
Genome Assembly of a Relict Arabian Species of Daphnia O. F. Müller (Crustacea: Cladocera) Adapted to the Desert Life

 , , ,
, , , 
Abstract
1. Introduction
2. Results
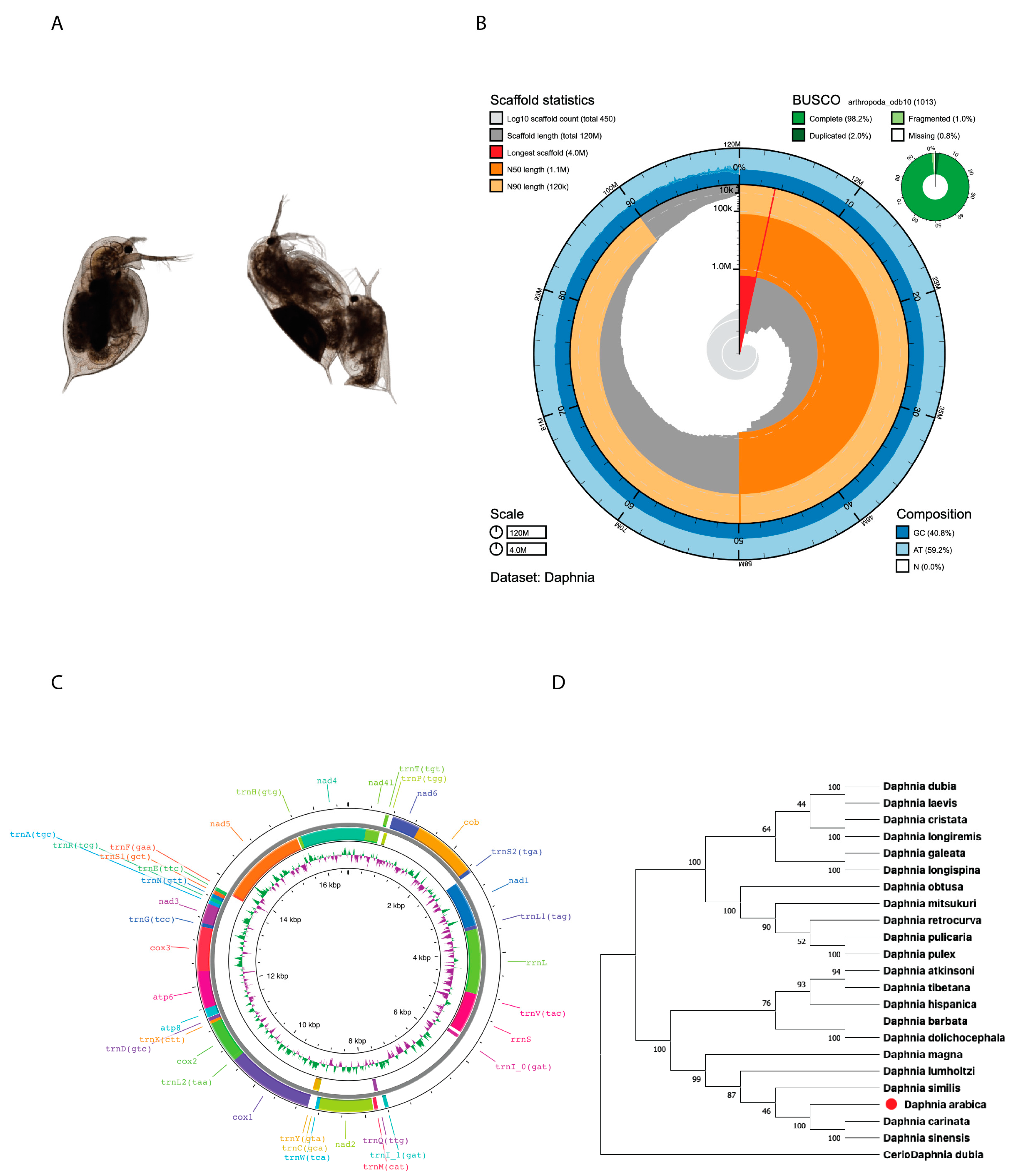
2.1. Genome Assembly and Characterization
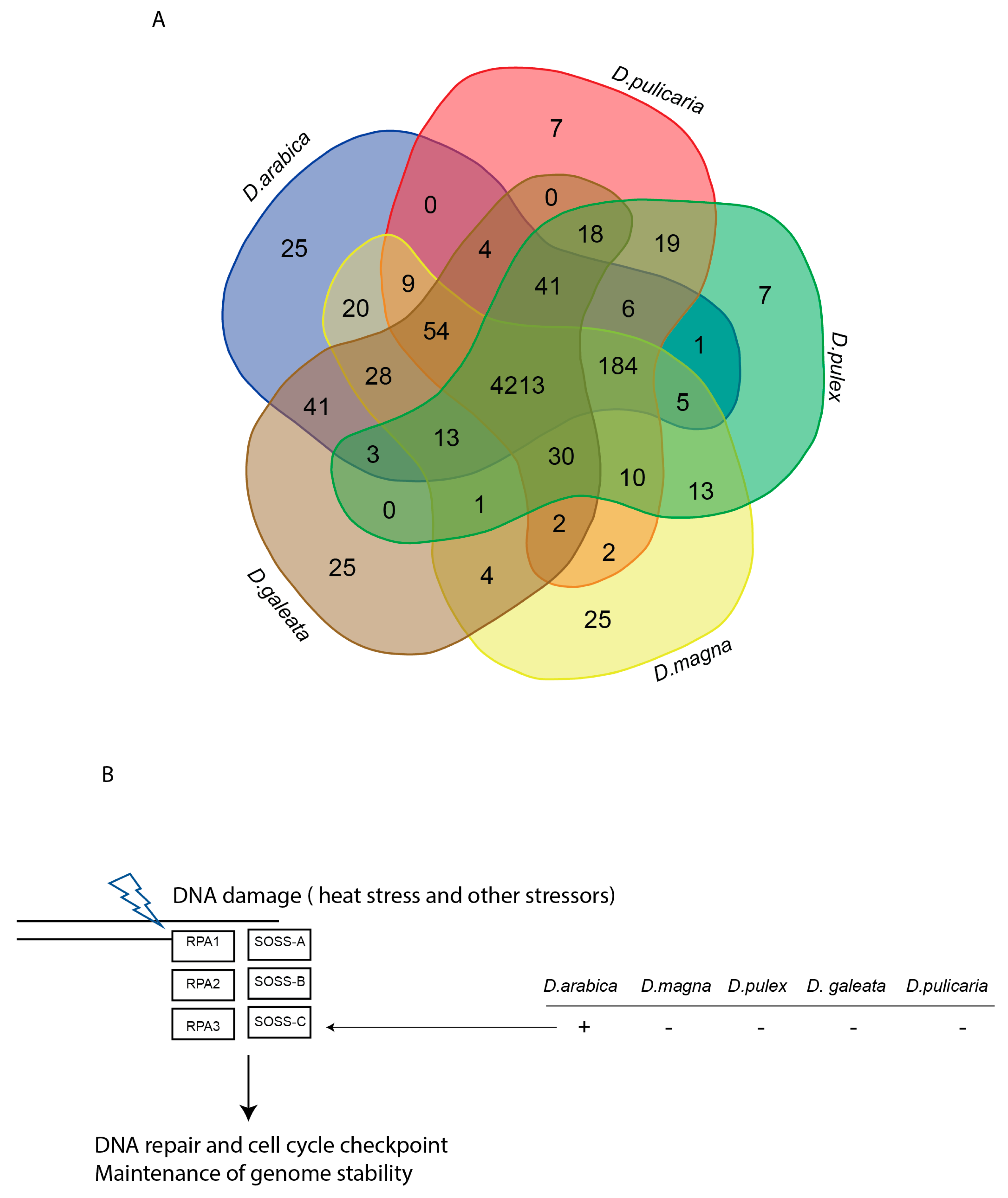
2.2. Diversity and Comparative Genome Analysis
2.3. Evolution and Demographic History
3. Discussion
4. Materials and Methods
4.1. D. arabica Isolation
4.2. Genomic DNA Isolation and QC
4.3. Whole Genome Sequencing Library Preparation
4.4. Transcriptome Sequencing
4.5. Sequencing Data Quality Check and Trimming
4.6. Genome Size Estimation Using Shot Gun Data
4.7. Genome Assembly and QC
4.8. Mitogenome Annotation and Phylogenetic Tree Construction
4.9. Gene Prediction and Annotation
4.10. Diversity and Comparative Genomic Analysis
4.11. Evolutionary and Demographic History
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Forró, L.; Korovchinsky, N.P.M.; Kotov, A.A.; Petrusek, A. Global diversity of cladocerans (Cladocera; Crustacea) in freshwater. Hydrobiologia 2007, 595, 177–184. [Google Scholar] [CrossRef]
- Dumont, H. Introduction to the class Branchiopoda. Guides Identif. Microinvertebr. Cont. Waters World 2002, 19, 1–398. [Google Scholar]
- Crease, T.J. The complete sequence of the mitochondrial genome of Daphnia pulex (Cladocera: Crustacea). Gene 1999, 233, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Colbourne, J.K.; Pfrender, M.E.; Gilbert, D.; Thomas, W.K.; Tucker, A.; Oakley, T.H.; Tokishita, S.; Aerts, A.; Arnold, G.J.; Basu, M.K. The ecoresponsive genome of Daphnia pulex. Science 2011, 331, 555–561. [Google Scholar] [CrossRef]
- Tokishita, S.-i.; Shibuya, H.; Kobayashi, T.; Sakamoto, M.; Ha, J.-Y.; Yokobori, S.-i.; Yamagata, H.; Hanazato, T. Diversification of mitochondrial genome of Daphnia galeata (Cladocera, Crustacea): Comparison with phylogenetic consideration of the complete sequences of clones isolated from five lakes in Japan. Gene 2017, 611, 38–46. [Google Scholar] [CrossRef]
- Routtu, J.; Jansen, B.; Colson, I.; De Meester, L.; Ebert, D. The first-generation Daphnia magna linkage map. BMC Genom. 2010, 11, 1–7. [Google Scholar] [CrossRef]
- Lee, J.-S.; Kim, D.-H.; Choi, B.-S.; Kato, Y.; Watanabe, H.; Lee, J.-S. Complete mitochondrial genome of the freshwater water flea Daphnia magna NIES strain (Cladocera, Daphniidae): Rearrangement of two ribosomal RNA genes. Mitochondrial DNA Part B 2020, 5, 1822–1823. [Google Scholar] [CrossRef]
- Cornetti, L.; Fields, P.D.; Van Damme, K.; Ebert, D. A fossil-calibrated phylogenomic analysis of Daphnia and the Daphniidae. Mol. Phylogenet. Evol. 2019, 137, 250–262. [Google Scholar] [CrossRef]
- Wei, W.; Zhang, K.; Shi, Q. Complete mitochondrial genome of Bosmina fatalis (Cladocera: Bosminidae) and its phylogenetic analysis. Mitochondrial DNA Part B 2021, 6, 2567–2568. [Google Scholar] [CrossRef]
- Gu, Y.-L.; Sun, C.-H.; Liu, P.; Zhang, X.; Sinev, A.Y.; Dumont, H.J.; Han, B.-P. Complete mitochondrial genome of Ovalona pulchella (Branchiopoda, Cladocera) as the first representative in the family Chydoridae: Gene rearrangements and phylogenetic analysis of Cladocera. Gene 2022, 818, 146230. [Google Scholar] [CrossRef]
- Liu, P.; Xu, S.; Huang, Q.; Dumont, H.J.; Lin, Q.; Han, B.-P. The mitochondrial genome of Diaphanosoma dubium with comparison with Daphnia magna. Mitochondrial DNA Part B 2017, 2, 926–927. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Choi, B.-S.; Lee, Y.H.; Kim, H.-J.; Hagiwara, A.; Lee, J.-S. Complete mitochondrial DNA of the marine water flea Diaphanosoma celebensis (Cladocera, Sididae). Mitochondrial DNA Part B 2020, 5, 2254–2255. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Liu, P.; Pajk, F.; Dumont, H.J.; Han, B.-P. The mitochondrial genome of Diaphanosoma excisum Sars, 1885 (Crustacea: Branchiopoda: Cladocera) from Hainan Island, China. Mitochondrial DNA Part B 2021, 6, 1279–1280. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.-L.; Han, B.-P.; Martínez, A.; Schwentner, M.; Fontaneto, D.; Dumont, H.J.; Kotov, A.A. Mitogenomics of Cladocera (Branchiopoda): Marked gene order rearrangements and independent predation roots. Mol. Phylogenet. Evol. 2021, 164, 107275. [Google Scholar] [CrossRef] [PubMed]
- Van Damme, K.; Cornetti, L.; Fields, P.D.; Ebert, D. Whole-genome phylogenetic reconstruction as a powerful tool to reveal homoplasy and ancient rapid radiation in waterflea evolution. Syst. Biol. 2022, 71, 777–787. [Google Scholar] [CrossRef] [PubMed]
- Gurney, R.X. On the Fresh-water Crustacea of Algeria and Tunisia. J. R. Microsc. Soc. 1909, 29, 273–305. [Google Scholar] [CrossRef]
- Gurney, R. List of Entomostraca collected in Seistan and the Baluch Desert. Rec. Zool. Surv. India 1920, 18, 145–146. [Google Scholar] [CrossRef]
- Harding, J. The Armstrong College Zoological Expedition to Siwa Oasis (Libyan Desert) 1935. Crustacea: Branchiopoda and Ostracoda. Proc. Egypti. Acad. Sci. 1955, 10, 58–68. [Google Scholar]
- Dumont, H.J.; Laureys, P.; Pensaert, J. Anostraca, Conchostraca, Cladocera and Copepoda from Tunisia. Hydrobiologia 1979, 66, 259–274. [Google Scholar] [CrossRef]
- Rocha, J.L.; Godinho, R.; Brito, J.C.; Nielsen, R. Life in deserts: The genetic basis of mammalian desert adaptation. Trends Ecol. Evol. 2021, 36, 637–650. [Google Scholar] [CrossRef]
- Tourenq, C.; Brook, M.; Knuteson, S.; Shuriqi, M.K.; Sawaf, M.; Perry, L. Hydrogeology of Wadi Wurayah, United Arab Emirates, and its importance for biodiversity and local communities. Hydrol. Sci. J. 2011, 56, 1407–1422. [Google Scholar] [CrossRef]
- Hamza, W.; Munawar, M. Protecting and managing the Arabian Gulf: Past, present and future. Aquat. Ecosyst. Health Manag. 2009, 12, 429–439. [Google Scholar] [CrossRef]
- Neubert, E.; Amr, Z.; Van Damme, D. The status and distribution of freshwater molluscs in the Arabian Peninsula. Status Distrib. Freshw. Biodivers. Arab. Penins. 2015, 30, 30–38. [Google Scholar]
- Odhiambo, G.O. Water scarcity in the Arabian Peninsula and socio-economic implications. Appl. Water Sci. 2017, 7, 2479–2492. [Google Scholar] [CrossRef]
- Van Damme, K.; Dumont, H.J. Further division of Alona Baird, 1843: Separation and position of Coronatella Dybowski & Grochowski and Ovalona gen. n.(Crustacea: Cladocera). Zootaxa 2008, 1960, 1–44. [Google Scholar]
- Hamza, W.; Ramadan, G.; AlKaabi, M. Morphological and molecular identification of first recorded Cladoceran organisms in the desert of Abu Dhabi, UAE. MOJ Eco. Environ. Sci. 2018, 3, 220–224. [Google Scholar] [CrossRef]
- Soesbergen, M. A preliminary investigation of plankton organisms of fresh and brackish inland waters in the northern United Arab Emirates. Tribulus 2018, 26, 47. [Google Scholar]
- Kotov, A.A.; Neretina, A.N.; Al Neyadi, S.E.S.; Karabanov, D.P.; Hamza, W. Cladocera (Crustacea: Branchiopoda) of Man-Made Lakes at the Northeast Part of the United Arab Emirates with a Hypothesis on Their Origin. Diversity 2022, 14, 688. [Google Scholar] [CrossRef]
- Hamza, W.; Neretina, A.N.; Al Neyadi, S.E.S.; Amiri, K.; Karabanov, D.P.; Kotov, A.A. Discovery of a New Species of Daphnia (Crustacea: Cladocera) from the Arabian Peninsula Revealed a Southern Origin of a Common Northern Eurasian Species Group. Water 2022, 14, 2350. [Google Scholar] [CrossRef]
- Ballinger, M.J.; Bruenn, J.A.; Kotov, A.A.; Taylor, D.J. Selectively maintained paleoviruses in Holarctic water fleas reveal an ancient origin for phleboviruses. Virology 2013, 446, 276–282. [Google Scholar] [CrossRef]
- Hebert, P.D.; Gregory, T.R. The promise of DNA barcoding for taxonomy. Syst. Biol. 2005, 54, 852–859. [Google Scholar] [CrossRef] [PubMed]
- Elías-Gutiérrez, M.; Hubert, N.; Collins, R.A.; Andrade-Sossa, C. Aquatic Organisms Research with DNA Barcodes. Diversity 2021, 13, 306. [Google Scholar] [CrossRef]
- Garibian, P.G.; Neretina, A.N.; Taylor, D.J.; Kotov, A.A. Partial revision of the neustonic genus Scapholeberis Schoedler, 1858 (Crustacea: Cladocera): Decoding of the barcoding results. PeerJ 2020, 8, e10410. [Google Scholar] [CrossRef] [PubMed]
- Rubinoff, D.; Holland, B.S. Between two extremes: Mitochondrial DNA is neither the panacea nor the nemesis of phylogenetic and taxonomic inference. Syst. Biol. 2005, 54, 952–961. [Google Scholar] [CrossRef] [PubMed]
- Galtier, N.; Nabholz, B.; Glémin, S.; Hurst, G. Mitochondrial DNA as a marker of molecular diversity: A reappraisal. Mol. Ecol. 2009, 18, 4541–4550. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, K.A.; Platt, R.N.; Bradley, R.D.; Ray, D.A. Whole mitochondrial genomes provide increased resolution and indicate paraphyly in deer mice. BMC Zool. 2017, 2, 1–6. [Google Scholar] [CrossRef]
- Ebach, M.C.; Holdrege, C. DNA barcoding is no substitute for taxonomy. Nature 2005, 434, 697-697. [Google Scholar] [CrossRef] [PubMed]
- Van Damme, K.; Kotov, A.A. The fossil record of the Cladocera (Crustacea: Branchiopoda): Evidence and hypotheses. Earth-Sci. Rev. 2016, 163, 162–189. [Google Scholar] [CrossRef]
- Parker, A.G. Pleistocene climate change in Arabia: Developing a framework for hominin dispersal over the last 350 ka. In The Evolution of Human Populations in Arabia; Springer: Dordrecht, The Netherlands, 2010; pp. 39–49. [Google Scholar]
- Rosenberg, T.; Preusser, F.; Fleitmann, D.; Schwalb, A.; Penkman, K.; Schmid, T.; Al-Shanti, M.; Kadi, K.; Matter, A. Humid periods in southern Arabia: Windows of opportunity for modern human dispersal. Geology 2011, 39, 1115–1118. [Google Scholar] [CrossRef]
- Kotov, A.A.; Garibian, P.G.; Bekker, E.I.; Taylor, D.J.; Karabanov, D.P. A new species group from the Daphnia curvirostris species complex (Cladocera: Anomopoda) from the eastern Palaearctic: Taxonomy, phylogeny and phylogeography. Zool. J. Linn. Soc. 2021, 191, 772–822. [Google Scholar] [CrossRef]
- Korovchinsky, N. The Cladocera (Crustacea: Branchiopoda) as a relict group. Zool. J. Linn. Soc. 2006, 147, 109–124. [Google Scholar] [CrossRef][Green Version]
- Dumont, H.J. Relict distribution patterns of aquatic animals: Another tool in evaluating late Pleistocene climate changes in the Sahara and Sahel. In Palaeoecology of Africa and the Surrounding Islands; Routledge: Oxfordshire, UK, 1982; pp. 1–24. [Google Scholar]
- Yampolsky, L.Y.; Zeng, E.; Lopez, J.; Williams, P.J.; Dick, K.B.; Colbourne, J.K.; Pfrender, M.E. Functional genomics of acclimation and adaptation in response to thermal stress in Daphnia. BMC Genom. 2014, 15, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Gong, Z.; Ghosal, G.; Chen, J. SOSS complexes participate in the maintenance of genomic stability. Mol. Cell 2009, 35, 384–393. [Google Scholar] [CrossRef]
- Nam, E.A.; Cortez, D. SOSS1/2: Sensors of single-stranded DNA at a break. Mol. Cell 2009, 35, 258–259. [Google Scholar] [CrossRef]
- Pfleiderer, M.M.; Galej, W.P. Emerging insights into the function and structure of the Integrator complex. Transcription 2021, 12, 251–265. [Google Scholar] [CrossRef] [PubMed]
- Partridge, L.; Barrie, B.; Fowler, K.; French, V. Evolution and development of body size and cell size in Drosophila melanogaster in response to temperature. Evolution 1994, 48, 1269–1276. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Sobeck, A.; Xu, C.; Meetei, A.R.; Hoatlin, M.; Li, L.; Wang, W. BLAP75, an essential component of Bloom’s syndrome protein complexes that maintain genome integrity. EMBO J. 2005, 24, 1465–1476. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data; Babraham Bioinformatics, Babraham Institute: Cambridge, UK, 2010. [Google Scholar]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef]
- Marcais, G.; Kingsford, C. Jellyfish: A fast k-mer counter. Tutor. Manuais 2012, 1, 1–8. [Google Scholar]
- Vurture, G.W.; Sedlazeck, F.J.; Nattestad, M.; Underwood, C.J.; Fang, H.; Gurtowski, J.; Schatz, M.C. GenomeScope: Fast reference-free genome profiling from short reads. Bioinformatics 2017, 33, 2202–2204. [Google Scholar] [CrossRef] [PubMed]
- Zimin, A.V.; Marçais, G.; Puiu, D.; Roberts, M.; Salzberg, S.L.; Yorke, J.A. The MaSuRCA genome assembler. Bioinformatics 2013, 29, 2669–2677. [Google Scholar] [CrossRef] [PubMed]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PloS ONE 2014, 9, e112963. [Google Scholar] [CrossRef]
- Seppey, M.; Manni, M.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness. In Gene Prediction; Springer: New York, NY, USA, 2019; pp. 227–245. [Google Scholar]
- Bernt, M.; Donath, A.; Jühling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Pütz, J.; Middendorf, M.; Stadler, P.F. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547. [Google Scholar] [CrossRef]
- Flynn, J.M.; Hubley, R.; Goubert, C.; Rosen, J.; Clark, A.G.; Feschotte, C.; Smit, A.F. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. USA 2020, 117, 9451–9457. [Google Scholar] [CrossRef]
- Chen, N. Using Repeat Masker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinform. 2004, 5, 4–10. [Google Scholar] [CrossRef]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef]
- Hoff, K.J.; Lomsadze, A.; Borodovsky, M.; Stanke, M. Whole-genome annotation with BRAKER. In Gene Prediction; Springer: New York, NY, USA, 2019; pp. 65–95. [Google Scholar]
- Stanke, M.; Keller, O.; Gunduz, I.; Hayes, A.; Waack, S.; Morgenstern, B. AUGUSTUS: Ab initio prediction of alternative transcripts. Nucleic Acids Res. 2006, 34, W435–W439. [Google Scholar] [CrossRef] [PubMed]
- Brůna, T.; Lomsadze, A.; Borodovsky, M. GeneMark-EP+: Eukaryotic gene prediction with self-training in the space of genes and proteins. NAR Genom. Bioinform. 2020, 2, lqaa026. [Google Scholar] [CrossRef] [PubMed]
- Haas, B.J.; Salzberg, S.L.; Zhu, W.; Pertea, M.; Allen, J.E.; Orvis, J.; White, O.; Buell, C.R.; Wortman, J.R. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 2008, 9, 1–22. [Google Scholar] [CrossRef]
- Korf, I. Gene finding in novel genomes. BMC Bioinform. 2004, 5, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Chan, P.P.; Lin, B.Y.; Mak, A.J.; Lowe, T.M. tRNAscan-SE 2.0: Improved detection and functional classification of transfer RNA genes. Nucleic Acids Res. 2021, 49, 9077–9096. [Google Scholar] [CrossRef]
- Lagesen, K.; Hallin, P.; Rødland, E.A.; Stærfeldt, H.-H.; Rognes, T.; Ussery, D.W. RNAmmer: Consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007, 35, 3100–3108. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Jones, P.; Binns, D.; Chang, H.-Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef]
- Moriya, Y.; Itoh, M.; Okuda, S.; Yoshizawa, A.C.; Kanehisa, M. KAAS: An automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007, 35, W182–W185. [Google Scholar] [CrossRef]
- Korneliussen, T.S.; Albrechtsen, A.; Nielsen, R. ANGSD: Analysis of next generation sequencing data. BMC Bioinform. 2014, 15, 1–13. [Google Scholar] [CrossRef]
- Fan, H.; Ives, A.R.; Surget-Groba, Y.; Cannon, C.H. An assembly and alignment-free method of phylogeny reconstruction from next-generation sequencing data. BMC Genom. 2015, 16, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Sanderson, M.J. r8s: Inferring absolute rates of molecular evolution and divergence times in the absence of a molecular clock. Bioinformatics 2003, 19, 301–302. [Google Scholar] [CrossRef]
- Kotov, A.A.; Taylor, D.J. Mesozoic fossils (>145 Mya) suggest the antiquity of the subgenera of Daphnia and their coevolution with chaoborid predators. BMC Evol. Biol. 2011, 11, 1–9. [Google Scholar] [CrossRef]
- Cabanettes, F.; Klopp, C. D-GENIES: Dot plot large genomes in an interactive, efficient and simple way. PeerJ 2018, 6, e4958. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 genome project data processing subgroup. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Ho, E.K.; Macrae, F.; Latta, L.C., 4th; McIlroy, P.; Ebert, D.; Fields, P.D.; Benner, M.J.; Schaack, S. High and highly variable spontaneous mutation rates in Daphnia. Mol. Biol. Evol. 2020, 37, 3258–3266. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| D. arabica Genome | D. arabica Mitogenome | |
|---|---|---|
| Total sequences | 453 | 1 |
| Genome size | 116,021,024 | 16,588 |
| A + T% | ~59.2 | ~69.7 |
| G + C% | ~40.7 | ~30.2 |
| n % | 0.0001 | 0 |
| Minimum sequence length | 1179 | 16,588 |
| Maximum sequence length | 4,005,661 | 16,588 |
| N50 length (bp) | 1,139,068 | 16,588 |
| L50 number | 30 | 1 |
| Length 1001–3000 bp | 9 | 0 |
| Length 3001–5000 bp | 10 | 0 |
| Length 5001–7000 bp | 7 | 0 |
| Length 7001–10,000 bp | 0 | 0 |
| Length 10,001–0.1 Mb bp | 249 | 1 |
| Length 100,001–1 Mb bp | 134 | 0 |
| Length > 1 Mb bp | 35 | 0 |
| Protein coding genes | 24,041 | 13 |
| tRNA genes | 5374 | 23 |
| rRNA genes | 643 | 2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hamza, W.; Hazzouri, K.M.; Sudalaimuthuasari, N.; Amiri, K.M.A.; Neretina, A.N.; Al Neyadi, S.E.S.; Kotov, A.A. Genome Assembly of a Relict Arabian Species of Daphnia O. F. Müller (Crustacea: Cladocera) Adapted to the Desert Life. Int. J. Mol. Sci. 2023, 24, 889. https://doi.org/10.3390/ijms24010889
Hamza W, Hazzouri KM, Sudalaimuthuasari N, Amiri KMA, Neretina AN, Al Neyadi SES, Kotov AA. Genome Assembly of a Relict Arabian Species of Daphnia O. F. Müller (Crustacea: Cladocera) Adapted to the Desert Life. International Journal of Molecular Sciences. 2023; 24(1):889. https://doi.org/10.3390/ijms24010889
Chicago/Turabian StyleHamza, Waleed, Khaled M. Hazzouri, Naganeeswaran Sudalaimuthuasari, Khaled M. A. Amiri, Anna N. Neretina, Shamma E. S. Al Neyadi, and Alexey A. Kotov. 2023. "Genome Assembly of a Relict Arabian Species of Daphnia O. F. Müller (Crustacea: Cladocera) Adapted to the Desert Life" International Journal of Molecular Sciences 24, no. 1: 889. https://doi.org/10.3390/ijms24010889
APA StyleHamza, W., Hazzouri, K. M., Sudalaimuthuasari, N., Amiri, K. M. A., Neretina, A. N., Al Neyadi, S. E. S., & Kotov, A. A. (2023). Genome Assembly of a Relict Arabian Species of Daphnia O. F. Müller (Crustacea: Cladocera) Adapted to the Desert Life. International Journal of Molecular Sciences, 24(1), 889. https://doi.org/10.3390/ijms24010889