
Exploring the Potential Energy Surface of Pt6 Sub-Nano Clusters Deposited over Graphene


Abstract
1. Introduction
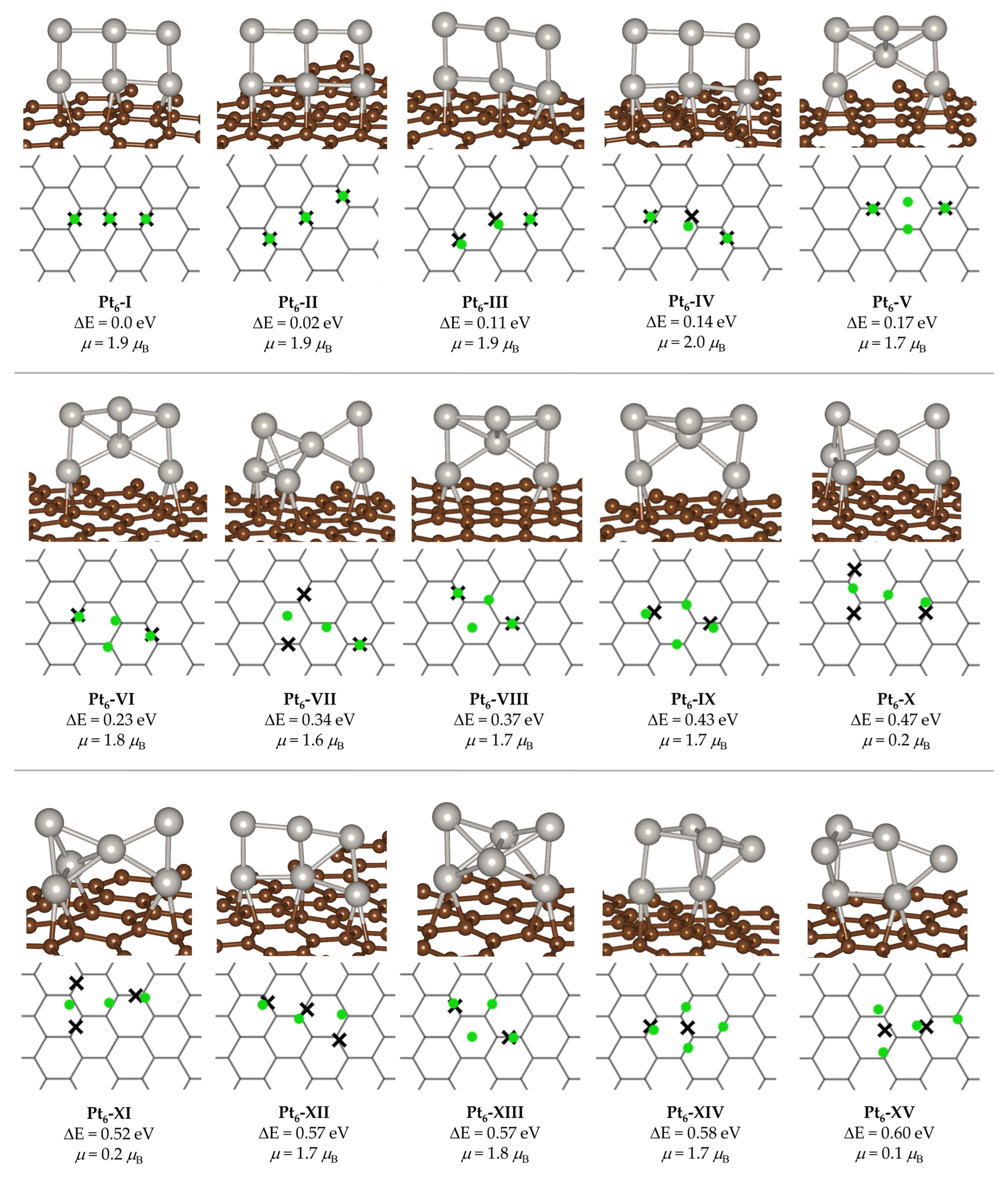
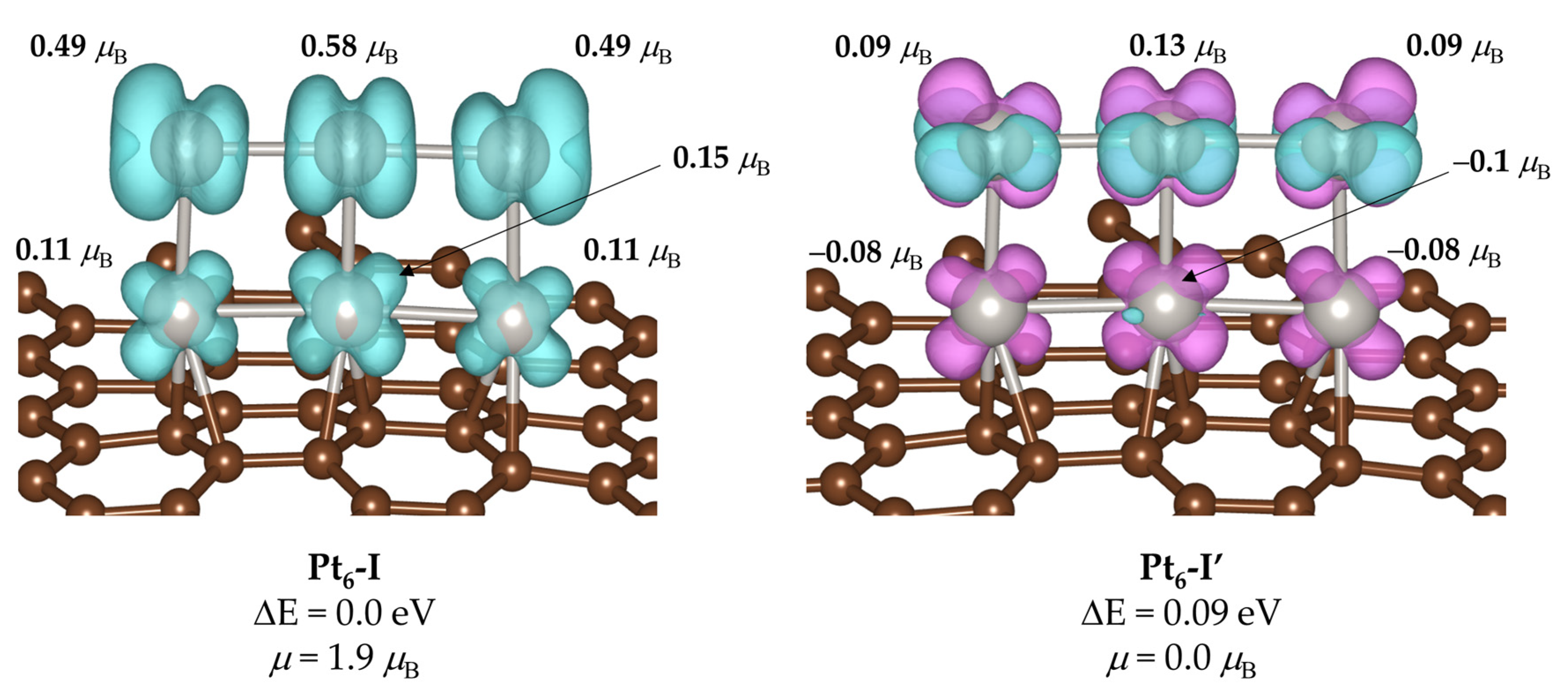
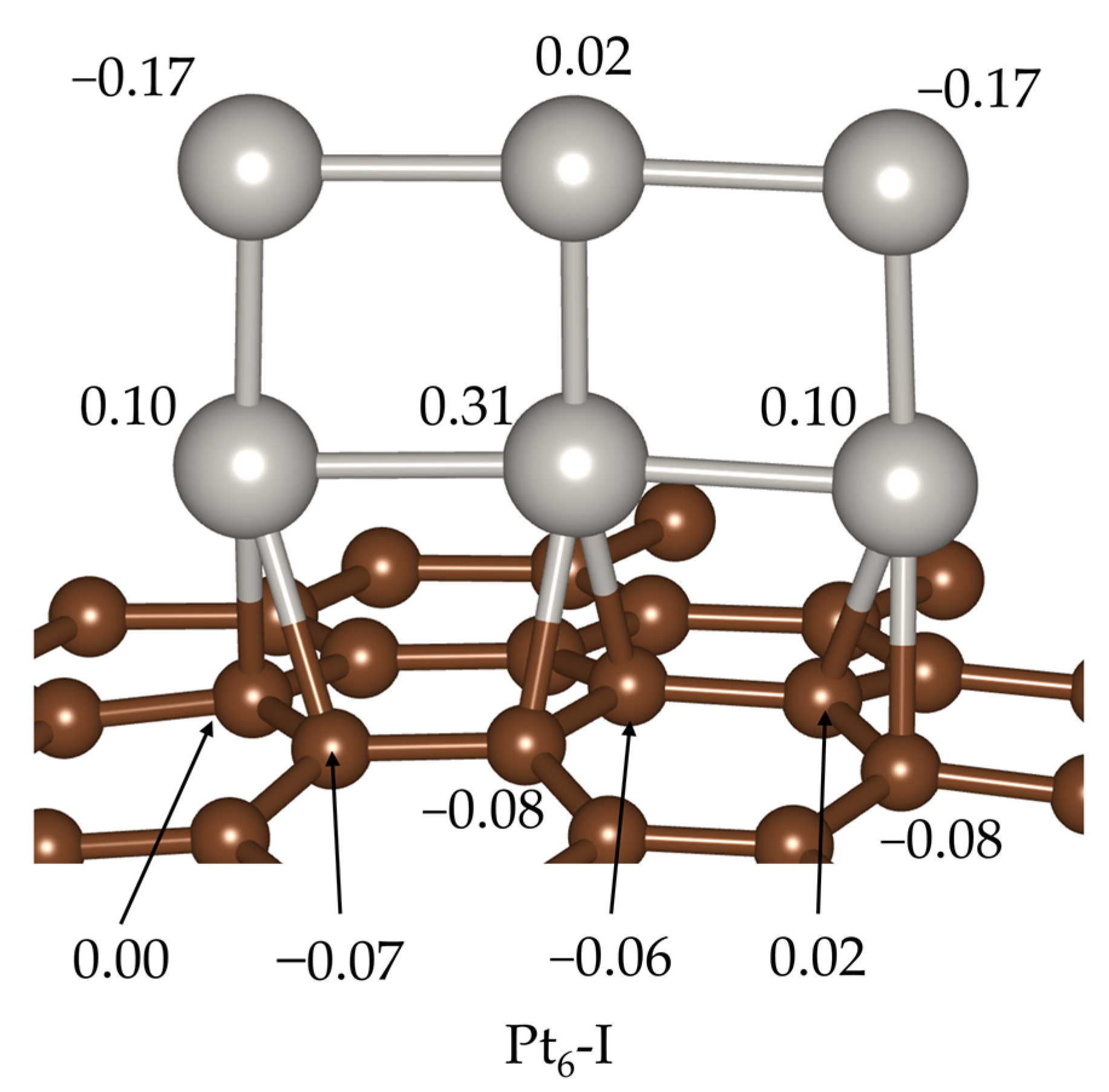
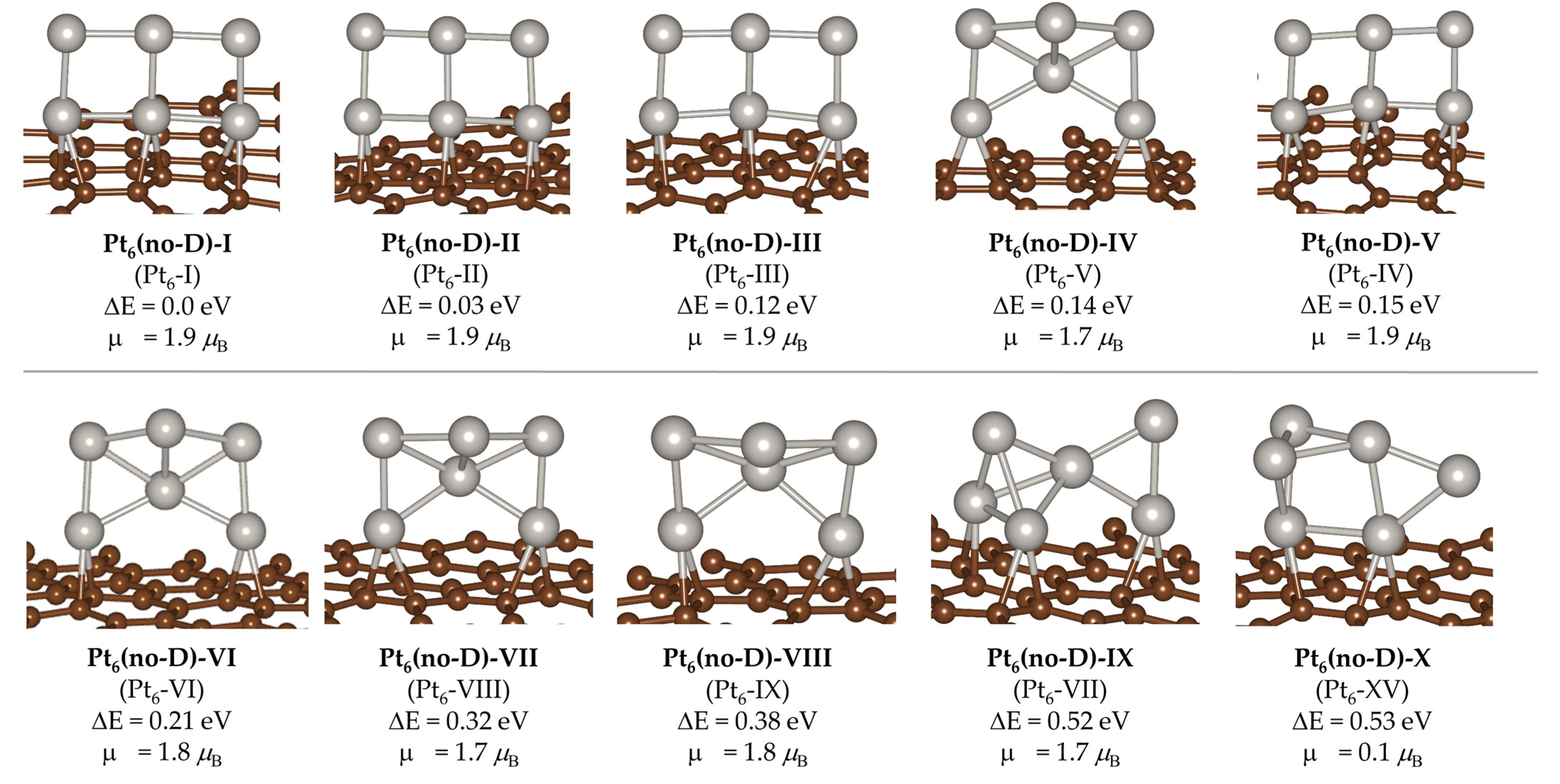
2. Results and Discussion
3. Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tyo, E.C.; Vajda, S. Catalysis by clusters with precise numbers of atoms. Nat. Nanotechnol. 2015, 10, 577–588. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, I.; Pradeep, T. Atomically Precise Clusters of Noble Metals: Emerging Link between Atoms and Nanoparticles. Chem. Rev. 2017, 117, 8208–8271. [Google Scholar] [CrossRef] [PubMed]
- Jin, R.; Zeng, C.; Zhou, M.; Chen, Y. Atomically Precise Colloidal Metal Nanoclusters and Nanoparticles: Fundamentals and Opportunities. Chem. Rev. 2016, 116, 10346–10413. [Google Scholar] [CrossRef]
- Liu, L.; Corma, A.A. Metal Catalysts for Heterogeneous Catalysis: From Single Atoms to Nanoclusters and Nanoparticles. Chem. Rev. 2018, 118, 4981–5079. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Kong, X.; Wang, F.; Fang, R.; Li, Y. Metal Sub-nanoclusters Confined within Hierarchical Porous Carbons with High Oxidation Activity. Angew. Chem. Int. Ed. 2021, 60, 10842–10849. [Google Scholar] [CrossRef]
- Halder, A.; Curtiss, L.A.; Fortunelli, A.; Vajda, S. Perspective: Size selected clusters for catalysis and electrochemistry. J. Chem. Phys. 2018, 148, 110901. [Google Scholar] [CrossRef]
- Von Weber, A.; Anderson, S.L. Electrocatalysis by Mass-Selected Ptn Clusters. Acc. Chem. Res. 2016, 49, 2632–2639. [Google Scholar] [CrossRef]
- Proch, S.; Wirth, M.; White, H.S.; Anderson, S.L. Strong Effects of Cluster Size and Air Exposure on Oxygen Reduction and Carbon Oxidation Electrocatalysis by Size-Selected Ptn (n ≤ 11) on Glassy Carbon Electrodes. J. Am. Chem. Soc. 2013, 135, 3073–3086. [Google Scholar] [CrossRef]
- Ohnuma, A.; Takahashi, K.; Tsunoyama, H.; Inoue, T.; Zhao, P.; Velloth, A.; Ehara, M.; Ichikuni, N.; Tabuchi, M.; Nakajima, A. Enhanced oxygen reduction activity of size-selected platinum subnanocluster catalysts: Ptn (n = 3–9). Catal. Sci. Technol. 2022, 12, 1400–1407. [Google Scholar] [CrossRef]
- Zhai, H.; Alexandrova, A.N. Fluxionality of Catalytic Clusters: When It Matters and How to Address It. ACS. Catal. 2017, 7, 1905–1911. [Google Scholar] [CrossRef]
- Zhang, Z.; Zandkarimi, B.; Alexandrova, A.N. Ensembles of Metastable States Govern Heterogeneous Catalysis on Dynamic Interfaces. Acc. Chem. Res. 2020, 53, 447–458. [Google Scholar] [CrossRef] [PubMed]
- Sun, G.; Sautet, P. Metastable Structures in Cluster Catalysis from First-Principles: Structural Ensemble in Reaction Conditions and Metastability Triggered Reactivity. J. Am. Chem. Soc. 2018, 140, 2812–2820. [Google Scholar] [CrossRef] [PubMed]
- Poidevin, C.; Paciok, P.; Heggen, M.; Auer, A.A. High resolution transmission electron microscopy and electronic structure theory investigation of platinum nanoparticles on carbon black. J. Chem. Phys. 2019, 150, 041705. [Google Scholar] [CrossRef]
- Campos-Roldaén, C.A.; Ramos-Saénchez, G.; Gonzalez-Huerta, R.G.; Vargas García, J.R.; Balbuena, P.B.; Alonso-Vante, N. Influence of sp3−sp2 Carbon Nanodomains on Metal/Support Interaction, Catalyst Durability, and Catalytic Activity for the Oxygen Reduction Reaction. ACS Appl. Mater. Interfaces 2016, 8, 23260–23269. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Kim, S.H.; Kwak, S.K.; Song, H.-K. Curvature-Induced Metal−Support Interaction of an Islands-by-Islands Composite of Platinum Catalyst and Carbon Nano-onion for Durable Oxygen Reduction. ACS Appl. Mater. Interfaces 2017, 9, 23302–23308. [Google Scholar] [CrossRef] [PubMed]
- Schneider, W.B.; Benedikt, U.; Auer, A.A. Interaction of Platinum Nanoparticles with Graphitic Carbon Structures: A Computational Study. ChemPhysChem 2013, 14, 2984–2989. [Google Scholar] [CrossRef] [PubMed]
- Jayabal, S.; Saranya, G.; Geng, D.; Lin, L.-Y.; Meng, X. Insight into the correlation of Pt–support interactions with electro-catalytic activity and durability in fuel cells. J. Mater. Chem. A 2020, 8, 9420–9446. [Google Scholar] [CrossRef]
- Ramos-Sanchez, G.; Balbuena, P.B. Interactions of platinum clusters with a graphite substrate. Phys. Chem. Chem. Phys. 2013, 15, 11950–11959. [Google Scholar] [CrossRef]
- Verga, L.G.; Aarons, J.; Sarwar, M.; Thompsett, D.; Russell, A.E.; Skylaris, C.-K. Effect of graphene support on large Pt nanoparticles. Phys. Chem. Chem. Phys. 2016, 18, 32713–32722. [Google Scholar] [CrossRef]
- Tsunoyama, H.; Ohnuma, A.; Takahashi, K.; Velloth, A.; Ehara, M.; Ichikuni, N.; Tabuchi, M.; Nakajima, A. Enhanced oxygen reduction activity of platinum subnanocluster catalysts through charge redistribution. Chem. Commun. 2019, 55, 12603–12606. [Google Scholar] [CrossRef]
- Wang, X.; Zhao, L.; Li, X.; Liu, Y.; Wang, Y.; Yao, Q.; Xie, J.; Xue, Q.; Yan, Z.; Yuan, X.; et al. Atomic-precision Pt6 nanoclusters for enhanced hydrogen electro-oxidation. Nat. Commun. 2022, 13, 1596. [Google Scholar] [CrossRef] [PubMed]
- Rêgo, C.R.C.; Tereshchuk, P.; Oliveira, L.N.; Da Silva, J.L.F. Graphene-supported small transition-metal clusters: A density functional theory investigation within van der Waals corrections. Phys. Rev. B 2017, 95, 235422. [Google Scholar] [CrossRef]
- Lavroff, R.H.; Morgan, H.W.T.; Zhang, Z.; Poths, P.; Alexandrova, A.N. Ensemble representation of catalytic interfaces: Soloists, orchestras, and everything in-between. Chem. Sci. 2022, 13, 8003–8016. [Google Scholar] [CrossRef]
- Nørskov, J.K.; Rossmeisl, J.; Logadottir, A.; Lindqvist, L.; Kitchin, J.R.; Bligaard, T.; Jónsson, H. Origin of the Overpotential for Oxygen Reduction at a Fuel-Cell Cathode. J. Phys. Chem. B 2004, 108, 17886–17892. [Google Scholar] [CrossRef]
- Sundararaman, R.; Goddard, W.A., III; Arias, T.A. Grand canonical electronic density-functional theory: Algorithms and applications to electrochemistry. J. Chem. Phys. 2017, 146, 114104. [Google Scholar] [CrossRef] [PubMed]
- Hutchison, P.; Rice, P.S.; Warburton, R.E.; Raugei, S.; Hammes-Schiffer, S. Multilevel Computational Studies Reveal the Importance of Axial Ligand for Oxygen Reduction Reaction on Fe−N−C Materials. J. Am. Chem. Soc. 2022, 144, 16524–16534. [Google Scholar] [CrossRef]
- Melander, M.M.; Kuisma, M.J.; Christensen, T.E.K.; Honkala, K. Grand-canonical approach to density functional theory of electrocatalytic systems: Thermodynamics of solid-liquid interfaces at constant ion and electrode potentials. J. Chem. Phys. 2019, 150, 041706. [Google Scholar] [CrossRef]
- Basdogan, Y.; Maldonado, A.M.; Keith, J.A. Advances and challenges in modeling solvated reaction mechanisms for re-newable fuels and chemicals. WIREs Comput. Mol. Sci. 2019, 10, e1446. [Google Scholar]
- Abidi, N.; Lim, K.R.G.; Seh, Z.W.; Steinmann, S.N. Atomistic modeling of electrocatalysis: Are we there yet? WIREs Comput. Mol. Sci. 2021, 11, e1499. [Google Scholar] [CrossRef]
- Gauthier, J.A.; Ringe, S.; Dickens, C.F.; Garza, A.J.; Bell, A.T.; Head-Gordon, M.; Nørskov, J.K.; Chan, K. Challenges in Modeling Electrochemical Reaction Energetics with Polarizable Continuum Models. ACS Catal. 2019, 9, 920–931. [Google Scholar] [CrossRef]
- Zhang, Z.; Zandkarimi, B.; Munarriz, J.; Dickerson, C.E.; Alexandrova, A.N. Fluxionality of Subnano Clusters Reshapes the Activity Volcano of Electrocatalysis. ChemCatChem 2022, 14, e202200345. [Google Scholar] [CrossRef]
- Munarriz, J.; Zhang, Z.; Sautet, P.; Alexandrova, A.N. Graphite-supported Ptn Cluster Electrocatalysts: Major Change of Active Sites as a Function of the Applied Potential. ACS Catal. 2022, 12, 14517–14526. [Google Scholar] [CrossRef]
- Duan, Z.; Henkelman, G. Atomic-Scale Mechanisms of Electrochemical Pt Dissolution. ACS Catal. 2021, 11, 14439–14447. [Google Scholar] [CrossRef]
- Hu, X.; Chen, S.; Chen, L.; Tian, Y.; Yao, S.; Lu, Z.; Zhang, X.; Zhou, Z. What is the Real Origin of the Activity of Fe−N−C Electrocatalysts in the O2 Reduction Reaction? Critical Roles of Coordinating Pyrrolic N and Axially Adsorbing Species. J. Am. Chem. Soc. 2022, 144, 18144–18152. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical GGA-type Density Functional Constructed with a Long-Range Dispersion Correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A Consistent and Accurate ab initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132, 154104–154119. [Google Scholar] [CrossRef] [PubMed]
- Gracia, J.; Sharpe, R.; Munarriz, J. Principles determining the activity of magnetic oxides for electron transfer reactions. J. Catal. 2018, 361, 331–338. [Google Scholar] [CrossRef]
- Biz, C.; Fianchini, M.; Gracia, J. Strongly Correlated Electrons in Catalysis: Focus on Quantum Exchange. ACS Catal. 2021, 11, 14249–14261. [Google Scholar] [CrossRef]
- Biz, C.; Fianchini, M.; Polo, V.; Gracia, J. Magnetism and Heterogeneous Catalysis: In Depth on the Quantum Spin-Exchange Interactions in Pt3M (M = V, Cr, Mn, Fe, Co, Ni, and Y)(111) Alloys. ACS Appl. Mater. Interfaces 2020, 12, 50484–50494. [Google Scholar] [CrossRef]
- Li, J.; Ma, J.; Ma, Z.; Zhao, E.; Du, K.; Guo, J.; Ling, T. Spin Effect on Oxygen Electrocatalysis. Adv. Energy Sustain. Res. 2021, 2, 2100034. [Google Scholar] [CrossRef]
- Sun, Y.; Ren, X.; Sun, S.; Liu, Z.; Xi, S.; Xu, Z.J. Engineering High-Spin State Cobalt Cations in Spinel Zinc Cobalt Oxide for Spin Channel Propagation and Active Site Enhancement in Water Oxidation. Angew. Chem. Int. Ed. 2021, 60, 14536–14544. [Google Scholar] [CrossRef] [PubMed]
- Pan, L.; Ai, M.; Huang, C.; Yin, L.; Liu, X.; Zhang, R.; Wang, S.; Jiang, Z.; Zhang, X.; Zou, J.-J.; et al. Manipulating spin polarization of titanium dioxide for efficient photocatalysis. Nat. Commun. 2020, 11, 418. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Cheng, Z.; Wang, X. Understanding the Mechanism of the Oxygen Evolution Reaction with Consideration of Spin. Electrochem. Energy Rev. 2021, 4, 136–145. [Google Scholar] [CrossRef]
- Groves, M.N.; Malardier-Jugroot, C.; Jugroot, M. Improving Platinum Catalyst Durability with a Doped Graphene Support. J. Phys. Chem. C 2012, 116, 10548–10556. [Google Scholar] [CrossRef]
- Li, R.; Odunlami, M.; Carbonnière, P. Low-lying Ptn cluster structures (n = 6–10) from global optimizations based on DFT potential energy surfaces: Sensitivity of the chemical ordering with the functional. Comput. Theor. Chem. 2017, 1107, 136–141. [Google Scholar] [CrossRef]
- Chaves, A.S.; Rondina, G.G.; Piotrowski, M.J.; Tereshchuk, P.; Da Silva, J.L.F. The Role of Charge States in the Atomic Structure of Cun and Ptn (n = 2−14 atoms) Clusters: A DFT Investigation. J. Phys. Chem. A 2004, 118, 10813–10821. [Google Scholar] [CrossRef]
- Xiao, L.; Wang, L. Structures of Platinum Clusters: Planar or Spherical? J. Phys. Chem. A 2004, 108, 8605–8614. [Google Scholar] [CrossRef]
- Kumar, V.; Kawazoe, Y. Evolution of atomic and electronic structure of Pt clusters: Planar, layered, pyramidal, cage, cubic, and octahedral growth. Phys. Rev. B 2008, 77, 205418. [Google Scholar] [CrossRef]
- Zandkarimi, B.; Alexandrova, A.N. Dynamics of Subnanometer Pt Clusters Can Break the Scaling Relationships in Catalysis. J. Phys. Chem. Lett. 2019, 10, 460–467. [Google Scholar] [CrossRef]
- Ignatov, S.K.; Razuvaev, A.G.; Loginova, A.S.; Masunov, A.E. Global Structure Optimization of Pt Clusters Based on the Modified Empirical Potentials, Calibrated using Density Functional Theory. J. Phys. Chem. C 2019, 123, 29024–29036. [Google Scholar] [CrossRef]
- Munarriz, J.; Polo, V.; Gracia, J. On the Role of Ferromagnetic Interactions in Highly Active Mo-Based Catalysts for Ammonia Synthesis. ChemPhysChem 2018, 19, 2843–2847. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B Condens. Matter Mater. Phys. 1993, 47, 558–561. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Hafner, J. Ab initio molecular-dynamics simulation of the liquid-metal–amorphous-semiconductor transition in germanium. Phys. Rev. B Condens. Matter Mater. Phys. 1994, 49, 14251–14269. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B Condens. Matter Mater. Phys. 1996, 54, 11169–11186. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B Condens. Matter Mater. Phys. 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B Condens. Matter Mater. Phys. 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Zhai, H.; Alexandrova, A.N. Ensemble-average representation of Pt clusters in conditions of catalysis accessed through GPU accelerated deep neural network fitting global optimization. J. Chem. Theory Comput. 2016, 12, 6213–6226. [Google Scholar] [CrossRef]
- Zhai, H.; Alexandrova, A.N. Local Fluxionality of Surface-Deposited Cluster Catalysts: The Case of Pt7 on Al2O3. J. Phys. Chem. Lett. 2018, 9, 1696–1702. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pt6(no-D)-# | Pt6-#’ | ∆E(Pt6(no-D)-#) | ∆E(Pt6-#’) | ∆(∆E) | Edisp |
|---|---|---|---|---|---|
| I | I | 0.00 | 0.00 | 0.00 | −7.920 |
| II | II | 0.03 | 0.02 | 0.01 | −7.930 |
| III | III | 0.12 | 0.11 | 0.01 | −7.928 |
| IV | V | 0.14 | 0.17 | −0.03 | −7.884 |
| V | IV | 0.15 | 0.14 | 0.01 | −7.936 |
| VI | VI | 0.21 | 0.23 | −0.02 | −7.900 |
| VII | VIII | 0.32 | 0.37 | −0.05 | −7.877 |
| VIII | IX | 0.38 | 0.43 | −0.05 | −7.872 |
| IX | VII | 0.52 | 0.34 | 0.18 | −8.115 |
| X | XV | 0.53 | 0.60 | −0.07 | −7.858 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barrena-Espés, D.; Boneta, S.; Polo, V.; Munárriz, J. Exploring the Potential Energy Surface of Pt6 Sub-Nano Clusters Deposited over Graphene. Int. J. Mol. Sci. 2023, 24, 870. https://doi.org/10.3390/ijms24010870
Barrena-Espés D, Boneta S, Polo V, Munárriz J. Exploring the Potential Energy Surface of Pt6 Sub-Nano Clusters Deposited over Graphene. International Journal of Molecular Sciences. 2023; 24(1):870. https://doi.org/10.3390/ijms24010870
Chicago/Turabian StyleBarrena-Espés, Daniel, Sergio Boneta, Victor Polo, and Julen Munárriz. 2023. "Exploring the Potential Energy Surface of Pt6 Sub-Nano Clusters Deposited over Graphene" International Journal of Molecular Sciences 24, no. 1: 870. https://doi.org/10.3390/ijms24010870
APA StyleBarrena-Espés, D., Boneta, S., Polo, V., & Munárriz, J. (2023). Exploring the Potential Energy Surface of Pt6 Sub-Nano Clusters Deposited over Graphene. International Journal of Molecular Sciences, 24(1), 870. https://doi.org/10.3390/ijms24010870