gut Microbiota in the Clinical Outcome of Septic Patients: State of the Art and Future Perspectives

 ,
,  , , ,
, , , 

Abstract
1. Introduction
2. Timely Diagnosis of Sepsis and the Role of Fast Microbiology
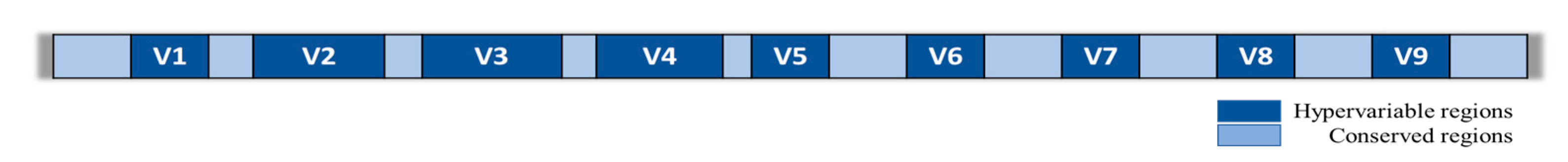
3. Gut microbiota Analysis via High-Throughput Methods
4. Interaction between Sepsis and Gut Microbiota: Clinical Point of View
5. The Impact of Antimicrobial Therapies and MDR Pathogens on Microbiota
6. Therapeutic Approach
- The microbiota is involved in nutrient metabolism, immune-modulation, and protection of the gastro-intestinal tract.
- During sepsis, there is an important disruption of the gut microbiota’s composition, characterized by an important reduction in microbial species diversity.
- Blood culture is the “gold standard” to diagnose sepsis but requires a long time. Biomarkers (CRP, PCT, and Presespin) can be very helpful for an earlier diagnosis.
- The use of antibiotics is mandatory but leads to a loss of important taxa, alters certain metabolic pathways, and induces microbiota to enter into a state of resilience against pathogens.
- Selective intestinal decontamination (SID); administration of probiotics, prebiotics, and symbiotics; and faecal microbiota transplants (FMTs) are therapeutic strategies effective in modulating the composition of the intestinal microbiota during infections or as prophylactic therapy.
7. Conclusions and Future Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Schnabl, B.; Brenner, D.A. Interactions between the intestinal microbiome and liver diseases. Gastroenterology 2014, 146, 1513–1524. [Google Scholar] [CrossRef] [PubMed]
- Rowland, I.; Gibson, G.; Heinken, A.; Scott, K.; Swann, J.; Thiele, I.; Tuohy, K. Gut microbiota functions: Metabolism of nutrients and other food components. Eur. J. Nutr. 2018, 57, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Thursby, E.; Juge, N. Introduction to the human gut microbiota. Biochem. J. 2017, 474, 1823–1836. [Google Scholar] [CrossRef] [PubMed]
- Nova, E.; Gómez-Martinez, S.; González-Soltero, R. The Influence of Dietary Factors on the Gut Microbiota. Microorganisms 2022, 10, 1368. [Google Scholar] [CrossRef]
- Jian, Z.; Zeng, L.; Xu, T.; Sun, S.; Yan, S.; Zhao, S.; Su, Z.; Ge, C.; Zhang, Y.; Jia, J.; et al. The intestinal microbiome associated with lipid metabolism and obesity in humans and animals. J. Appl. Microbiol. 2022, 133, 2915–2930. [Google Scholar] [CrossRef]
- Jandhyala, S.M.; Talukdar, R.; Subramanyam, C.; Vuyyuru, H.; Sasikala, M.; Nageshwar Reddy, D. Role of the normal gut microbiota. World J. Gastroenterol. 2015, 21, 8787–8803. [Google Scholar] [CrossRef]
- Konturek, P.C.; Harsch, I.A.; Konturek, K.; Schink, M.; Konturek, T.; Neurath, M.F.; Zopf, Y. Gut−Liver Axis: How Do Gut Bacteria Influence the Liver? Med. Sci. 2018, 6, 79. [Google Scholar] [CrossRef]
- Anand, S.; Mande, S.S. Host-microbiome interactions: Gut-Liver axis and its connection with other organs. NPJ Biofilms Microbiomes 2022, 8, 89. [Google Scholar] [CrossRef]
- Abenavoli, L.; Scarlata, G.G.M.; Scarpellini, E.; Boccuto, L.; Spagnuolo, R.; Tilocca, B.; Roncada, P.; Luzza, F. Metabolic-Dysfunction-Associated Fatty Liver Disease and Gut Microbiota: From Fatty Liver to Dysmetabolic Syndrome. Medicina 2023, 59, 594. [Google Scholar] [CrossRef]
- Marascio, N.; De Caro, C.; Quirino, A.; Mazzitelli, M.; Russo, E.; Torti, C.; Matera, G. The Role of the Microbiota Gut-Liver Axis during HCV Chronic Infection: A Schematic Overview. J. Clin. Med. 2022, 11, 5936. [Google Scholar] [CrossRef]
- Russo, A.; Serapide, F.; Quirino, A.; Tarsitano, M.G.; Marascio, N.; Serraino, R.; Rotundo, S.; Matera, G.; Trecarichi, E.M.; Torti, C. Microbiological and Clinical Findings of SARS-CoV-2 Infection after 2 Years of Pandemic: From Lung to Gut Microbiota. Diagnostics 2022, 12, 2143. [Google Scholar] [CrossRef]
- Lankelma, J.M.; van Vught, L.A.; Belzer, C.; Schultz, M.J.; van der Poll, T.; de Vos, W.M.; Wiersinga, W.J. Critically ill patients demonstrate large interpersonal variation in intestinal microbiota dysregulation: A pilot study. Intensive Care Med. 2017, 43, 59–68. [Google Scholar] [CrossRef]
- Singer, M.; Deutschman, C.S.; Seymour, C.W.; Shankar-Hari, M.; Annane, D.; Bauer, M.; Bellomo, R.; Bernard, G.R.; Chiche, J.D.; Coopersmith, C.M.; et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA 2016, 315, 801–810. [Google Scholar] [CrossRef]
- Huang, M.; Cai, S.; Su, J. The Pathogenesis of Sepsis and Potential Therapeutic Targets. Int. J. Mol. Sci. 2019, 20, 5376. [Google Scholar] [CrossRef]
- Global Report on the Epidemiology and Burden of Sepsis. Available online: https://apps.who.int/iris/bitstream/handle/10665/334216/9789240010789-eng.pdf (accessed on 20 April 2023).
- Li, A.T.; Moussa, A.; Gus, E.; Paul, E.; Yii, E.; Romero, L.; Lin, Z.C.; Padiglione, A.; Lo, C.H.; Cleland, H.; et al. Biomarkers for the Early Diagnosis of Sepsis in Burns: Systematic Review and Meta-analysis. Ann. Surg. 2022, 275, 654–662. [Google Scholar] [CrossRef]
- Hotchkiss, R.S.; Moldawer, L.L.; Opal, S.M.; Reinhart, K.; Turnbull, I.R.; Vincent, J.L. Sepsis and septic shock. Nat. Rev. Dis. Primers 2016, 2, 16045. [Google Scholar] [CrossRef]
- Serapide, F.; Quirino, A.; Scaglione, V.; Morrone, H.L.; Longhini, F.; Bruni, A.; Garofalo, E.; Matera, G.; Marascio, N.; Scarlata, G.G.M.; et al. Is the Pendulum of Antimicrobial Drug Resistance Swinging Back after COVID-19? Microorganisms 2022, 10, 957. [Google Scholar] [CrossRef]
- Bongiorno, D.; Bivona, D.A.; Cicino, C.; Trecarichi, E.M.; Russo, A.; Marascio, N.; Mezzatesta, M.L.; Musso, N.; Privitera, G.F.; Quirino, A.; et al. Omic insights into various ceftazidime-avibactam-resistant Klebsiella pneumoniae isolates from two southern Italian regions. Front. Cell. Infect. Microbiol. 2023, 12, 1010979. [Google Scholar] [CrossRef]
- Wang, M.; Liu, H.; Ren, J.; Huang, Y.; Deng, Y.; Liu, Y.; Chen, Z.; Chow, F.W.; Leung, P.H.; Li, S. Enzyme-Assisted Nucleic Acid Amplification in Molecular Diagnosis: A Review. Biosensors 2023, 13, 160. [Google Scholar] [CrossRef]
- Bassetti, M.; Russo, A.; Righi, E.; Dolso, E.; Merelli, M.; Cannarsa, N.; D’Aurizio, F.; Sartor, A.; Curcio, F. Comparison between procalcitonin and C-reactive protein to predict blood culture results in ICU patients. Crit. Care 2018, 22, 252. [Google Scholar] [CrossRef]
- Bassetti, M.; Russo, A.; Righi, E.; Dolso, E.; Merelli, M.; D’Aurizio, F.; Sartor, A.; Curcio, F. Role of procalcitonin in bacteremic patients and its potential use in predicting infection etiology. Expert Rev. Anti-Infect. Ther. 2019, 17, 99–105. [Google Scholar] [CrossRef]
- Bassetti, M.; Russo, A.; Righi, E.; Dolso, E.; Merelli, M.; D’Aurizio, F.; Sartor, A.; Curcio, F. Role of procalcitonin in predicting etiology in bacteremic patients: Report from a large single-center experience. J. Infect. Public Health 2020, 13, 40–45. [Google Scholar] [CrossRef] [PubMed]
- Russo, A.; Venditti, M.; Ceccarelli, G.; Mastroianni, C.M.; d’Ettorre, G. Procalcitonin in daily clinical practice: An evergreen tool also during a pandemic. Intern. Emerg. Med. 2021, 16, 541–543. [Google Scholar] [CrossRef] [PubMed]
- Alessandri, F.; Pugliese, F.; Angeletti, S.; Ciccozzi, M.; Russo, A.; Mastroianni, C.M.; d’Ettorre, G.; Venditti, M.; Ceccarelli, G. Procalcitonin in the Assessment of Ventilator Associated Pneumonia: A Systematic Review. Adv. Exp. Med. Biol. 2021, 1323, 103–114. [Google Scholar] [PubMed]
- Bargieł, W.; Cierpiszewska, K.; Maruszczak, K.; Pakuła, A.; Szwankowska, D.; Wrzesińska, A.; Gutowski, Ł.; Formanowicz, D. Recognized and Potentially New Biomarkers-Their Role in Diagnosis and Prognosis of Cardiovascular Disease. Medicina 2021, 57, 701. [Google Scholar] [CrossRef]
- Matera, G.; Quirino, A.; Giancotti, A.; Pulicari, M.C.; Rametti, L.; Rodríguez, M.L.; Liberto, M.C.; Focà, A. Procalcitonin neutralizes bacterial LPS and reduces LPS-induced cytokine release in human peripheral blood mononuclear cells. BMC Microbiol. 2012, 12, 68. [Google Scholar] [CrossRef]
- Matera, G.; Quirino, A.; Peronace, C.; Settembre, P.; Marano, V.; Loria, M.T.; Marascio, N.; Galati, L.; Barreca, G.S.; Giancotti, A.; et al. Soluble CD14 Subtype—A New Biomarker in Predicting the Outcome of Critically Ill Septic Patients. Am. J. Med. Sci. 2017, 353, 543–551. [Google Scholar] [CrossRef]
- Quirino, A.; Scaglione, V.; Marascio, N.; Mazzitelli, M.; Garofalo, E.; Divenuto, F.; Serapide, F.; Bruni, A.; Lionello, R.; Pavia, G.; et al. Role of the T2Dx magnetic resonance assay in patients with suspected bloodstream infection: A single-centre real-world experience. BMC Infect. Dis. 2022, 22, 113. [Google Scholar] [CrossRef]
- Quirino, A.; Marascio, N.; Peronace, C.; Gallo, L.; Barreca, G.S.; Giancotti, A.; Lamberti, A.G.; Colosimo, M.; Minchella, P.; Trecarichi, E.M.; et al. Direct antimicrobial susceptibility testing (AST) from positive blood cultures using Microscan system for early detection of bacterial resistance phenotypes. Diagn. Microbiol. Infect. Dis. 2021, 101, 115485. [Google Scholar] [CrossRef]
- Brenner, T.; Decker, S.O.; Grumaz, S.; Stevens, P.; Bruckner, T.; Schmoch, T.; Pletz, M.W.; Bracht, H.; Hofer, S.; Marx, G.; et al. Next-generation sequencing diagnostics of bacteremia in sepsis (Next GeneSiS-Trial): Study protocol of a prospective, observational, noninterventional, multicenter, clinical trial. Medicine 2018, 97, e9868. [Google Scholar] [CrossRef]
- Mandal, R.S.; Saha, S.; Das, S. Metagenomic surveys of gut microbiota. Genom. Proteom. Bioinform. 2015, 13, 148–158. [Google Scholar] [CrossRef]
- Janda, J.M.; Abbott, S.L. 16S rRNA gene sequencing for bacterial identification in the diagnostic laboratory: Pluses, perils, and pitfalls. J. Clin. Microbiol. 2007, 45, 2761–2764. [Google Scholar] [CrossRef]
- Winand, R.; Bogaerts, B.; Hoffman, S.; Lefevre, L.; Delvoye, M.; Van Braekel, J.; Fu, Q.; Roosens, N.H.; De Keersmaecker, S.C.; Vanneste, K. Targeting The 16S rRNA Gene For Bacterial Identification In Complex Mixed Samples: Comparative Evaluation Of Second (Illumina) And Third (Oxford Nanopore Technologies) Generation Sequencing Technologies. Int. J. Mol. Sci. 2019, 21, 298. [Google Scholar] [CrossRef]
- Jo, J.H.; Kennedy, E.A.; Kong, H.H. Research Techniques Made Simple: Bacterial 16S Ribosomal RNA Gene Sequencing in Cutaneous Research. J. Investig. Dermatol. 2016, 136, e23–e27. [Google Scholar] [CrossRef]
- Katiraei, S.; Anvar, Y.; Hoving, L.; Berbée, J.F.P.; van Harmelen, V.; Willems van Dijk, K. Evaluation of Full-Length Versus V4-Region 16S rRNA Sequencing for Phylogenetic Analysis of Mouse Intestinal Microbiota After a Dietary Intervention. Curr. Microbiol. 2022, 79, 276. [Google Scholar] [CrossRef]
- Wensel, C.R.; Pluznick, J.L.; Salzberg, S.L.; Sears, C.L. Next-generation sequencing: Insights to advance clinical investigations of the microbiome. J. Clin. Investig. 2022, 132, e154944. [Google Scholar] [CrossRef]
- Xia, Y.; Sun, J.; Chen, D.G. Statistical Analysis of Microbiome Data with R; Springer: Singapore, 2018. [Google Scholar]
- DeSantis, T.Z.; Hugenholtz, P.; Larsen, N.; Rojas, M.; Brodie, E.L.; Keller, K.; Huber, T.; Dalevi, D.; Hu, P.; Andersen, G.L. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 2006, 72, 5069–5072. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- Walters, K.E.; Martiny, J.B.H. Alpha-, beta-, and gamma-diversity of bacteria varies across habitats. PLoS ONE 2020, 15, e0233872. [Google Scholar] [CrossRef]
- Portincasa, P.; Bonfrate, L.; Vacca, M.; De Angelis, M.; Farella, I.; Lanza, E.; Khalil, M.; Wang, D.Q.; Sperandio, M.; Di Ciaula, A. Gut Microbiota and Short Chain Fatty Acids: Implications in Glucose Homeostasis. Int. J. Mol. Sci. 2022, 23, 1105. [Google Scholar] [CrossRef]
- Guo, Y.; Zhu, G.; Wang, F.; Zhang, H.; Chen, X.; Mao, Y.; Lv, Y.; Xia, F.; Jin, Y.; Ding, G.; et al. Distinct Serum and Fecal Metabolite Profiles Linking With Gut Microbiome in Older Adults With Frailty. Front. Med. 2022, 9, 827174. [Google Scholar] [CrossRef] [PubMed]
- Whon, T.W.; Chung, W.H.; Lim, M.Y.; Song, E.J.; Kim, P.S.; Hyun, D.W.; Shin, N.R.; Bae, J.W.; Nam, Y.D. The effects of sequencing platforms on phylogenetic resolution in 16 S rRNA gene profiling of human feces. Sci. Data 2018, 5, 180068. [Google Scholar] [CrossRef]
- Abellan-Schneyder, I.; Matchado, M.S.; Reitmeier, S.; Sommer, A.; Sewald, Z.; Baumbach, J.; List, M.; Neuhaus, K. Primer, Pipelines, Parameters: Issues in 16S rRNA Gene Sequencing. mSphere 2021, 6, e01202-20. [Google Scholar] [CrossRef] [PubMed]
- Kameoka, S.; Motooka, D.; Watanabe, S.; Kubo, R.; Jung, N.; Midorikawa, Y.; Shinozaki, N.O.; Sawai, Y.; Takeda, A.K.; Nakamura, S. Benchmark of 16S rRNA gene amplicon sequencing using Japanese gut microbiome data from the V1-V2 and V3-V4 primer sets. BMC Genom. 2021, 22, 527. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Hui, P.C.; Hui, M.; Yeoh, Y.K.; Wong, P.Y.; Chan, M.C.; Wong, M.C.; Ng, S.C.; Chan, F.K.; Chan, P.K. Impact of Preservation Method and 16S rRNA Hypervariable Region on Gut Microbiota Profiling. mSystems 2019, 4, e00271-18. [Google Scholar] [CrossRef]
- Wan, Y.D.; Zhu, R.X.; Wu, Z.Q.; Lyu, S.Y.; Zhao, L.X.; Du, Z.J.; Pan, X.T. Gut Microbiota Disruption in Septic Shock Patients: A Pilot Study. Med. Sci. Monit. 2018, 24, 8639–8646. [Google Scholar] [CrossRef]
- Du, B.; Shen, N.; Tao, Y.; Sun, S.; Zhang, F.; Ren, H.; Cao, Q.; Mo, X. Analysis of gut microbiota alteration and application as an auxiliary prognostic marker for sepsis in children: A pilot study. Transl. Pediatr. 2021, 10, 1647–1657. [Google Scholar] [CrossRef]
- Liu, W.; Cheng, M.; Li, J.; Zhang, P.; Fan, H.; Hu, Q.; Han, M.; Su, L.; He, H.; Tong, Y.; et al. Classification of the Gut Microbiota of Patients in Intensive Care Units During Development of Sepsis and Septic Shock. Genom. Proteom. Bioinform. 2020, 18, 696–707. [Google Scholar] [CrossRef]
- Ricci, V.; Carcione, D.; Messina, S.; Colombo, G.I.; D’Alessandra, Y. Circulating 16S RNA in Biofluids: Extracellular Vesicles as Mirrors of Human Microbiome? Int. J. Mol. Sci. 2020, 21, 8959. [Google Scholar] [CrossRef]
- Miller, W.D.; Keskey, R.; Alverdy, J.C. Sepsis and the Microbiome: A Vicious Cycle. J. Infect. Dis. 2021, 223, S264–S269. [Google Scholar] [CrossRef]
- Meng, J.; Banerjee, S.; Li, D.; Sindberg, G.M.; Wang, F.; Ma, J.; Roy, S. Opioid exacerbation of gram-positive sepsis, induced by gut microbial modulation, is rescued by IL-17A neutralization. Sci. Rep. 2015, 5, 10918. [Google Scholar] [CrossRef]
- Belizário, J.E.; Faintuch, J.; Garay-Malpartida, M. Gut Microbiome Dysbiosis and Immunometabolism: New Frontiers for Treatment of Metabolic Diseases. Mediat. Inflamm. 2018, 2018, 2037838. [Google Scholar] [CrossRef]
- Matera, G.; Puccio, R.; Giancotti, A.; Quirino, A.; Pulicari, M.C.; Zicca, E.; Caroleo, S.; Renzulli, A.; Liberto, M.C.; Focà, A. Impact of interleukin-10, soluble CD25 and interferon-γ on the prognosis and early diagnosis of bacteremic systemic inflammatory response syndrome: A prospective observational study. Crit. Care 2013, 17, R64. [Google Scholar] [CrossRef]
- Kennedy, E.A.; King, K.Y.; Baldridge, M.T. Mouse Microbiota Models: Comparing Germ-Free Mice and Antibiotics Treatment as Tools for Modifying Gut Bacteria. Front. Physiol. 2018, 9, 1534. [Google Scholar] [CrossRef]
- Im, Y.; Kang, D.; Ko, R.E.; Lee, Y.J.; Lim, S.Y.; Park, S.; Na, S.J.; Chung, C.R.; Park, M.H.; Oh, D.K.; et al. Time-to-antibiotics and clinical outcomes in patients with sepsis and septic shock: A prospective nationwide multicenter cohort study. Crit. Care 2022, 26, 19. [Google Scholar] [CrossRef]
- Blaser, M.J. Antibiotic use and its consequences for the normal microbiome. Science 2016, 352, 544–545. [Google Scholar] [CrossRef]
- Ianiro, G.; Tilg, H.; Gasbarrini, A. Antibiotics as deep modulators of gut microbiota: Between good and evil. Gut 2016, 65, 1906–1915. [Google Scholar] [CrossRef]
- Lange, K.; Buerger, M.; Stallmach, A.; Bruns, T. Effects of Antibiotics on Gut Microbiota. Dig. Dis. 2016, 34, 260–268. [Google Scholar] [CrossRef]
- Ramirez, J.; Guarner, F.; Bustos Fernandez, L.; Maruy, A.; Sdepanian, V.L.; Cohen, H. Antibiotics as Major Disruptors of Gut Microbiota. Front. Cell. Infect. Microbiol. 2020, 10, 572912. [Google Scholar] [CrossRef]
- Becattini, S.; Taur, Y.; Pamer, E.G. Antibiotic-Induced Changes in the Intestinal Microbiota and Disease. Trends Mol. Med. 2016, 22, 458–478. [Google Scholar] [CrossRef]
- Guo, Y.; Yang, X.; Qi, Y.; Wen, S.; Liu, Y.; Tang, S.; Huang, R.; Tang, L. Long-term use of ceftriaxone sodium induced changes in gut microbiota and immune system. Sci. Rep. 2017, 7, 43035. [Google Scholar] [CrossRef] [PubMed]
- de Lastours, V.; Goulenok, T.; Guérin, F.; Jacquier, H.; Eyma, C.; Chau, F.; Cattoir, V.; Fantin, B. Ceftriaxone promotes the emergence of AmpC-overproducing Enterobacteriaceae in gut microbiota from hospitalized patients. Eur. J. Clin. Microbiol. Infect. Dis. 2018, 37, 417–421. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, P.; Curtis, N. The effect of antibiotics on the composition of the intestinal microbiota—A systematic review. J. Infect. 2019, 79, 471–489. [Google Scholar] [CrossRef] [PubMed]
- Smits, W.K.; Lyras, D.; Lacy, D.B.; Wilcox, M.H.; Kuijper, E.J. Clostridium difficile infection. Nat. Rev. Dis. Primers 2016, 2, 16020. [Google Scholar] [CrossRef]
- Gu, S.L.; Gong, Y.; Zhang, J.; Chen, Y.; Wu, Z.; Xu, Q.; Fang, Y.; Wang, J.; Tang, L.L. Effect of the Short-Term Use of Fluoroquinolone and β-Lactam Antibiotics on Mouse Gut Microbiota. Infect. Drug Resist. 2020, 13, 4547–4558. [Google Scholar] [CrossRef]
- Liou, J.M.; Chen, C.C.; Chang, C.M.; Fang, Y.J.; Bair, M.J.; Chen, P.Y.; Chang, C.Y.; Hsu, Y.C.; Chen, M.J.; Chen, C.C.; et al. Long-term changes of gut microbiota, antibiotic resistance, and metabolic parameters after Helicobacter pylori eradication: A multicentre, open-label, randomised trial. Lancet Infect. Dis. 2019, 19, 1109–1120. [Google Scholar] [CrossRef]
- Huang, C.; Feng, S.; Huo, F.; Liu, H. Effects of Four Antibiotics on the Diversity of the Intestinal Microbiota. Microbiol. Spectr. 2022, 10, e0190421. [Google Scholar] [CrossRef]
- Clarke, T.B. Early innate immunity to bacterial infection in the lung is regulated systemically by the commensal microbiota via nod-like receptor ligands. Infect. Immun. 2014, 82, 4596–4606. [Google Scholar] [CrossRef]
- Feng, Y.; Huang, Y.; Wang, Y.; Wang, P.; Wang, F. Severe burn injury alters intestinal microbiota composition and impairs intestinal barrier in mice. Burns Trauma 2019, 7, 20. [Google Scholar] [CrossRef]
- Barsuk, A.L.; Nekaeva, E.S.; Lovtsova, L.V.; Urakov, A.L. Selective Intestinal Decontamination as a Method for Preventing Infectious Complications (Review). Sovrem. Tekhnologii Med. 2021, 12, 86–95. [Google Scholar] [CrossRef]
- Van Saene, J.J.M.; Stoutenbeek, C.P.; van Saene, H.K.F.; Matera, G.; Martinez Pellus, A.E.; Ramsay, G. Reduction of the intestinal endotoxin pool by three different SDD regimens in human volunteers. J. Endotoxin Res. 1996, 3, 337–343. [Google Scholar] [CrossRef]
- Markowiak, P.; Śliżewska, K. Effects of Probiotics, Prebiotics, and Synbiotics on Human Health. Nutrients 2017, 9, 1021. [Google Scholar] [CrossRef]
- Ooijevaar, R.E.; Terveer, E.M.; Verspaget, H.W.; Kuijper, E.J.; Keller, J.J. Clinical Application and Potential of Fecal Microbiota Transplantation. Annu. Rev. Med. 2019, 70, 335–351. [Google Scholar] [CrossRef]
- Pamer, E.G. Resurrecting the intestinal microbiota to combat antibiotic-resistant pathogens. Science 2016, 352, 535–538. [Google Scholar] [CrossRef]
- Vincent, J.L.; Rello, J.; Marshall, J.; Silva, E.; Anzueto, A.; Martin, C.D.; Moreno, R.; Lipman, J.; Gomersall, C.; Sakr, Y.; et al. International study of the prevalence and outcomes of infection in intensive care units. JAMA 2009, 302, 2323–2329. [Google Scholar] [CrossRef]
- Hemarajata, P.; Versalovic, J. Effects of probiotics on gut microbiota: Mechanisms of intestinal immunomodulation and neuromodulation. Therap. Adv. Gastroenterol. 2013, 6, 39–51. [Google Scholar] [CrossRef]
- Shen, Z.H.; Zhu, C.X.; Quan, Y.S.; Yang, Z.Y.; Wu, S.; Luo, W.W.; Tan, B.; Wang, X.Y. Relationship between intestinal microbiota and ulcerative colitis: Mechanisms and clinical application of probiotics and fecal microbiota transplantation. World J. Gastroenterol. 2018, 24, 5–14. [Google Scholar] [CrossRef]
- Hou, K.; Wu, Z.X.; Chen, X.Y.; Wang, J.Q.; Zhang, D.; Xiao, C.; Zhu, D.; Koya, J.B.; Wei, L.; Li, J.; et al. Microbiota in health and diseases. Sig. Transduct. Target Ther. 2022, 7, 135. [Google Scholar] [CrossRef]
- Farias, D.P.; Araújo, F.F.; Neri-Numa, I.A.; Pastore, G.M. Prebiotics: Trends in food, health and technological applications. Trends Food Sci. Technol. 2019, 93, 23–35. [Google Scholar] [CrossRef]
- Mohanty, D.; Misra, S.; Mohapatra, S.; Sahu, P.S. Prebiotics and synbiotics: Recent concepts in nutrition. Food Biosci. 2018, 26, 152–160. [Google Scholar] [CrossRef]
- Chi, C.; Buys, N.; Li, C.; Sun, J.; Yin, C. Effects of prebiotics on sepsis, necrotizing enterocolitis, mortality, feeding intolerance, time to full enteral feeding, length of hospital stay, and stool frequency in preterm infants: A meta-analysis. Eur. J. Clin. Nutr. 2019, 73, 657–670. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Xu, L.; Yang, Z.; Wang, D.; Li, T.; Yang, F.; Li, Z.; Bai, X.; Wang, Y. Gut-muscle axis and sepsis-induced myopathy: The potential role of gut microbiota. Biomed. Pharmacother. 2023, 163, 114837. [Google Scholar] [CrossRef] [PubMed]
- Veneziano, C.; Marascio, N.; De Marco, C.; Quaresima, B.; Biamonte, F.; Trecarichi, E.M.; Santamaria, G.; Quirino, A.; Torella, D.; Quattrone, A.; et al. The Spread of SARS-CoV-2 Omicron Variant in CALABRIA: A Spatio-Temporal Report of Viral Genome Evolution. Viruses 2023, 15, 408. [Google Scholar] [CrossRef] [PubMed]
- Shoji, F.; Yamashita, T.; Kinoshita, F.; Takamori, S.; Fujishita, T.; Toyozawa, R.; Ito, K.; Yamazaki, K.; Nakashima, N.; Okamoto, T. Artificial intelligence-derived gut microbiome as a predictive biomarker for therapeutic response to immunotherapy in lung cancer: Protocol for a multicentre, prospective, observational study. BMJ Open 2022, 12, e061674. [Google Scholar] [CrossRef] [PubMed]
- Iadanza, E.; Fabbri, R.; Bašić-ČiČak, D.; Amedei, A.; Telalovic, J.H. Gut microbiota and artificial intelligence approaches: A scoping review. Health Technol. 2020, 10, 1343–1358. [Google Scholar] [CrossRef]
- Vilne, B.; Ķibilds, J.; Siksna, I.; Lazda, I.; Valciņa, O.; Krūmiņa, A. Could Artificial Intelligence/Machine Learning and Inclusion of Diet-Gut Microbiome Interactions Improve Disease Risk Prediction? Case Study: Coronary Artery Disease. Front. Microbiol. 2022, 13, 627892. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Size | Variable Region Sequenced | NGS Platform | Sequencing Performance by Taxonomy | References |
|---|---|---|---|---|
| 172 healthy subjects | V1-V3, V3-V4 and V4. | MiSeq (Illumina, CA, USA); PacBio (Pacific Biosciences, CA, USA). | Higher discrimination of Ruminococcaceae and Sphingomonas (V1-3), Akkermansia (V3-4), Haemophilus, Methanobrevibacter, and Citrobacter taxa (V4). | Whon TW. et al., 2018 [44]. |
| 33 healthy subjects | V3-V4 and V4-V5. | MiSeq (Illumina, CA, USA). | Higher discrimination of Actinomyces, Alistipes, Bacteroides, Cellulosimicrobium, Parabacteroides, and Flavonifractor genera (V3-V4). | Abellan-Schneyder I. et al., 2021 [45]. |
| 192 healthy subjects | V1-V2 and V3-V4. | MiSeq (Illumina, CA, USA). | Higher discrimination of Bacteroidetes, Proteobacteria, and Actinobacteria phyla (V1-V2). | Kameoka S. et al., 2021 [46]. |
| 5 healthy subjects | V1-V2, V3-V4 and V4. | MiSeq (Illumina, CA, USA). | Lower discrimination of Bifidobacteriales and higher discrimination of Enterobacteriales and Erysipelotrichales (V1-V2); higher discrimination of Clostridiales and lower discrimination of Bacteroidales, Betaproteobacteriales, Choriobacteriales, and Pasteurellales (V3-V4); discrimination of MollicutesRF39 (V4). | Chen Z. et al., 2019 [47]. |
| 15 septic shock patients vs. 15 healthy control subjects | V3-V4. | MiSeq (Illumina, CA, USA). | Higher abundance of Proteobacteria and Fusobacteria in septic shock patients compared to healthy control subjects. | Wan YD et al., 2018 [48]. |
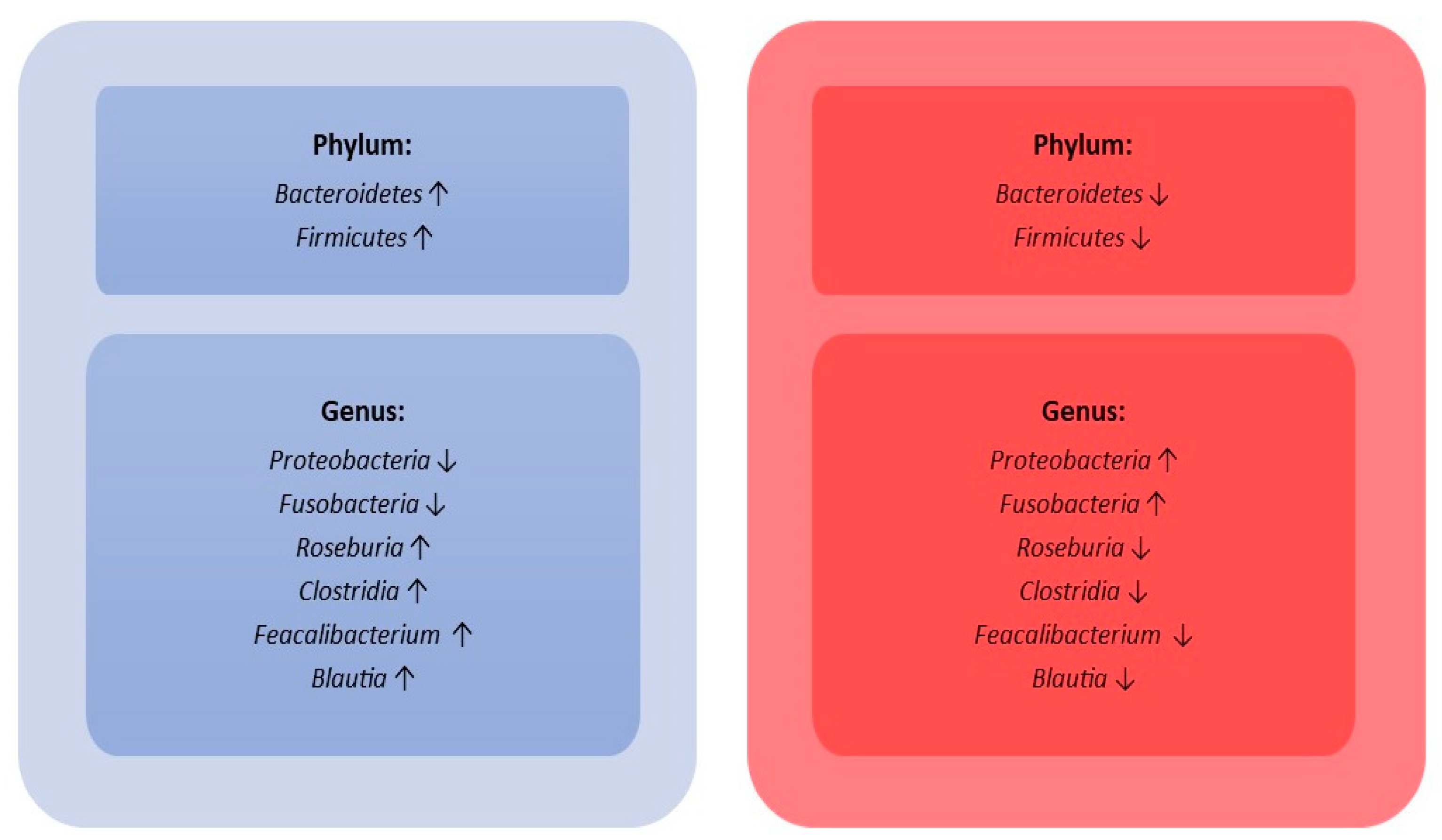
| 25 septic children vs. 15 healthy control subjects | V3-V4. | HiSeq (Illumina, CA, USA). | Gut microbiota diversity in septic children lower than healthy control subjects. Higher abundance of Acinetobacter and Enterococcus and lower abundance of Roseburia, Bacteroides, Clostridia, Faecalibacterium, and Blautia in septic children compared to healthy control subjects. | Du B. et al., 2021 [49] |
| 34 critically ill patients (25 septic patients and 9 without septic diagnosis) vs. 15 healthy control subjects | V1-V2. | MiSeq (Illumina, CA, USA). | Firmicutes and Bacteroidetes Phyla constituted <89% of all bacteria. Faecalibacterium, Blautia, Ruminococcus, Subdoligranulum, and Pseudobutyrivibrio were the most dominant genera. | Lankelma JM et al., 2017 [12] |
| 131 septic patients vs. 264 healthy control subjects (E1 group); 129 septic patients vs. 26 healthy control subjects (E2 group). | V3-V4. | MiSeq (Illumina, CA, USA). | Higher abundance of Bacteroides in septic patients of E1 group with respect to E2 group and higher abundance of Enterococcus in septic patients of E2 group with respect to E1 group. | Liu W. et al., 2020 [50] |
| Antibiotics | Microbiota Composition | ||
|---|---|---|---|
| Gram-Positive | Gram-Negative | Anaerobes | |
| Cetriaxone | increase | largest decrease | no changes |
| Amoxicillin | increase | increase | no changes |
| Clindamycin | increase | increase | largest decrease |
| Fluoroquinolones | no changes | largest decrease | no changes |
| Metronidazole | no changes | no changes | no changes |
| Macrolide | decrease | increase | decrease |
| Vancomycin | increase or decrease | no changes | decrease |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marascio, N.; Scarlata, G.G.M.; Romeo, F.; Cicino, C.; Trecarichi, E.M.; Quirino, A.; Torti, C.; Matera, G.; Russo, A. The Role of Gut Microbiota in the Clinical Outcome of Septic Patients: State of the Art and Future Perspectives. Int. J. Mol. Sci. 2023, 24, 9307. https://doi.org/10.3390/ijms24119307
Marascio N, Scarlata GGM, Romeo F, Cicino C, Trecarichi EM, Quirino A, Torti C, Matera G, Russo A. The Role of Gut Microbiota in the Clinical Outcome of Septic Patients: State of the Art and Future Perspectives. International Journal of Molecular Sciences. 2023; 24(11):9307. https://doi.org/10.3390/ijms24119307
Chicago/Turabian StyleMarascio, Nadia, Giuseppe Guido Maria Scarlata, Francesco Romeo, Claudia Cicino, Enrico Maria Trecarichi, Angela Quirino, Carlo Torti, Giovanni Matera, and Alessandro Russo. 2023. "The Role of Gut Microbiota in the Clinical Outcome of Septic Patients: State of the Art and Future Perspectives" International Journal of Molecular Sciences 24, no. 11: 9307. https://doi.org/10.3390/ijms24119307
APA StyleMarascio, N., Scarlata, G. G. M., Romeo, F., Cicino, C., Trecarichi, E. M., Quirino, A., Torti, C., Matera, G., & Russo, A. (2023). The Role of Gut Microbiota in the Clinical Outcome of Septic Patients: State of the Art and Future Perspectives. International Journal of Molecular Sciences, 24(11), 9307. https://doi.org/10.3390/ijms24119307