


Transcriptome Profiling of Prostate Cancer, Considering Risk Groups and the TMPRSS2-ERG Molecular Subtype

 ,
,  ,
,  , , , , , and
, , , , , and
Abstract
1. Introduction
2. Results
2.1. Differentially Expressed Genes among Risk Groups within Localized PCa
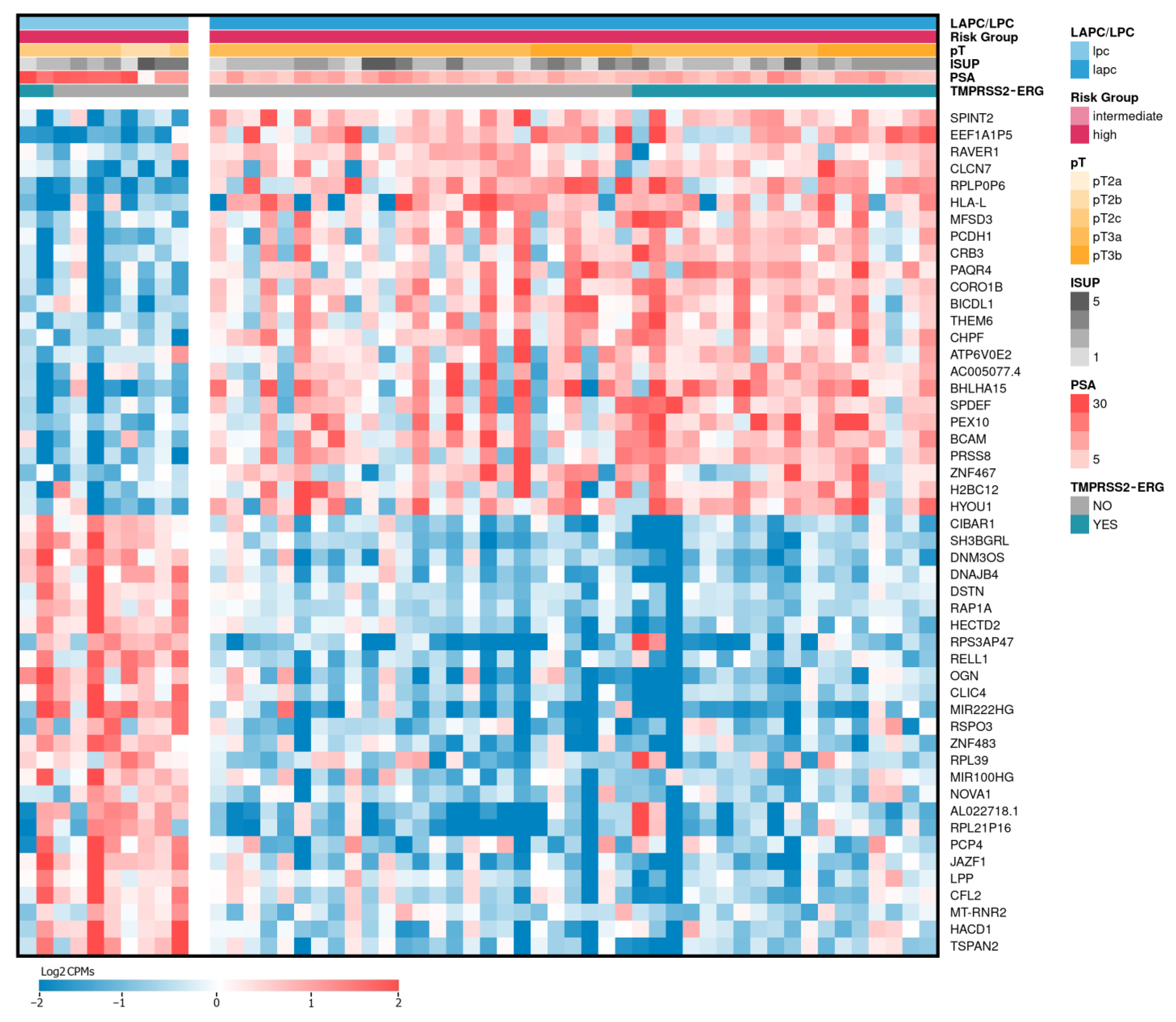
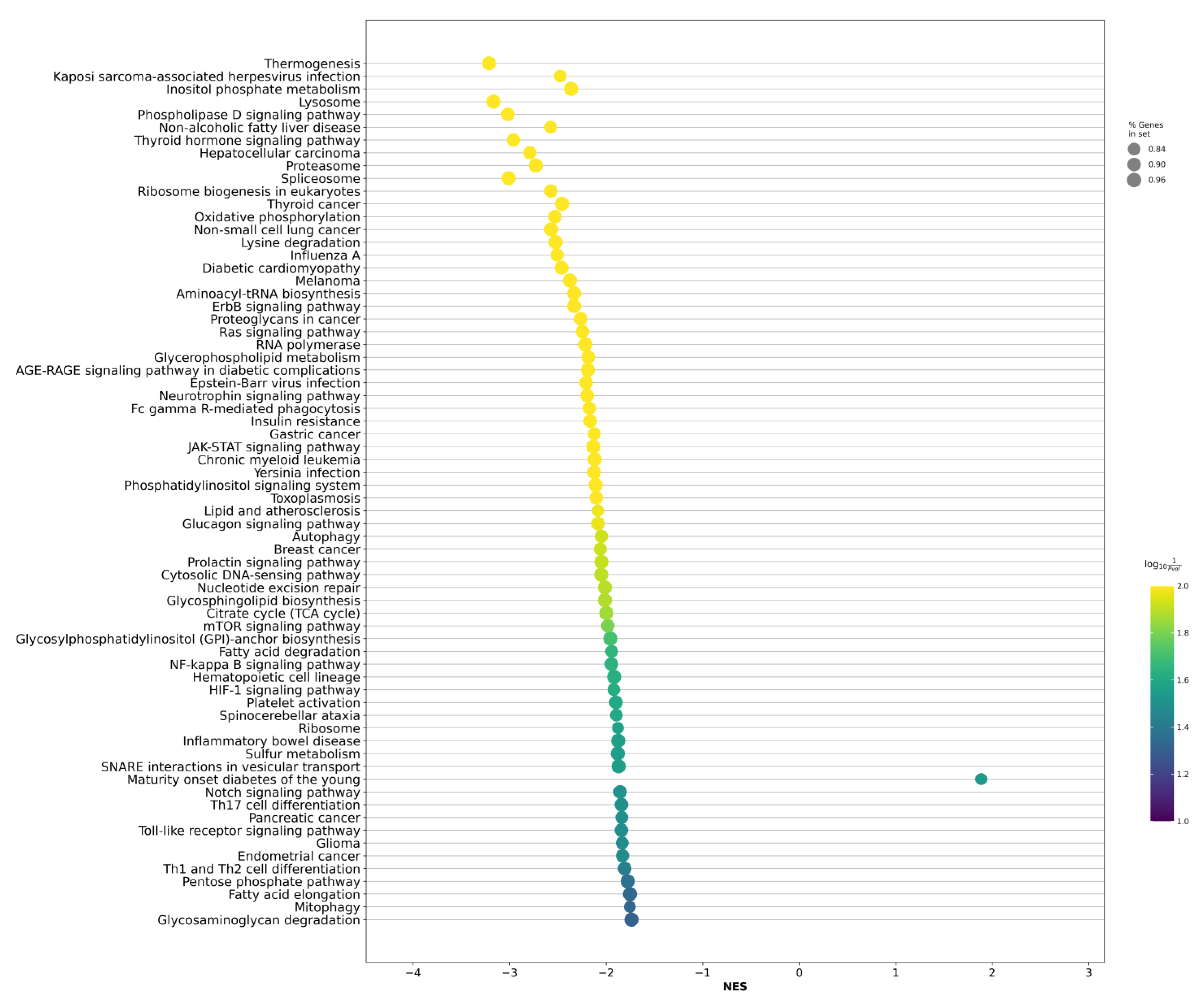
2.2. Differentially Expressed Genes between LAPCa and LPCa for High-Risk Group
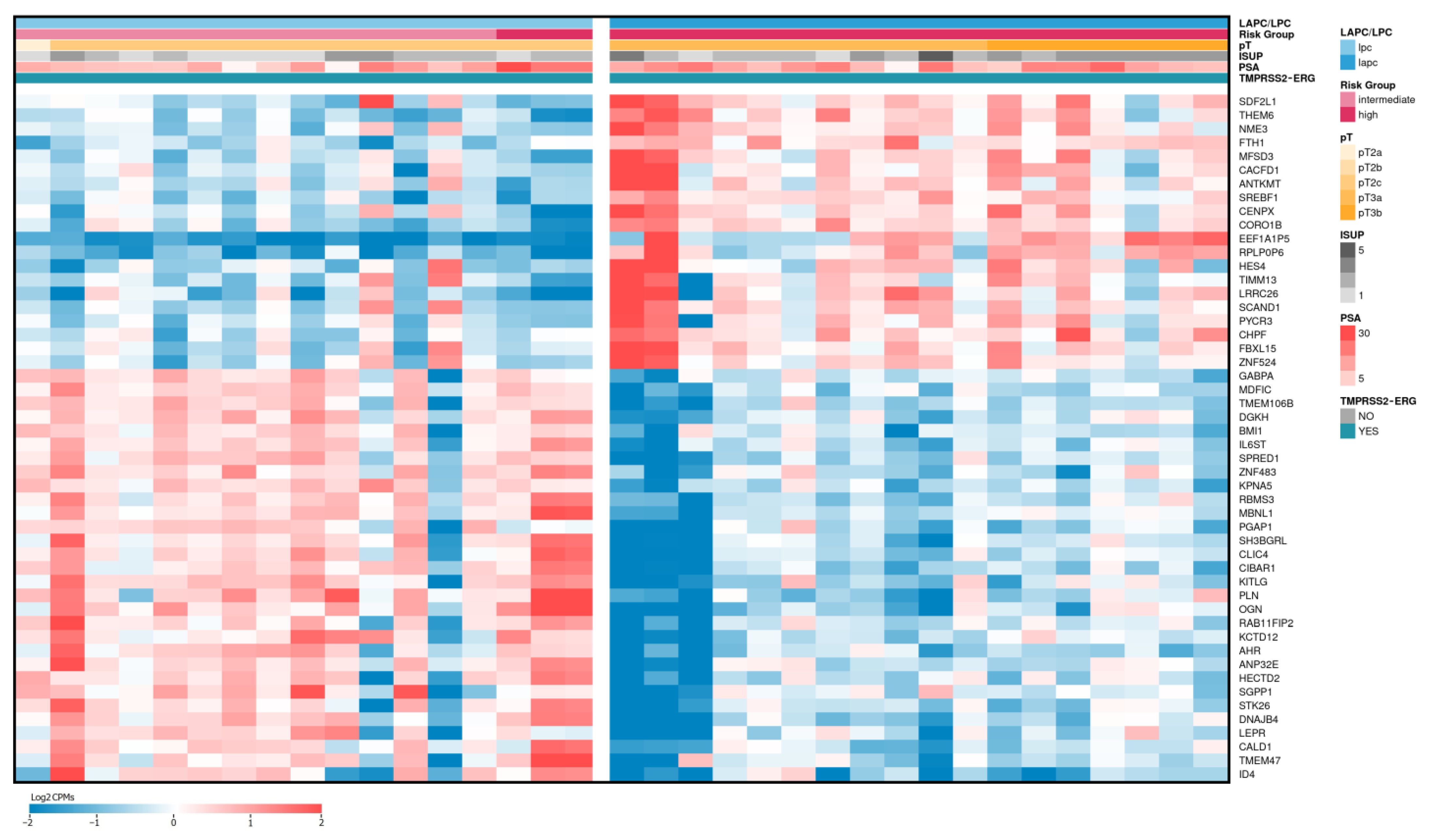
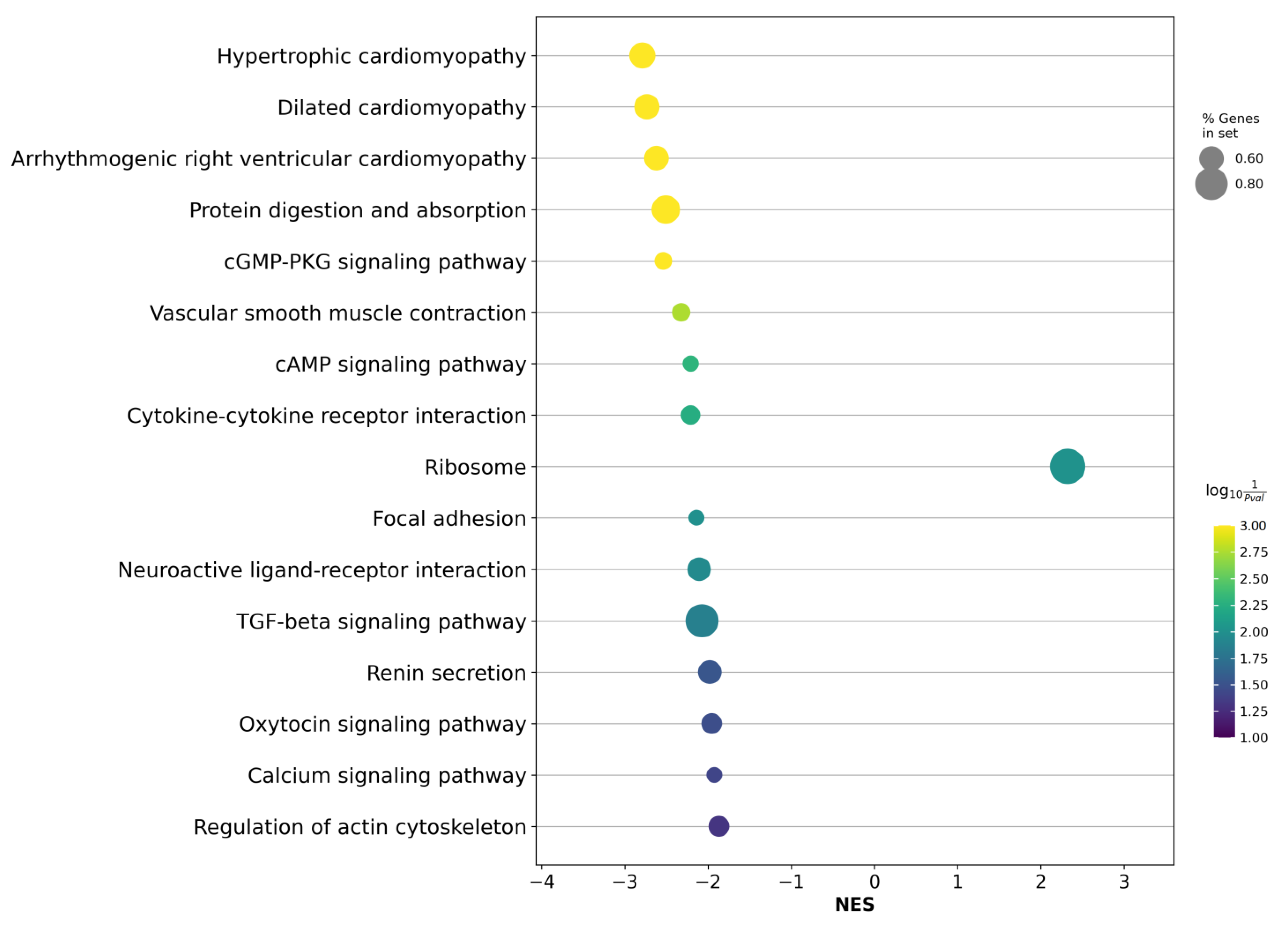
2.3. Differentially Expressed Genes between LAPCa and LPCa within the TMPRSS2-ERG Molecular Subtype
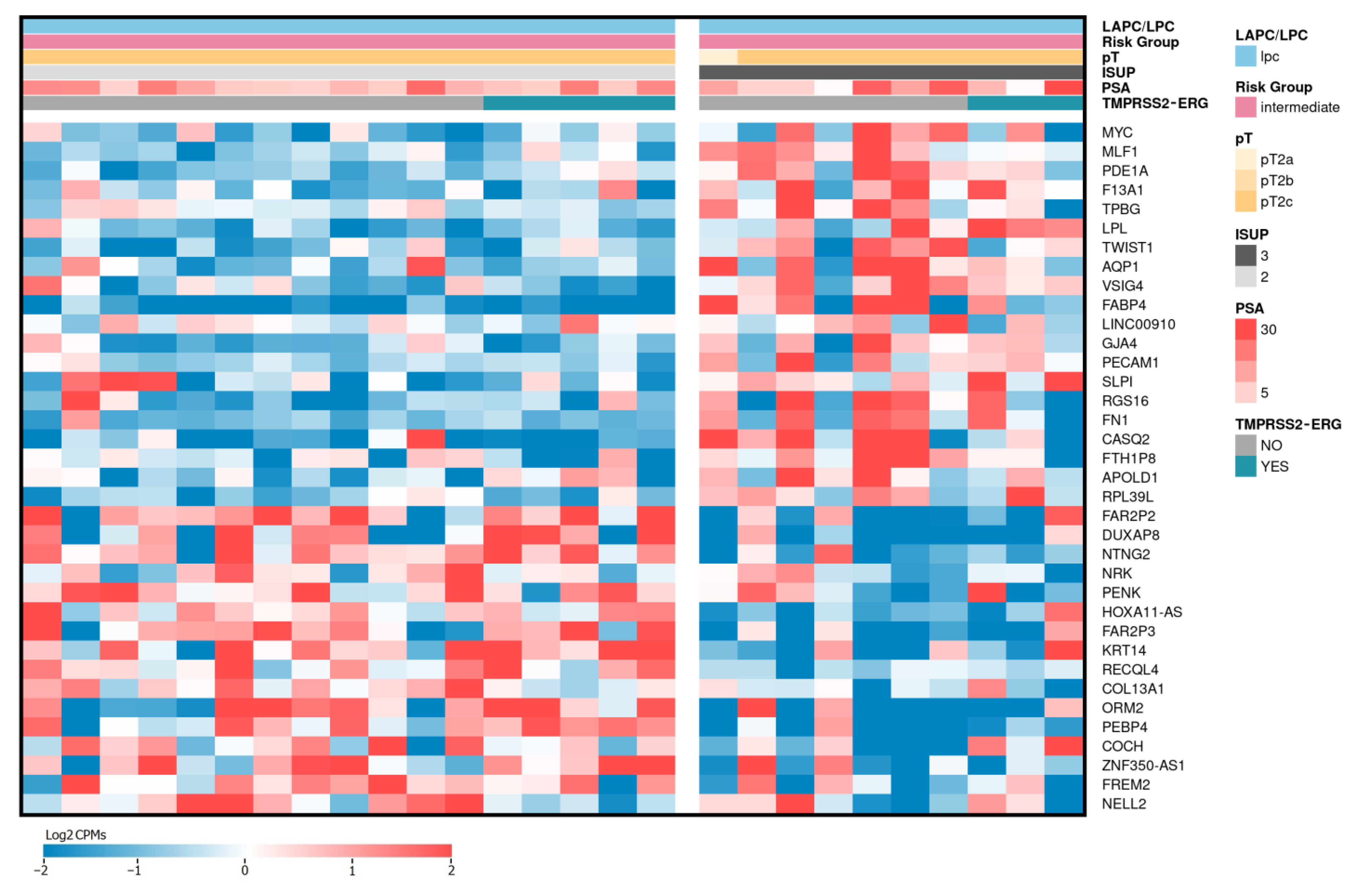
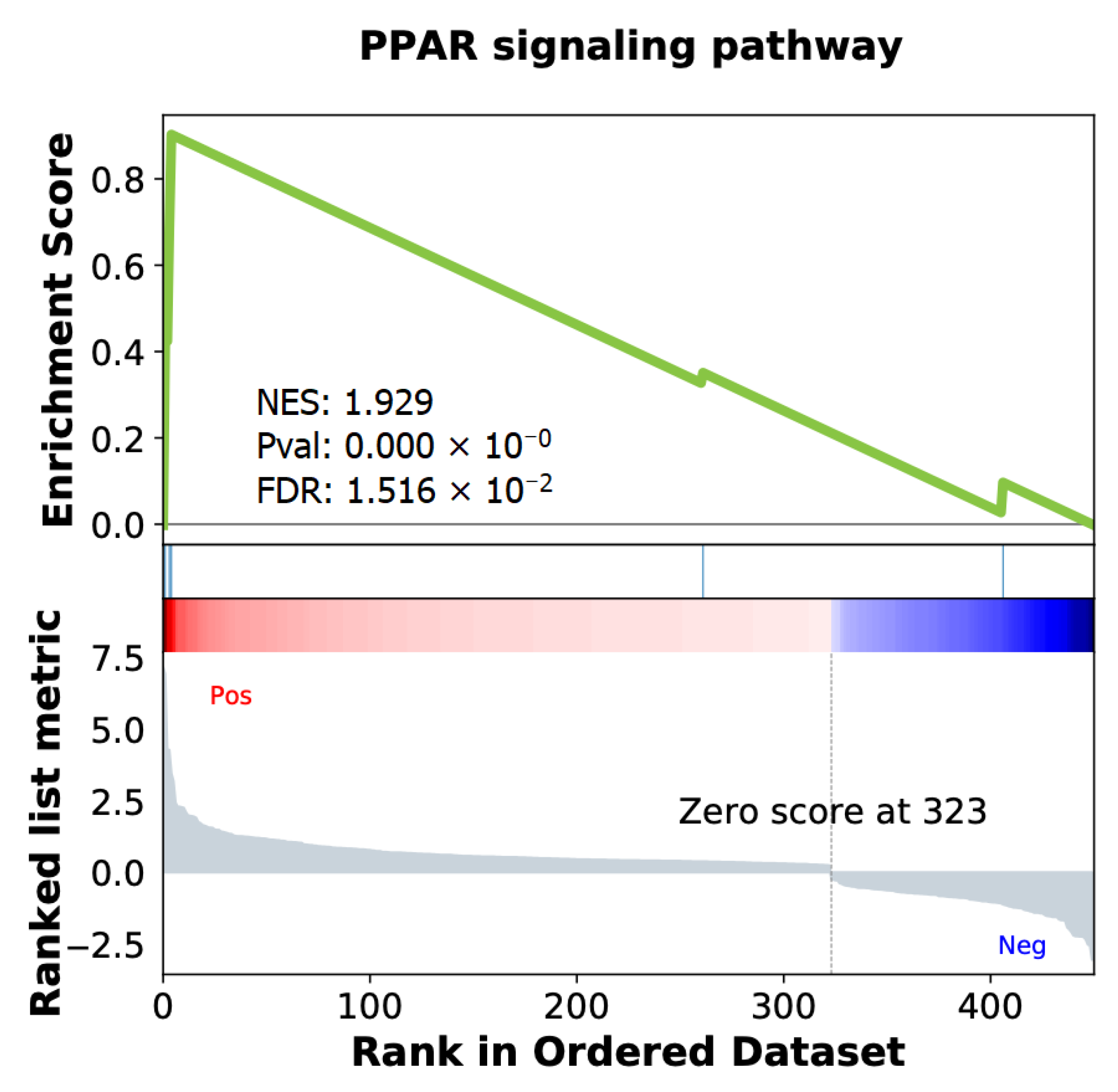
2.4. Differentially Expressed Genes Associated with ISUP 3 at Intermediate Risk for Localized PCa
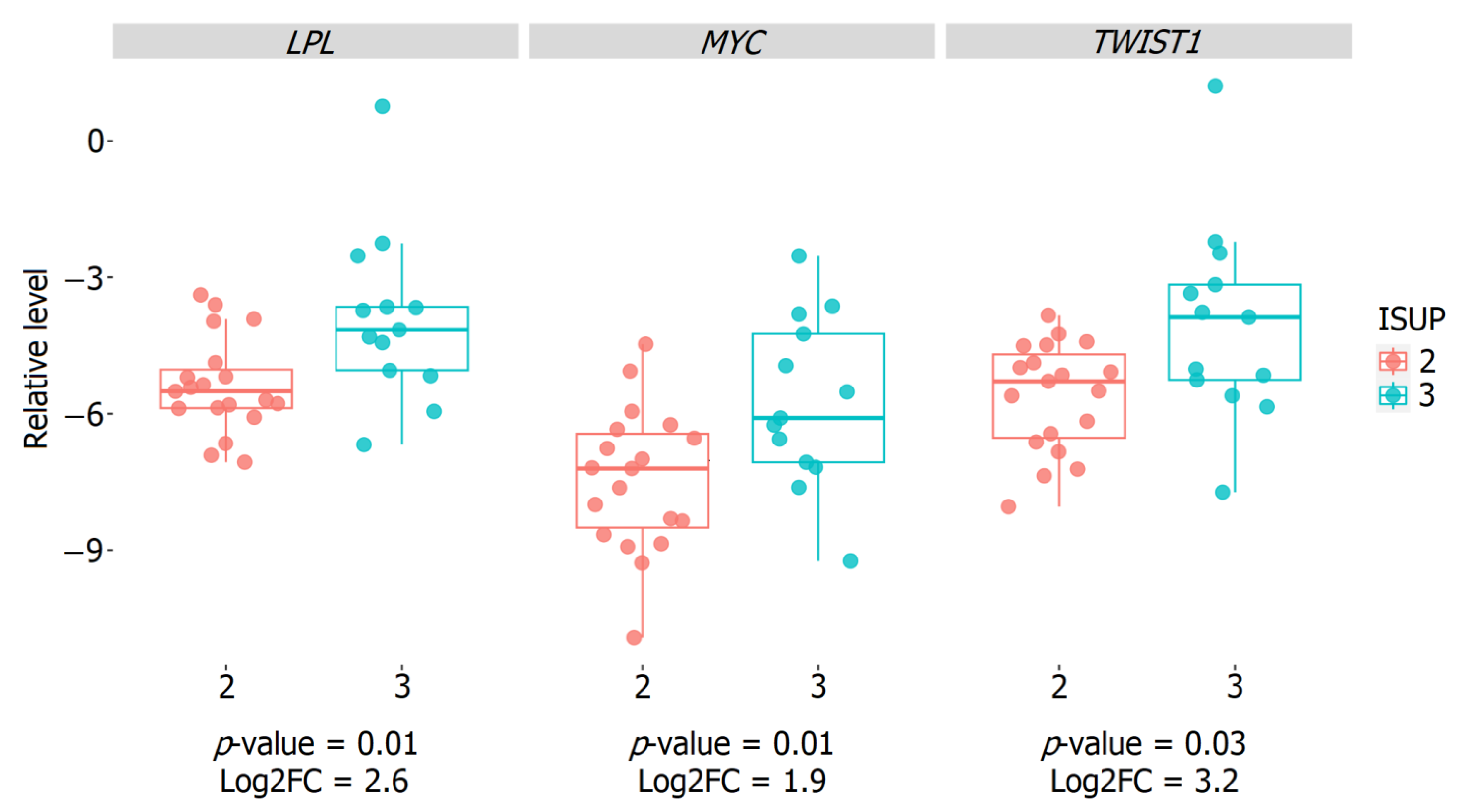
2.5. Validation of the Relative Expression of Genes Associated with the ISUP 3 Group in the Intermediate-Risk LPCa Group of Russian Patients
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Methods
4.2.1. Total RNA Isolation, Library Preparation, and Next Generation Sequencing (NGS)
4.2.2. Bioinformatic Analysis
4.2.3. Reverse Transcription and Quantitative PCR (qPCR)
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pernar, C.H.; Ebot, E.M.; Wilson, K.M.; Mucci, L.A. The Epidemiology of Prostate Cancer. Cold Spring Harb. Perspect. Med. 2018, 8, a030361. [Google Scholar] [CrossRef] [PubMed]
- D’Amico, A.V.; Whittington, R.; Malkowicz, S.B.; Schultz, D.; Blank, K.; Broderick, G.A.; Tomaszewski, J.E.; Renshaw, A.A.; Kaplan, I.; Beard, C.J.; et al. Biochemical outcome after radical prostatectomy, external beam radiation therapy, or interstitial radiation therapy for clinically localized prostate cancer. JAMA 1998, 280, 969–974. [Google Scholar] [CrossRef] [PubMed]
- Bostrom, P.J.; Bjartell, A.S.; Catto, J.W.; Eggener, S.E.; Lilja, H.; Loeb, S.; Schalken, J.; Schlomm, T.; Cooperberg, M.R. Genomic Predictors of Outcome in Prostate Cancer. Eur. Urol. 2015, 68, 1033–1044. [Google Scholar] [CrossRef] [PubMed]
- Grasso, C.S.; Wu, Y.M.; Robinson, D.R.; Cao, X.; Dhanasekaran, S.M.; Khan, A.P.; Quist, M.J.; Jing, X.; Lonigro, R.J.; Brenner, J.C.; et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature 2012, 487, 239–243. [Google Scholar] [CrossRef]
- Taylor, B.S.; Schultz, N.; Hieronymus, H.; Gopalan, A.; Xiao, Y.; Carver, B.S.; Arora, V.K.; Kaushik, P.; Cerami, E.; Reva, B.; et al. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2010, 18, 11–22. [Google Scholar] [CrossRef]
- Barbieri, C.E.; Bangma, C.H.; Bjartell, A.; Catto, J.W.; Culig, Z.; Gronberg, H.; Luo, J.; Visakorpi, T.; Rubin, M.A. The mutational landscape of prostate cancer. Eur. Urol. 2013, 64, 567–576. [Google Scholar] [CrossRef]
- Haffner, M.C.; Mosbruger, T.; Esopi, D.M.; Fedor, H.; Heaphy, C.M.; Walker, D.A.; Adejola, N.; Gurel, M.; Hicks, J.; Meeker, A.K.; et al. Tracking the clonal origin of lethal prostate cancer. J. Clin. Investig. 2013, 123, 4918–4922. [Google Scholar] [CrossRef]
- Kumar, A.; White, T.A.; MacKenzie, A.P.; Clegg, N.; Lee, C.; Dumpit, R.F.; Coleman, I.; Ng, S.B.; Salipante, S.J.; Rieder, M.J.; et al. Exome sequencing identifies a spectrum of mutation frequencies in advanced and lethal prostate cancers. Proc. Natl. Acad. Sci. USA 2011, 108, 17087–17092. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. The Molecular Taxonomy of Primary Prostate Cancer. Cell 2015, 163, 1011–1025. [Google Scholar] [CrossRef]
- Arora, K.; Barbieri, C.E. Molecular Subtypes of Prostate Cancer. Curr. Oncol. Rep. 2018, 20, 58. [Google Scholar] [CrossRef]
- Kobelyatskaya, A.A.; Kudryavtsev, A.A.; Kudryavtseva, A.V.; Snezhkina, A.V.; Fedorova, M.S.; Kalinin, D.V.; Pavlov, V.S.; Guvatova, Z.G.; Naberezhnev, P.A.; Nyushko, K.M.; et al. ALDH3A2, ODF2, QSOX2, and MicroRNA-503-5p Expression to Forecast Recurrence in TMPRSS2-ERG-Positive Prostate Cancer. Int. J. Mol. Sci. 2022, 23, 11695. [Google Scholar] [CrossRef]
- Perner, S.; Mosquera, J.M.; Demichelis, F.; Hofer, M.D.; Paris, P.L.; Simko, J.; Collins, C.; Bismar, T.A.; Chinnaiyan, A.M.; De Marzo, A.M.; et al. TMPRSS2-ERG fusion prostate cancer: An early molecular event associated with invasion. Am. J. Surg. Pathol. 2007, 31, 882–888. [Google Scholar] [CrossRef]
- Adamo, P.; Ladomery, M.R. The oncogene ERG: A key factor in prostate cancer. Oncogene 2016, 35, 403–414. [Google Scholar] [CrossRef]
- Hagglof, C.; Hammarsten, P.; Stromvall, K.; Egevad, L.; Josefsson, A.; Stattin, P.; Granfors, T.; Bergh, A. TMPRSS2-ERG expression predicts prostate cancer survival and associates with stromal biomarkers. PLoS ONE 2014, 9, e86824. [Google Scholar] [CrossRef]
- Kim, S.H.; Kim, S.H.; Joung, J.Y.; Lee, G.K.; Hong, E.K.; Kang, K.M.; Yu, A.; Nam, B.H.; Chung, J.; Seo, H.K.; et al. Overexpression of ERG and Wild-Type PTEN Are Associated with Favorable Clinical Prognosis and Low Biochemical Recurrence in Prostate Cancer. PLoS ONE 2015, 10, e0122498. [Google Scholar] [CrossRef]
- Gerke, J.S.; Orth, M.F.; Tolkach, Y.; Romero-Perez, L.; Wehweck, F.S.; Stein, S.; Musa, J.; Knott, M.M.L.; Holting, T.L.B.; Li, J.; et al. Integrative clinical transcriptome analysis reveals TMPRSS2-ERG dependency of prognostic biomarkers in prostate adenocarcinoma. Int. J. Cancer 2020, 146, 2036–2046. [Google Scholar] [CrossRef]
- Zimta, A.A.; Sigurjonsson, O.E.; Gulei, D.; Tomuleasa, C. The Malignant Role of Exosomes as Nanocarriers of Rare RNA Species. Int. J. Mol. Sci. 2020, 21, 5866. [Google Scholar] [CrossRef]
- Carvalho, A.S.; Baeta, H.; Silva, B.C.; Moraes, M.C.S.; Bodo, C.; Beck, H.C.; Rodriguez, M.S.; Saraswat, M.; Pandey, A.; Matthiesen, R. Extra-cellular vesicles carry proteome of cancer hallmarks. Front. Biosci. 2020, 25, 398–436. [Google Scholar] [CrossRef]
- Sun, T.; Wang, X.; He, H.H.; Sweeney, C.J.; Liu, S.X.; Brown, M.; Balk, S.; Lee, G.S.; Kantoff, P.W. MiR-221 promotes the development of androgen independence in prostate cancer cells via downregulation of HECTD2 and RAB1A. Oncogene 2014, 33, 2790–2800. [Google Scholar] [CrossRef]
- Mirzaei, S.; Paskeh, M.D.A.; Saghari, Y.; Zarrabi, A.; Hamblin, M.R.; Entezari, M.; Hashemi, M.; Aref, A.R.; Hushmandi, K.; Kumar, A.P.; et al. Transforming growth factor-beta (TGF-beta) in prostate cancer: A dual function mediator? Int. J. Biol. Macromol. 2022, 206, 435–452. [Google Scholar] [CrossRef]
- Pudova, E.A.; Krasnov, G.S.; Nyushko, K.M.; Kobelyatskaya, A.A.; Savvateeva, M.V.; Poloznikov, A.A.; Dolotkazin, D.R.; Klimina, K.M.; Guvatova, Z.G.; Simanovsky, S.A.; et al. miRNAs expression signature potentially associated with lymphatic dissemination in locally advanced prostate cancer. BMC Med. Genom. 2020, 13, 129. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Wang, W.; Shi, G.; Hao, M.; Wang, Y.; Yao, M.; Huang, Y.; Du, L.; Zhang, X.; Ye, D.; et al. Targeting HIC1/TGF-beta axis-shaped prostate cancer microenvironment restrains its progression. Cell. Death Dis. 2022, 13, 624. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Kong, Q.; Wang, J.; Jiang, Y.; Hua, H. Complex roles of cAMP-PKA-CREB signaling in cancer. Exp. Hematol. Oncol. 2020, 9, 32. [Google Scholar] [CrossRef]
- Merkle, D.; Hoffmann, R. Roles of cAMP and cAMP-dependent protein kinase in the progression of prostate cancer: Cross-talk with the androgen receptor. Cell. Signal 2011, 23, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Rahman, M.A.; Chen, Z.G.; Shin, D.M. Multiple biological functions of Twist1 in various cancers. Oncotarget 2017, 8, 20380–20393. [Google Scholar] [CrossRef]
- Gajula, R.P.; Chettiar, S.T.; Williams, R.D.; Thiyagarajan, S.; Kato, Y.; Aziz, K.; Wang, R.; Gandhi, N.; Wild, A.T.; Vesuna, F.; et al. The twist box domain is required for Twist1-induced prostate cancer metastasis. Mol. Cancer Res. 2013, 11, 1387–1400. [Google Scholar] [CrossRef]
- Madden, S.K.; de Araujo, A.D.; Gerhardt, M.; Fairlie, D.P.; Mason, J.M. Taking the Myc out of cancer: Toward therapeutic strategies to directly inhibit c-Myc. Mol. Cancer 2021, 20, 3. [Google Scholar] [CrossRef]
- Rebello, R.J.; Pearson, R.B.; Hannan, R.D.; Furic, L. Therapeutic Approaches Targeting MYC-Driven Prostate Cancer. Genes 2017, 8, 71. [Google Scholar] [CrossRef]
- Pingitore, P.; Lepore, S.M.; Pirazzi, C.; Mancina, R.M.; Motta, B.M.; Valenti, L.; Berge, K.E.; Retterstol, K.; Leren, T.P.; Wiklund, O.; et al. Identification and characterization of two novel mutations in the LPL gene causing type I hyperlipoproteinemia. J. Clin. Lipidol. 2016, 10, 816–823. [Google Scholar] [CrossRef]
- Kuemmerle, N.B.; Rysman, E.; Lombardo, P.S.; Flanagan, A.J.; Lipe, B.C.; Wells, W.A.; Pettus, J.R.; Froehlich, H.M.; Memoli, V.A.; Morganelli, P.M.; et al. Lipoprotein lipase links dietary fat to solid tumor cell proliferation. Mol. Cancer Ther. 2011, 10, 427–436. [Google Scholar] [CrossRef]
- Wang, W.; Bai, L.; Li, W.; Cui, J. The Lipid Metabolic Landscape of Cancers and New Therapeutic Perspectives. Front. Oncol. 2020, 10, 605154. [Google Scholar] [CrossRef]
- Gouw, A.M.; Margulis, K.; Liu, N.S.; Raman, S.J.; Mancuso, A.; Toal, G.G.; Tong, L.; Mosley, A.; Hsieh, A.L.; Sullivan, D.K.; et al. The MYC Oncogene Cooperates with Sterol-Regulated Element-Binding Protein to Regulate Lipogenesis Essential for Neoplastic Growth. Cell. Metab. 2019, 30, 556–572.e55. [Google Scholar] [CrossRef]
- Moch, H.; Cubilla, A.L.; Humphrey, P.A.; Reuter, V.E.; Ulbright, T.M. The 2016 WHO Classification of Tumours of the Urinary System and Male Genital Organs-Part A: Renal, Penile, and Testicular Tumours. Eur. Urol. 2016, 70, 93–105. [Google Scholar] [CrossRef]
- Kobelyatskaya, A.A.; Pudova, E.A.; Snezhkina, A.V.; Fedorova, M.S.; Pavlov, V.S.; Guvatova, Z.G.; Savvateeva, M.V.; Melnikova, N.V.; Dmitriev, A.A.; Trofimov, D.Y.; et al. Impact TMPRSS2-ERG Molecular Subtype on Prostate Cancer Recurrence. Life 2021, 11, 588. [Google Scholar] [CrossRef]
- Pudova, E.A.; Kobelyatskaya, A.A.; Katunina, I.V.; Snezhkina, A.V.; Fedorova, M.S.; Pavlov, V.S.; Bakhtogarimov, I.R.; Lantsova, M.S.; Kokin, S.P.; Nyushko, K.M.; et al. Lymphatic Dissemination in Prostate Cancer: Features of the Transcriptomic Profile and Prognostic Models. Int. J. Mol. Sci. 2023, 24, 2418. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Fang, Z.; Liu, X.; Peltz, G. GSEApy: A comprehensive package for performing gene set enrichment analysis in Python. Bioinformatics 2023, 39, btac757. [Google Scholar] [CrossRef]
- Senchenko, V.N.; Krasnov, G.S.; Dmitriev, A.A.; Kudryavtseva, A.V.; Anedchenko, E.A.; Braga, E.A.; Pronina, I.V.; Kondratieva, T.T.; Ivanov, S.V.; Zabarovsky, E.R.; et al. Differential expression of CHL1 gene during development of major human cancers. PLoS ONE 2011, 6, e15612. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name (Biotype) | Description | LAPCa vs. LPCa | LAPCa vs. LPCa (TMPRSS2-ERG) | ||
|---|---|---|---|---|---|
| Log2FC | rs | Log2FC | rs | ||
| BHLHA15 (protein coding) | basic helix-loop-helix family member a15 | 1.79 | 0.57 | 1.40 | 0.68 |
| CIBAR1 (protein coding) | CBY1 interacting BAR domain containing 1 | −1.05 | −0.65 | −1.20 | −0.81 |
| CLIC4 (protein coding) | chloride intracellular channel 4 | −1.02 | −0.53 | −1.14 | −0.81 |
| CORO1B (protein coding) | coronin 1B | 1.17 | 0.63 | 1.04 | 0.77 |
| CRB3 (protein coding) | crumbs cell polarity complex component 3 | 1.15 | 0.58 | 1.03 | 0.72 |
| DNAJB4 (protein coding) | DnaJ heat shock protein family (Hsp40) member B4 | −1.13 | −0.62 | −1.22 | −0.80 |
| DNM3OS (lncRNA) | DNM3 opposite strand/antisense RNA | −1.22 | −0.62 | −1.58 | −0.75 |
| EEF1A1P5 (processed pseudogene) | eukaryotic translation elongation factor 1 alpha 1 pseudogene 5 | 2.45 | 0.77 | 2.70 | 0.87 |
| HECTD2 (protein coding) | HECT domain E3 ubiquitin protein ligase 2 | −1.05 | −0.63 | −1.04 | −0.77 |
| ID4 (protein coding) | inhibitor of DNA binding 4, HLH protein | −1.34 | −0.55 | 5.85 | −0.67 |
| MFSD3 (protein coding) | major facilitator superfamily domain containing 3 | 1.10 | 0.61 | 1.09 | 0.73 |
| MIR222HG (lncRNA) | miR222/221 cluster host gene | −1.27 | −0.56 | −1.27 | −0.60 |
| OGN (protein coding) | osteoglycin | −1.10 | −0.54 | −1.62 | −0.76 |
| RPLP0P6 (processed pseudogene) | ribosomal protein lateral stalk subunit P0 pseudogene 6 | 1.83 | 0.70 | 1.94 | 0.77 |
| SH3BGRL (protein coding) | SH3 domain binding glutamate rich protein like | −1.06 | −0.63 | −1.12 | −0.76 |
| ZNF483 (protein coding) | zinc finger protein 483 | −1.02 | −0.58 | −1.15 | −0.78 |
| Gene Name | LAPCa vs. LPCa | LAPCa vs. LPCa (TMPRSS2-ERG) | ||||
|---|---|---|---|---|---|---|
| AUC (CI, 95%) | Accuracy | p-Value | AUC (CI, 95%) | Accuracy | p-Value | |
| BHLHA15 | 0.864 (0.581–0.964) | 0.750 | 6.0 × 10−6 | 0.867 (0.820–1.000) | 0.708 | 1.2 × 10−2 |
| CIBAR1 | 0.821 (0.701–1.000) | 0.708 | 3.0 × 10−6 | 0.979 (0.877–1.000) | 0.917 | 8.8 × 10−3 |
| CLIC4 | 0.711 (0.611–0.985) | 0.750 | 8.1 × 10−5 | 0.930 (0.891–1.000) | 0.833 | 8.5 × 10−3 |
| CORO1B | 0.830 (0.701–1.000) | 0.750 | 1.0 × 10−6 | 0.889 (0.897–1.000) | 0.792 | 2.6 × 10−2 |
| CRB3 | 0.798 (0.661–1.000) | 0.750 | 2.0 × 10−6 | 0.734 (0.686–1.000) | 0.542 | 6.0 × 10−3 |
| DNAJB4 | 0.867 (0.654–0.992) | 0.792 | 1.0 × 10−5 | 0.824 (0.790–1.000) | 0.708 | 8.4 × 10−3 |
| DNM3OS | 0.867 (0.718–1.000) | 0.792 | 1.2 × 10−5 | 0.824 (0.807–1.000) | 0.708 | 9.7 × 10−3 |
| EEF1A1P5 | 0.979 (0.904–1.000) | 0.917 | 6.5 × 10−5 | 1.000 (0.977–1.000) | 0.875 | 2.9 × 10−2 |
| HECTD2 | 0.815 (0.691–1.000) | 0.708 | 6.0 × 10−6 | 0.977 (0.947–1.000) | 0.875 | 5.7 × 10−3 |
| ID4 | 0.832 (0.486–0.924) | 0.750 | 3.9 × 10−5 | 0.811 (0.592–1.000) | 0.750 | 8.3 × 10−3 |
| MFSD3 | 0.837 (0.604–0.992) | 0.667 | 2.0 × 10−5 | 0.874 (0.754–1.000) | 0.708 | 8.5 × 10−3 |
| MIR222HG | 0.778 (0.512–0.950) | 0.708 | 1.3 × 10−4 | 0.879 (0.763–1.000) | 0.750 | 4.1 × 10−2 |
| OGN | 0.764 (0.541–0.988) | 0.708 | 3.9 × 10−5 | 0.963 (0.797–1.000) | 0.875 | 1.6 × 10−2 |
| RPLP0P6 | 0.930 (0.745–1.000) | 0.792 | 5.2 × 10−6 | 0.984 (0.764–1.000) | 0.958 | 2.3 × 10−2 |
| SH3BGRL | 0.857 (0.581–0.981) | 0.792 | 6.0 × 10−6 | 0.818 (0.757–1.000) | 0.792 | 9.2 × 10−3 |
| ZNF483 | 0.879 (0.501–0.944) | 0.750 | 1.5 × 10−5 | 0.914 (0.891–1.000) | 0.708 | 7.3 × 10−3 |
| Criterion | LPCa, n | LAPCa, n | |
|---|---|---|---|
| PCa samples | 58 | 43 | |
| Age, years | 64 (41–78) | 64 (46–77) | |
| pT | pT2a | 3 | 0 |
| pT2b | 5 | 0 | |
| pT2c | 50 | 0 | |
| pT3a | 0 | 30 | |
| pT3b | 0 | 13 | |
| pT4 | 0 | 0 | |
| pN | pN0 | 58 | 43 |
| pN1 | 0 | 0 | |
| cM | cM0 | 58 | 43 |
| cM1 | 0 | 0 | |
| Gleason score | 6 | 22 | 6 |
| 7 | 32 | 29 | |
| 8 | 2 | 5 | |
| 9 | 1 | 3 | |
| 10 | 0 | 0 | |
| ISUP grade | 1 | 22 | 6 |
| 2 | 20 | 17 | |
| 3 | 12 | 12 | |
| 4 | 2 | 5 | |
| 5 | 1 | 3 | |
| PSA, ng/mL | 11.1 (0.3–30) | 12.8 (3.4–27.6) | |
| Risk group | Low | 1 | 0 |
| Intermediate | 47 | 0 | |
| High | 10 | 43 | |
| Molecular subtype TMPRSS2-ERG | Yes | 16 | 18 |
| No | 42 | 25 | |
| mRNA | Primer Sequence (5′→3′) | Product Length, b.p. |
|---|---|---|
| LPL | F: CAGCCCTACCCTTGTTAGTTATT R: ACGTTGGAGGATGTGCTATTT | 103 |
| MYC | F: ATCTCTGGGAGGAATGCTACTA R: ATCTGCGTGGCTACAGATAAG | 95 |
| TWIST | F: CGGAGACCTAGATGTCATTGTTT R: ACGCCCTGTTTCTTTGAATTTG | 146 |
| PUM1 | F: TGGACCATTTCGCCCTTTAG R: CAGAGAGTTGTTGCCGTAGAA | 103 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kobelyatskaya, A.A.; Pudova, E.A.; Katunina, I.V.; Snezhkina, A.V.; Fedorova, M.S.; Pavlov, V.S.; Kotelnikova, A.O.; Nyushko, K.M.; Alekseev, B.Y.; Krasnov, G.S.; et al. Transcriptome Profiling of Prostate Cancer, Considering Risk Groups and the TMPRSS2-ERG Molecular Subtype. Int. J. Mol. Sci. 2023, 24, 9282. https://doi.org/10.3390/ijms24119282
Kobelyatskaya AA, Pudova EA, Katunina IV, Snezhkina AV, Fedorova MS, Pavlov VS, Kotelnikova AO, Nyushko KM, Alekseev BY, Krasnov GS, et al. Transcriptome Profiling of Prostate Cancer, Considering Risk Groups and the TMPRSS2-ERG Molecular Subtype. International Journal of Molecular Sciences. 2023; 24(11):9282. https://doi.org/10.3390/ijms24119282
Chicago/Turabian StyleKobelyatskaya, Anastasiya A., Elena A. Pudova, Irina V. Katunina, Anastasiya V. Snezhkina, Maria S. Fedorova, Vladislav S. Pavlov, Anastasiya O. Kotelnikova, Kirill M. Nyushko, Boris Y. Alekseev, George S. Krasnov, and et al. 2023. "Transcriptome Profiling of Prostate Cancer, Considering Risk Groups and the TMPRSS2-ERG Molecular Subtype" International Journal of Molecular Sciences 24, no. 11: 9282. https://doi.org/10.3390/ijms24119282
APA StyleKobelyatskaya, A. A., Pudova, E. A., Katunina, I. V., Snezhkina, A. V., Fedorova, M. S., Pavlov, V. S., Kotelnikova, A. O., Nyushko, K. M., Alekseev, B. Y., Krasnov, G. S., & Kudryavtseva, A. V. (2023). Transcriptome Profiling of Prostate Cancer, Considering Risk Groups and the TMPRSS2-ERG Molecular Subtype. International Journal of Molecular Sciences, 24(11), 9282. https://doi.org/10.3390/ijms24119282