
The Inhibitory Properties of a Novel, Selective LMTK3 Kinase Inhibitor

 ,
,  , ,
, ,  , , and
, , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Selectivity Profile of C36 Inhibitor

2.2. Biochemical/Mechanistic Investigation of C36 Binding to LMTK3
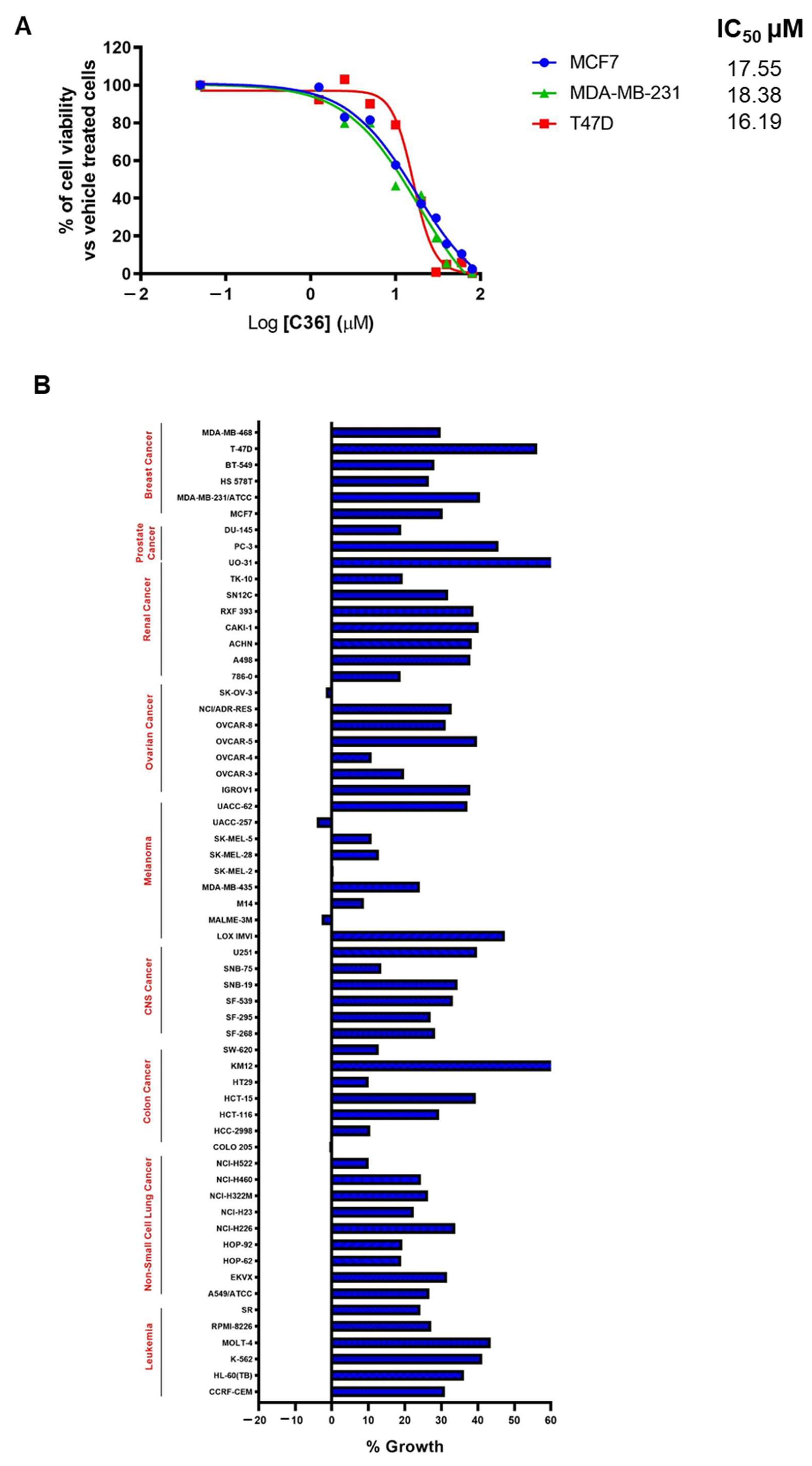
2.3. C36 Exhibits Potent Anticancer Activity in Different Human Cancer Cell Lines
2.4. Pharmacological Properties of C36
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. Cell Death and Apoptosis
4.3. Cell Viability Assay
4.4. In Vitro Kinase Assay
4.5. Kinase Inhibitor Competition Binding Assay
4.6. Microscale Thermophoresis (MST)
4.7. Thermal Shift Assay
4.8. CD Spectroscopy
4.9. Molecular Modelling of LMTK3 with Bound C36
4.10. Caco-2 Permeability Assay
4.11. Compound Stability in Mouse Hepatic Microsomes
4.12. NCI-60 Human Tumor Cell Line Screen
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cicenas, J.; Zalyte, E.; Bairoch, A.; Gaudet, P. Kinases and Cancer. Cancers 2018, 10, 63. [Google Scholar] [CrossRef] [PubMed]
- Ditsiou, A.; Cilibrasi, C.; Simigdala, N.; Papakyriakou, A.; Milton-Harris, L.; Vella, V.; Nettleship, J.E.; Lo, J.H.; Soni, S.; Smbatyan, G.; et al. The structure-function relationship of oncogenic LMTK3. Sci. Adv. 2020, 6, eabc3099. [Google Scholar] [CrossRef] [PubMed]
- Bhullar, K.S.; Lagarón, N.O.; McGowan, E.M.; Parmar, I.; Jha, A.; Hubbard, B.P.; Rupasinghe, H.P.V. Kinase-targeted cancer therapies: Progress, challenges and future directions. Mol. Cancer 2018, 17, 48. [Google Scholar] [CrossRef]
- Blume-Jensen, P.; Hunter, T. Oncogenic kinase signalling. Nature 2001, 411, 355–365. [Google Scholar] [CrossRef] [PubMed]
- Krause, D.S.; Van Etten, R.A. Tyrosine kinases as targets for cancer therapy. N. Engl. J. Med. 2005, 353, 172–187. [Google Scholar] [CrossRef]
- Ditsiou, A.; Gagliano, T.; Samuels, M.; Vella, V.; Tolias, C.; Giamas, G. The multifaceted role of lemur tyrosine kinase 3 in health and disease. Open Biol. 2021, 11, 210218. [Google Scholar] [CrossRef]
- Montrose, K.; Kobayashi, S.; Manabe, T.; Yamamoto, T. Lmtk3-KO Mice Display a Range of Behavioral Abnormalities and Have an Impairment in GluA1 Trafficking. Neuroscience 2019, 414, 154–167. [Google Scholar] [CrossRef]
- Giamas, G.; Filipović, A.; Jacob, J.; Messier, W.; Zhang, H.; Yang, D.; Zhang, W.; Shifa, B.A.; Photiou, A.; Tralau-Stewart, C.; et al. Kinome screening for regulators of the estrogen receptor identifies LMTK3 as a new therapeutic target in breast cancer. Nat. Med. 2011, 17, 715–719. [Google Scholar] [CrossRef]
- Jacob, J.; Favicchio, R.; Karimian, N.; Mehrabi, M.; Harding, V.; Castellano, L.; Stebbing, J.; Giamas, G. LMTK3 escapes tumour suppressor miRNAs via sequestration of DDX5. Cancer Lett. 2016, 372, 137–146. [Google Scholar] [CrossRef]
- Li, Z.; Wu, J.; Ji, M.; Shi, L.; Xu, B.; Jiang, J.; Wu, C. Prognostic role of lemur tyrosine kinase 3 in postoperative gastric cancer. Mol. Clin. Oncol. 2014, 2, 756–760. [Google Scholar] [CrossRef]
- Shi, H.; Wu, J.; Ji, M.; Zhou, Q.; Li, Z.; Zheng, X.; Xu, B.; Deng, H.; Zhao, W.; Wu, C.; et al. Serum lemur tyrosine kinase 3 expression in colorectal cancer patients predicts cancer progression and prognosis. Med. Oncol. 2013, 30, 754. [Google Scholar] [CrossRef] [PubMed]
- Stebbing, J.; Filipovic, A.; Ellis, I.O.; Green, A.R.; D’Silva, T.R.; Lenz, H.J.; Coombes, R.C.; Wang, T.; Lee, S.C.; Giamas, G. LMTK3 expression in breast cancer: Association with tumor phenotype and clinical outcome. Breast Cancer Res. Treat. 2012, 132, 537–544. [Google Scholar] [CrossRef]
- Stebbing, J.; Filipovic, A.; Lit, L.C.; Blighe, K.; Grothey, A.; Xu, Y.; Miki, Y.; Chow, L.W.; Coombes, R.C.; Sasano, H.; et al. LMTK3 is implicated in endocrine resistance via multiple signaling pathways. Oncogene 2013, 32, 3371–3380. [Google Scholar] [CrossRef] [PubMed]
- Stebbing, J.; Shah, K.; Lit, L.C.; Gagliano, T.; Ditsiou, A.; Wang, T.; Wendler, F.; Simon, T.; Szabó, K.S.; O’Hanlon, T.; et al. LMTK3 confers chemo-resistance in breast cancer. Oncogene 2018, 37, 3113–3130. [Google Scholar] [CrossRef]
- Wakatsuki, T.; LaBonte, M.J.; Bohanes, P.O.; Zhang, W.; Yang, D.; Azuma, M.; Barzi, A.; Ning, Y.; Loupakis, F.; Saadat, S.; et al. Prognostic role of lemur tyrosine kinase-3 germline polymorphisms in adjuvant gastric cancer in Japan and the United States. Mol. Cancer Ther. 2013, 12, 2261–2272. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Zhang, H.; Lit, L.C.; Grothey, A.; Athanasiadou, M.; Kiritsi, M.; Lombardo, Y.; Frampton, A.E.; Green, A.R.; Ellis, I.O.; et al. The kinase LMTK3 promotes invasion in breast cancer through GRB2-mediated induction of integrin β1. Sci. Signal. 2014, 7, ra58. [Google Scholar] [CrossRef]
- Xu, Y.; Zhang, H.; Nguyen, V.T.; Angelopoulos, N.; Nunes, J.; Reid, A.; Buluwela, L.; Magnani, L.; Stebbing, J.; Giamas, G. LMTK3 Represses Tumor Suppressor-like Genes through Chromatin Remodeling in Breast Cancer. Cell Rep. 2015, 12, 837–849. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Chen, L.; Deng, H.; Zou, Y.; Liu, J.; Shi, H.; Xu, B.; Lu, M.; Li, C.; Jiang, J.; et al. Serum lemur tyrosine kinase-3: A novel biomarker for screening primary non-small cell lung cancer and predicting cancer progression. Int. J. Clin. Exp. Pathol. 2015, 8, 629–635. [Google Scholar]
- Lu, L.; Yuan, X.; Zhang, Q.; Zhang, H.; Shen, B. LMTK3 knockdown retards cell growth and invasion and promotes apoptosis in thyroid cancer. Mol. Med. Rep. 2017, 15, 2015–2022. [Google Scholar] [CrossRef]
- Klug, L.R.; Bannon, A.E.; Javidi-Sharifi, N.; Town, A.; Fleming, W.H.; VanSlyke, J.K.; Musil, L.S.; Fletcher, J.A.; Tyner, J.W.; Heinrich, M.C. LMTK3 is essential for oncogenic KIT expression in KIT-mutant GIST and melanoma. Oncogene 2019, 38, 1200–1210. [Google Scholar] [CrossRef]
- Jiang, T.; Lu, X.; Yang, F.; Wang, M.; Yang, H.; Xing, N. LMTK3 promotes tumorigenesis in bladder cancer via the ERK/MAPK pathway. FEBS Open Bio 2020, 10, 2107–2121. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Qi, X.; Zhang, X.; Yu, L. Preoperative serum LMTK3 as a novel biomarker in non-small cell lung cancer. Tumour Biol. 2014, 35, 5007–5011. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Yang, M.; Gu, X.; Gu, Y. Lemur tyrosine kinase-3 (LMTK3) induces chemoresistance to cetuximab in colorectal cancer via the ERK/MAPK pathway. Bioengineered 2021, 12, 6594–6605. [Google Scholar] [CrossRef] [PubMed]
- Kinase Screen. Available online: https://www.kinase-screen.mrc.ac.uk/ (accessed on 1 September 2022).
- Eurofins DiscoverX. Available online: https://www.discoverx.com/home (accessed on 1 September 2022).
- Rainard, J.M.; Pandarakalam, G.C.; McElroy, S.P. Using Microscale Thermophoresis to Characterize Hits from High-Throughput Screening: A European Lead Factory Perspective. SLAS Discov. 2018, 23, 225–241. [Google Scholar] [CrossRef]
- Entzian, C.; Schubert, T. Studying small molecule-aptamer interactions using MicroScale Thermophoresis (MST). Methods 2016, 97, 27–34. [Google Scholar] [CrossRef]
- Magnez, R.; Bailly, C.; Thuru, X. Microscale Thermophoresis as a Tool to Study Protein Interactions and Their Implication in Human Diseases. Int. J. Mol. Sci. 2022, 23, 7672. [Google Scholar] [CrossRef]
- Gao, K.; Oerlemans, R.; Groves, M.R. Theory and applications of differential scanning fluorimetry in early-stage drug discovery. Biophys. Rev. 2020, 12, 85–104. [Google Scholar] [CrossRef]
- Khrapunov, S. Circular dichroism spectroscopy has intrinsic limitations for protein secondary structure analysis. Anal. Biochem. 2009, 389, 174–176. [Google Scholar] [CrossRef]
- Wu, J.; Tseng, Y.D.; Xu, C.F.; Neubert, T.A.; White, M.F.; Hubbard, S.R. Structural and biochemical characterization of the KRLB region in insulin receptor substrate-2. Nat. Struct. Mol. Biol. 2008, 15, 251–258. [Google Scholar] [CrossRef]
- DTP Developmental Therapeutics Program. Available online: https://dtp.cancer.gov/discovery_development/nci-60/ (accessed on 1 September 2022).
- Han, K.C.; Kim, S.Y.; Yang, E.G. Recent advances in designing substrate-competitive protein kinase inhibitors. Curr. Pharm. Des. 2012, 18, 2875–2882. [Google Scholar] [CrossRef]
- Perryman, A.L.; Stratton, T.P.; Ekins, S.; Freundlich, J.S. Predicting Mouse Liver Microsomal Stability with “Pruned” Machine Learning Models and Public Data. Pharm. Res. 2016, 33, 433–449. [Google Scholar] [CrossRef] [PubMed]
- Quintieri, L.; Fantin, M.; Palatini, P.; De Martin, S.; Rosato, A.; Caruso, M.; Geroni, C.; Floreani, M. In vitro hepatic conversion of the anticancer agent nemorubicin to its active metabolite PNU-159682 in mice, rats and dogs: A comparison with human liver microsomes. Biochem. Pharmacol. 2008, 76, 784–795. [Google Scholar] [CrossRef] [PubMed]
- Palmer, B.D.; Thompson, A.M.; Sutherland, H.S.; Blaser, A.; Kmentova, I.; Franzblau, S.G.; Wan, B.; Wang, Y.; Ma, Z.; Denny, W.A. Synthesis and Structure−Activity Studies of Biphenyl Analogues of the Tuberculosis Drug (6S)-2-Nitro-6-{[4-(trifluoromethoxy)benzyl]oxy}-6,7-dihydro-5H-imidazo[2,1-b][1,3]oxazine (PA-824). J. Med. Chem. 2010, 53, 282–294. [Google Scholar] [CrossRef]
- Slamon, D.J.; Neven, P.; Chia, S.; Fasching, P.A.; De Laurentiis, M.; Im, S.A.; Petrakova, K.; Bianchi, G.V.; Esteva, F.J.; Martín, M.; et al. Phase III Randomized Study of Ribociclib and Fulvestrant in Hormone Receptor-Positive, Human Epidermal Growth Factor Receptor 2-Negative Advanced Breast Cancer: MONALEESA-3. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2018, 36, 2465–2472. [Google Scholar] [CrossRef] [PubMed]
- Sledge, G.W., Jr.; Toi, M.; Neven, P.; Sohn, J.; Inoue, K.; Pivot, X.; Burdaeva, O.; Okera, M.; Masuda, N.; Kaufman, P.A.; et al. MONARCH 2: Abemaciclib in Combination With Fulvestrant in Women With HR+/HER2- Advanced Breast Cancer Who Had Progressed While Receiving Endocrine Therapy. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2017, 35, 2875–2884. [Google Scholar] [CrossRef]
- Hashemzadeh, N.; Dolatkhah, M.; Adibkia, K.; Aghanejad, A.; Barzegar-Jalali, M.; Omidi, Y.; Barar, J. Recent advances in breast cancer immunotherapy: The promising impact of nanomedicines. Life Sci. 2021, 271, 119110. [Google Scholar] [CrossRef]
- Giamas, G.; Hirner, H.; Shoshiashvili, L.; Grothey, A.; Gessert, S.; Kühl, M.; Henne-Bruns, D.; Vorgias, C.E.; Knippschild, U. Phosphorylation of CK1delta: Identification of Ser370 as the major phosphorylation site targeted by PKA in vitro and in vivo. Biochem. J. 2007, 406, 389–398. [Google Scholar] [CrossRef]
- Sali, A.; Blundell, T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Case, D.A.; Cheatham, T.E., 3rd; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M., Jr.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Agnarelli, A.; Lauer Betrán, A.; Papakyriakou, A.; Vella, V.; Samuels, M.; Papanastasopoulos, P.; Giamas, C.; Mancini, E.J.; Stebbing, J.; Spencer, J.; et al. The Inhibitory Properties of a Novel, Selective LMTK3 Kinase Inhibitor. Int. J. Mol. Sci. 2023, 24, 865. https://doi.org/10.3390/ijms24010865
Agnarelli A, Lauer Betrán A, Papakyriakou A, Vella V, Samuels M, Papanastasopoulos P, Giamas C, Mancini EJ, Stebbing J, Spencer J, et al. The Inhibitory Properties of a Novel, Selective LMTK3 Kinase Inhibitor. International Journal of Molecular Sciences. 2023; 24(1):865. https://doi.org/10.3390/ijms24010865
Chicago/Turabian StyleAgnarelli, Alessandro, Andrea Lauer Betrán, Athanasios Papakyriakou, Viviana Vella, Mark Samuels, Panagiotis Papanastasopoulos, Christina Giamas, Erika J. Mancini, Justin Stebbing, John Spencer, and et al. 2023. "The Inhibitory Properties of a Novel, Selective LMTK3 Kinase Inhibitor" International Journal of Molecular Sciences 24, no. 1: 865. https://doi.org/10.3390/ijms24010865
APA StyleAgnarelli, A., Lauer Betrán, A., Papakyriakou, A., Vella, V., Samuels, M., Papanastasopoulos, P., Giamas, C., Mancini, E. J., Stebbing, J., Spencer, J., Cilibrasi, C., Ditsiou, A., & Giamas, G. (2023). The Inhibitory Properties of a Novel, Selective LMTK3 Kinase Inhibitor. International Journal of Molecular Sciences, 24(1), 865. https://doi.org/10.3390/ijms24010865