
The Role of Hyperexcitability in Gliomagenesis


{kind=link}
Abstract
1. Introduction
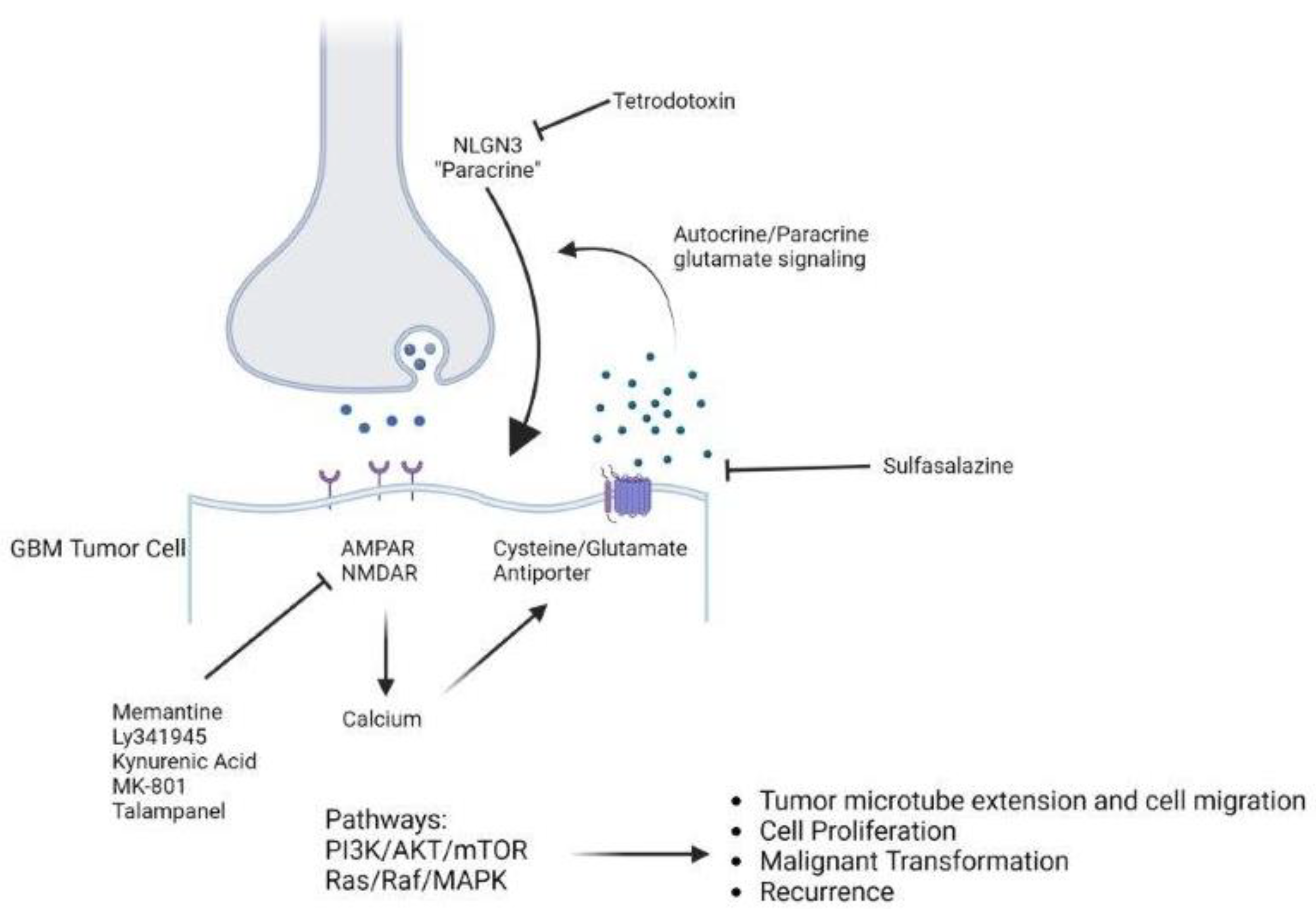
2. Neuron–Tumor Synapses and Glioma Cell Networks
3. The Hyperexcitable Microenvironment
4. Cell Migration and Invasion
5. Proliferation, Tumor Growth, and Recurrence
6. Progression from Low-Grade to High-Grade Glioma
7. Downstream Pathways
8. Therapeutic Targets
9. Conclusions
10. Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Radin, D.P.; Tsirka, S.E. Interactions between Tumor Cells, Neurons, and Microglia in the Glioma Microenvironment. Int. J. Mol. Sci. 2020, 21, 8476. [Google Scholar] [CrossRef] [PubMed]
- Omuro, A.; DeAngelis, L.M. Glioblastoma and other malignant gliomas: A clinical review. JAMA 2013, 310, 1842–1850. [Google Scholar] [CrossRef] [PubMed]
- Lange, F.; Hörnschemeyer, J.; Kirschstein, T. Glutamatergic Mechanisms in Glioblastoma and Tumor-Associated Epilepsy. Cells 2021, 10, 1226. [Google Scholar] [CrossRef]
- Venkataramani, V.; Tanev, D.I.; Kuner, T.; Wick, W.; Winkler, F. Synaptic input to brain tumors: Clinical implications. Neuro-Oncology 2020, 23, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Campbell, S.L.; Buckingham, S.C.; Sontheimer, H. Human glioma cells induce hyperexcitability in cortical networks. Epilepsia 2012, 53, 1360–1370. [Google Scholar] [CrossRef] [PubMed]
- Yu, K.; Lin, C.-C.J.; Hatcher, A.; Lozzi, B.; Kong, K.; Huang-Hobbs, E.; Cheng, Y.-T.; Beechar, V.B.; Zhu, W.; Zhang, Y.; et al. PIK3CA variants selectively initiate brain hyperactivity during gliomagenesis. Nature 2020, 578, 166–171. [Google Scholar] [CrossRef]
- Chaunsali, L.; Tewari, B.P.; Gallucci, A.; Thompson, E.G.; Savoia, A.; Feld, N.; Campbell, S.L. Glioma-induced peritumoral hyperexcitability in a pediatric glioma model. Physiol. Rep. 2020, 8, e14567. [Google Scholar] [CrossRef]
- Tobochnik, S.; Lapinskas, E.; Vogelzang, J.; Ligon, K.L.; Lee, J.W. Early EEG hyperexcitability is associated with decreased survival in newly diagnosed IDH-wildtype glioma. J. Neurooncol. 2022, 159, 211–218. [Google Scholar] [CrossRef]
- Venkatesh, H.S.; Morishita, W.; Geraghty, A.C.; Silverbush, D.; Gillespie, S.M.; Arzt, M.; Tam, L.T.; Espenel, C.; Ponnuswami, A.; Ni, L.; et al. Electrical and synaptic integration of glioma into neural circuits. Nature 2019, 573, 539–545. [Google Scholar] [CrossRef]
- Venkataramani, V.; Tanev, D.I.; Strahle, C.; Studier-Fischer, A.; Fankauser, L.; Kessler, T.; Korber, C.; Kardorff, M.; Ratliff, M.; Xie, R.; et al. Glutamatergic synaptic input to glioma cells drives brain tumour progression. Nature 2019, 573, 532–538. [Google Scholar] [CrossRef]
- Losada-Pérez, M.; Hernández García-Moreno, M.; García-Ricote, I.; Casas-Tintó, S. Synaptic components are required for glioblastoma progression in Drosophila. PLoS Genet. 2022, 18, e1010329. [Google Scholar] [CrossRef]
- Polewski, M.D.; Reveron-Thornton, R.F.; Cherryholmes, G.A.; Marinov, G.K.; Cassady, K.; Aboody, K.S. Increased expression of system xc− in glioblastoma confers an altered metabolism and chemoresistance. Mol. Cancer Res. MCR 2016, 14, 1229–1242. [Google Scholar] [CrossRef] [PubMed]
- Hua, T.; Shi, H.; Zhu, M.; Chen, C.; Su, Y.; Wen, S.; Zhang, X.; Chen, J.; Huang, Q.; Wang, H. Glioma-neuronal interactions in tumor progression: Mechanism, therapeutic strategies and perspectives (Review). Int. J. Oncol. 2022, 61, 104. [Google Scholar] [CrossRef]
- Huberfeld, G.; Vecht, C.J. Seizures and gliomas—Towards a single therapeutic approach. Nat. Rev. Neurol. 2016, 12, 204–216. [Google Scholar] [CrossRef] [PubMed]
- Hatcher, A.; Yu, K.; Meyer, J.; Aiba, I.; Deneen, B.; Noebels, J.L. Pathogenesis of peritumoral hyperexcitability in an immunocompetent CRISPR-based glioblastoma model. J. Clin. Investig. 2020, 130, 2286–2300. [Google Scholar] [CrossRef]
- Pei, Z.; Lee, K.C.; Khan, A.; Erisnor, G.; Wang, H.Y. Pathway analysis of glutamate-mediated, calcium-related signaling in glioma progression. Biochem. Pharmacol. 2020, 176, 113814. [Google Scholar] [CrossRef]
- de Groot, J.; Sontheimer, H. Glutamate and the Biology of Gliomas. Glia 2011, 59, 1181–1189. [Google Scholar] [CrossRef]
- Hills, K.E.; Kostarelos, K.; Wykes, R.C. Converging Mechanisms of Epileptogenesis and Their Insight in Glioblastoma. Front. Mol. Neurosci. 2022, 15, 903115. [Google Scholar] [CrossRef]
- Venkataramani, V.; Yang, Y.; Schubert, M.C.; Reyhan, E.; Tetzlaff, S.K.; Wissman, N.; Botz, M.; Soyka, S.J.; Beretta, C.A.; Pramatarov, R.L.; et al. Glioblastoma hijacks neuronal mechanisms for brain invasion. Cell 2022, 185, 2899–2917.e31. [Google Scholar] [CrossRef] [PubMed]
- Ishiuchi, S.; Tsuzuki, K.; Yoshida, Y.; Tamada, N.; Hagimura, N.; Okado, H.; Miwa, A.; Kurihara, H.; Nakazato, Y.; Tamura, M.; et al. Blockage of Ca2+-permeable AMPA receptors suppresses migration and induces apoptosis in human glioblastoma cells. Nat. Med. 2002, 8, 971–978. [Google Scholar] [CrossRef] [PubMed]
- Piao, Y.; Lu, L.; de Groot, J. AMPA receptors promote perivascular glioma invasion via β1 integrin–dependent adhesion to the extracellular matrix. Neuro-Oncology 2009, 11, 260. [Google Scholar] [CrossRef]
- Suina, K.; Tsuchihashi, K.; Yamasaki, J.; Kamenori, S.; Shintani, S.; Hirata, Y.; Okazaki, S.; Sampetrean, O.; Baba, E.; Akashi, K.; et al. Epidermal growth factor receptor promotes glioma progression by regulating xCT and GluN2B-containing N-methyl-d-aspartate-sensitive glutamate receptor signaling. Cancer Sci. 2018, 109, 3874–3882. [Google Scholar] [CrossRef] [PubMed]
- Nandakumar, D.N.; Ramaswamy, P.; Prasad, C.; Srinivas, D.; Goswami, K. Glioblastoma invasion and NMDA receptors: A novel prospect. Physiol. Int. 2019, 106, 250–260. [Google Scholar] [CrossRef]
- Müller-Längle, A.; Lutz, H.; Hehlgans, S.; Rödel, F.; Rau, K.; Laube, B. NMDA Receptor-Mediated Signaling Pathways Enhance Radiation Resistance, Survival and Migration in Glioblastoma Cells—A Potential Target for Adjuvant Radiotherapy. Cancers 2019, 11, 503. [Google Scholar] [CrossRef] [PubMed]
- Chung, W.J.; Lyons, S.A.; Nelson, G.M.; Hamza, H.; Gladson, C.L.; Gillespie, G.Y.; Sontheimer, H. Inhibition of Cystine Uptake Disrupts the Growth of Primary Brain Tumors. J. Neurosci. 2005, 25, 7101–7110. [Google Scholar] [CrossRef]
- Oh, M.C.; Kim, J.M.; Safaee, M.; Kaur, G.; Sun, M.Z.; Kaur, R.; Celli, A.; Mauro, T.M.; Parsa, A.T. Overexpression of Calcium-Permeable Glutamate Receptors in Glioblastoma Derived Brain Tumor Initiating Cells. PLoS ONE 2012, 7, e47846. [Google Scholar] [CrossRef] [PubMed]
- Ramaswamy, P.; Aditi Devi, N.; Hurmath Fathima, K.; Dalavaikodihalli Nanjaiah, N. Activation of NMDA receptor of glutamate influences MMP-2 activity and proliferation of glioma cells. Neurol. Sci. 2014, 35, 823–829. [Google Scholar] [CrossRef]
- Tsuji, S.; Nakamura, S.; Shoda, K.; Yamada, T.; Shimazawa, M.; Nakayama, N.; Iwama, T.; Hara, H. NMDA receptor signaling induces the chemoresistance of temozolomide via upregulation of MGMT expression in glioblastoma cells. J. Neurooncol. 2022, 160, 375–388. [Google Scholar] [CrossRef]
- Arcella, A.; Carpinelli, G.; Battaglia, G.; D’Onofrio, M.; Santoro, F.; Ngomba, R.T.; Bruno, V.; Casolini, P.; Giangaspero, F.; Nicoletti, F. Pharmacological blockade of group II metabotropic glutamate receptors reduces the growth of glioma cells in vivo. Neuro-Oncology 2005, 7, 236–245. [Google Scholar] [CrossRef]
- Wang, L.; Liu, Y.; Yu, Z.; Gong, J.; Deng, Z.; Ren, N.; Zhong, Z.; Cai, H.; Tang, Z.; Cheng, H.; et al. Mir-139-5p inhibits glioma cell proliferation and progression by targeting GABRA1. J. Transl. Med. 2021, 19, 213. [Google Scholar] [CrossRef]
- Ferretti, M.; Fabbiano, C.; Di Bari, M.; Ponti, D.; Calogero, A.; Tata, A.M. M2 muscarinic receptors inhibit cell proliferation in human glioblastoma cell lines. Life Sci. 2012, 91, 1134–1137. [Google Scholar] [CrossRef] [PubMed]
- Ferretti, M.; Fabbiano, C.; Bari, M.D.; Ponti, D.; Calogero, A.; Tata, A.M. M2 receptor activation inhibits cell cycle progression and survival in human glioblastoma cells. J. Cell. Mol. Med. 2013, 17, 552–566. [Google Scholar] [CrossRef] [PubMed]
- Venkatesh, H.S.; Johung, T.B.; Caretti, V.; Noll, A.; Tang, Y.; Nagaraja, S.; Gibson, E.M.; Mount, C.W.; Polepalli, J.; Mitra, S.S.; et al. Neuronal Activity Promotes Glioma Growth through Neuroligin-3 Secretion. Cell 2015, 161, 803–816. [Google Scholar] [CrossRef]
- Venkatesh, H.S.; Tam, L.T.; Woo, P.J.; Lennon, J.; Nagaraja, S.; Gillespie, S.M.; Ni, J.; Duveau, D.Y.; Morris, P.J.; Zhao, J.J.; et al. Targeting neuronal activity-regulated neuroligin-3 dependency in high-grade glioma. Nature 2017, 549, 533–537. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Xu, Y.; Zhu, H.; Wang, Y.; Li, P.; Wang, D. The dark side of synaptic proteins in tumours. Br. J. Cancer 2022, 127, 1184–1192. [Google Scholar] [CrossRef]
- Liu, R.; Qin, X.; Zhuang, Y.; Zhang, Y.; Liao, H.; Tang, J.; Pan, M.; Zeng, F.; Lei, Y.; Lei, R.; et al. Glioblastoma recurrence correlates with NLGN3 levels. Cancer Med. 2018, 7, 2848–2859. [Google Scholar] [CrossRef]
- Pauletto, G.; Nilo, A.; Lettieri, C.; Verriello, L.; Tomasino, B.; Gigli, G.L.; Skrap, M.; Ius, T. Pre- and Post-surgical Poor Seizure Control as Hallmark of Malignant Progression in Patients With Glioma? Front. Neurol. 2022, 13, 890857. [Google Scholar] [CrossRef]
- de Groot, J.F.; Liu, T.J.; Fuller, G.; Yung, W.K.A. The excitatory amino acid transporter-2 induces apoptosis and decreases glioma growth in vitro and in vivo. Cancer Res. 2005, 65, 1934–1940. [Google Scholar] [CrossRef]
- Marcus, H.J.; Carpenter, K.L.H.; Price, S.J.; Hutchinson, P.J. In vivo assessment of high-grade glioma biochemistry using microdialysis: A study of energy-related molecules, growth factors and cytokines. J. Neurooncol. 2010, 97, 11–23. [Google Scholar] [CrossRef]
- Sonoda, Y.; Ozawa, T.; Aldape, K.D.; Deen, D.F.; Berger, M.S.; Pieper, R.O. Akt pathway activation converts anaplastic astrocytoma to glioblastoma multiforme in a human astrocyte model of glioma. Cancer Res. 2001, 61, 6674–6678. [Google Scholar]
- Wang, J.; Wang, H.; Li, Z.; Wu, Q.; Lathia, J.D.; McLendon, R.E.; Hjelmeland, A.B.; Rich, J.N. c-Myc Is Required for Maintenance of Glioma Cancer Stem Cells. PLoS ONE 2008, 3, e3769. [Google Scholar] [CrossRef] [PubMed]
- Corsi, L.; Mescola, A.; Alessandrini, A. Glutamate Receptors and Glioblastoma Multiforme: An Old “Route” for New Perspectives. Int. J. Mol. Sci. 2019, 20, 1796. [Google Scholar] [CrossRef]
- Li, X.; Wu, C.; Chen, N.; Gu, H.; Yen, A.; Cao, L.; Wang, E.; Wang, L. PI3K/Akt/mTOR signaling pathway and targeted therapy for glioblastoma. Oncotarget 2016, 7, 33440–33450. [Google Scholar] [CrossRef] [PubMed]
- Gao, Q.; Lei, T.; Ye, F. Therapeutic targeting of EGFR-activated metabolic pathways in glioblastoma. Expert Opin. Investig. Drugs 2013, 22, 1023–1040. [Google Scholar] [CrossRef]
- Schunemann, D.P.; Grivicich, I.; Regner, A.; Leal, L.F.; de Araujo, D.R.; Jotz, G.P.; Fedrigo, C.A.; Simon, D.; da Rocha, A.B. Glutamate promotes cell growth by EGFR signaling on U-87MG human glioblastoma cell line. Pathol. Oncol. Res. POR 2010, 16, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Ishiuchi, S.; Yoshida, Y.; Sugawara, K.; Aihara, M.; Ohtani, T.; Watanabe, T.; Saito, N.; Tsuzuki, K.; Okaro, H.; Miwa, A.; et al. Ca2+-Permeable AMPA Receptors Regulate Growth of Human Glioblastoma via Akt Activation. J. Neurosci. 2007, 27, 7987–8001. [Google Scholar] [CrossRef]
- Lutz, H.; Nguyen, T.A.; Joswig, J.; Rau, K.; Laube, B. NMDA Receptor Signaling Mediates cFos Expression via Top2β-Induced DSBs in Glioblastoma Cells. Cancers. 2019, 11, 306. [Google Scholar] [CrossRef]
- Liu, Z.G.; Jiang, G.; Tang, J.; Wang, H.; Feng, G.; Chen, F.; Tu, Z.; Liu, G.; Zhao, Y.; Peng, M.; et al. c-Fos over-expression promotes radioresistance and predicts poor prognosis in malignant glioma. Oncotarget 2016, 7, 65946. [Google Scholar] [CrossRef]
- Ignarro, R.S.; Facchini, G.; Vieira, A.S.; De Melo, D.R.; Lopes-Cendes, I.; Castilho, R.F.; Rogerio, F. Sulfasalazine intensifies temozolomide cytotoxicity in human glioblastoma cells. Mol. Cell Biochem. 2016, 418, 167–178. [Google Scholar] [CrossRef]
- Robe, P.A.; Martin, D.H.; Nguyen-Khac, M.T.; Artesi, M.; Deprez, M.; Albert, A.; Vanbelle, S.; Califice, S.; Bredel, M.; Bours, V. Early termination of ISRCTN45828668, a phase 1/2 prospective, randomized study of sulfasalazine for the treatment of progressing malignant gliomas in adults. BMC Cancer 2009, 9, 372. [Google Scholar] [CrossRef]
- Takeuchi, S.; Wada, K.; Nagatani, K.; Otani, N.; Osada, H.; Nawashiro, H. Sulfasalazine and temozolomide with radiation therapy for newly diagnosed glioblastoma. Neurol. India 2014, 62, 42. [Google Scholar] [CrossRef] [PubMed]
- Walczak, K.; Deneka-Hannemann, S.; Jarosz, B.; Zgrajka, W.; Stoma, F.; Trojanowski, T.; Turski, W.A.; Rzeski, W. Kynurenic acid inhibits proliferation and migration of human glioblastoma T98G cells. Pharmacol. Rep. PR 2014, 66, 130–136. [Google Scholar] [CrossRef]
- Grossman, S.A.; Ye, X.; Chamberlain, M.; Mikkelsen, T.; Bathcelor, T.; Desideri, S.; Piantadosi, S.; Fisher, J.; Fine, H.A. Talampanel With Standard Radiation and Temozolomide in Patients with Newly Diagnosed Glioblastoma: A Multicenter Phase II Trial. J. Clin. Oncol. 2009, 27, 4155–4161. [Google Scholar] [CrossRef] [PubMed]
- Iwamoto, F.M.; Kreisl, T.N.; Kim, L.; Duic, J.P.; Butman, J.A.; Albert, P.S.; Fine, H.A. Phase 2 trial of talampanel, a glutamate receptor inhibitor, for adults with recurrent malignant gliomas. Cancer 2010, 116, 1776–1782. [Google Scholar] [CrossRef]
- Trinka, E.; Steinhoff, B.J.; Nikanorova, M.; Brodie, M.J. Perampanel for focal epilepsy: Insights from early clinical experience. Acta Neurol. Scand. 2016, 133, 160–172. [Google Scholar] [CrossRef]
- Meador, K.J.; Yang, H.; Piña-Garza, J.E.; Laurenza, A.; Kumar, D.; Wesnes, K.A. Cognitive effects of adjunctive perampanel for partial-onset seizures: A randomized trial. Epilepsia 2016, 57, 243–251. [Google Scholar] [CrossRef]
- Eskilsson, E.; Røsland, G.V.; Solecki, G.; Wang, Q.; Harter, P.N.; Graziani, G.; Verhaak, R.G.W.; Winkler, F.; Bjerkvig, R.; Miletic, H. EGFR heterogeneity and implications for therapeutic intervention in glioblastoma. Neuro-Oncology 2018, 20, 743–752. [Google Scholar] [CrossRef] [PubMed]
- Thorne, A.H.; Zanca, C.; Furnari, F. Epidermal growth factor receptor targeting and challenges in glioblastoma. Neuro-Oncology 2016, 18, 914–918. [Google Scholar] [CrossRef]
- Westphal, M.; Maire, C.L.; Lamszus, K. EGFR as a Target for Glioblastoma Treatment: An Unfulfilled Promise. CNS Drugs 2017, 31, 723–735. [Google Scholar] [CrossRef]
- Nicoletti, N.F.; Erig, T.C.; Zanin, R.F.; Rozo, M.R.; Ferreira, N.P.; Gomez, M.V.; Morrone, F.B.; Campos, M.M. Pre-clinical evaluation of voltage-gated calcium channel blockers derived from the spider P. nigriventer in glioma progression. Toxicon Off. J. Int. Soc. Toxinology 2017, 129, 58–67. [Google Scholar] [CrossRef]
- De Meulenaere, V.; Bonte, E.; Verhoeven, J.; Kalala Okito, J.; Pieters, L.; Vral, A.; De Wever, O.; Leybaert, L.; Goethals, I.; Vanhove, C.; et al. Adjuvant therapeutic potential of tonabersat in the standard treatment of glioblastoma: A preclinical F98 glioblastoma rat model study. PLoS ONE 2019, 14, e0224130. [Google Scholar] [CrossRef] [PubMed]
- Happold, C.; Gorlia, T.; Chinot, O.; Gilbert, M.R.; Nabors, L.B.; Wick, W.; Pugh, S.L.; Hegi, M.; Cloughesy, T.; Roth, P.; et al. Does Valproic Acid or Levetiracetam Improve Survival in Glioblastoma? A Pooled Analysis of Prospective Clinical Trials in Newly Diagnosed Glioblastoma. J. Clin. Oncol. 2016, 34, 731–739. [Google Scholar] [CrossRef] [PubMed]
- Pallud, J.; Huberfeld, G.; Dezamis, E.; Peeters, S.; Moiraghi, A.; Gavaret, M.; Guinard, E.; Dhermain, F.; Varlet, P.; Oppenheim, C.; et al. Effect of Levetiracetam Use Duration on Overall Survival of Isocitrate Dehydrogenase Wild-Type Glioblastoma in Adults: An Observational Study. Neurology 2022, 98, e125–e140. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Goethe, E.A.; Deneen, B.; Noebels, J.; Rao, G. The Role of Hyperexcitability in Gliomagenesis. Int. J. Mol. Sci. 2023, 24, 749. https://doi.org/10.3390/ijms24010749
Goethe EA, Deneen B, Noebels J, Rao G. The Role of Hyperexcitability in Gliomagenesis. International Journal of Molecular Sciences. 2023; 24(1):749. https://doi.org/10.3390/ijms24010749
Chicago/Turabian StyleGoethe, Eric A., Benjamin Deneen, Jeffrey Noebels, and Ganesh Rao. 2023. "The Role of Hyperexcitability in Gliomagenesis" International Journal of Molecular Sciences 24, no. 1: 749. https://doi.org/10.3390/ijms24010749
APA StyleGoethe, E. A., Deneen, B., Noebels, J., & Rao, G. (2023). The Role of Hyperexcitability in Gliomagenesis. International Journal of Molecular Sciences, 24(1), 749. https://doi.org/10.3390/ijms24010749