

Transcriptome Profile in the Mouse Brain of Hepatic Encephalopathy and Alzheimer’s Disease


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
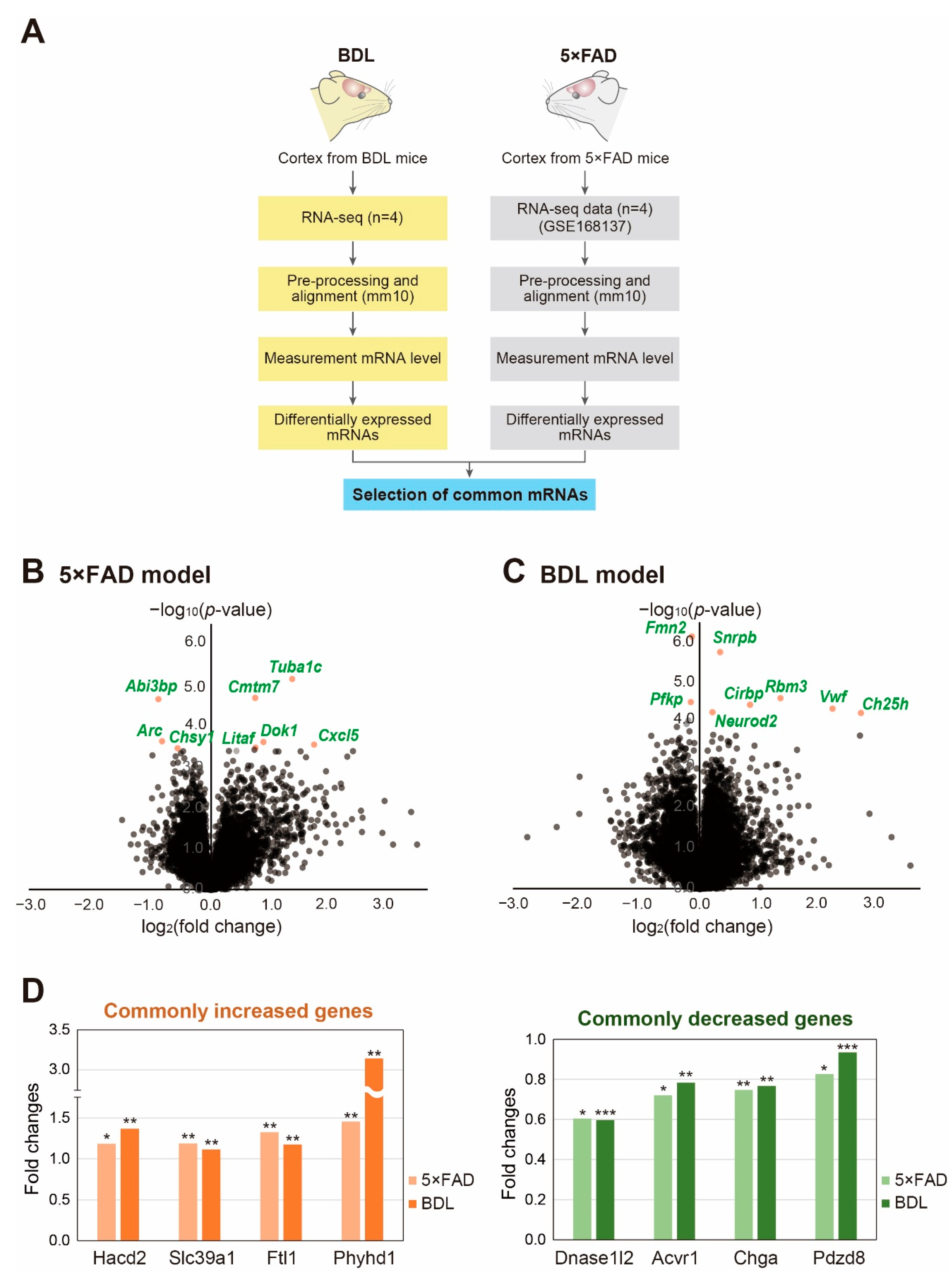
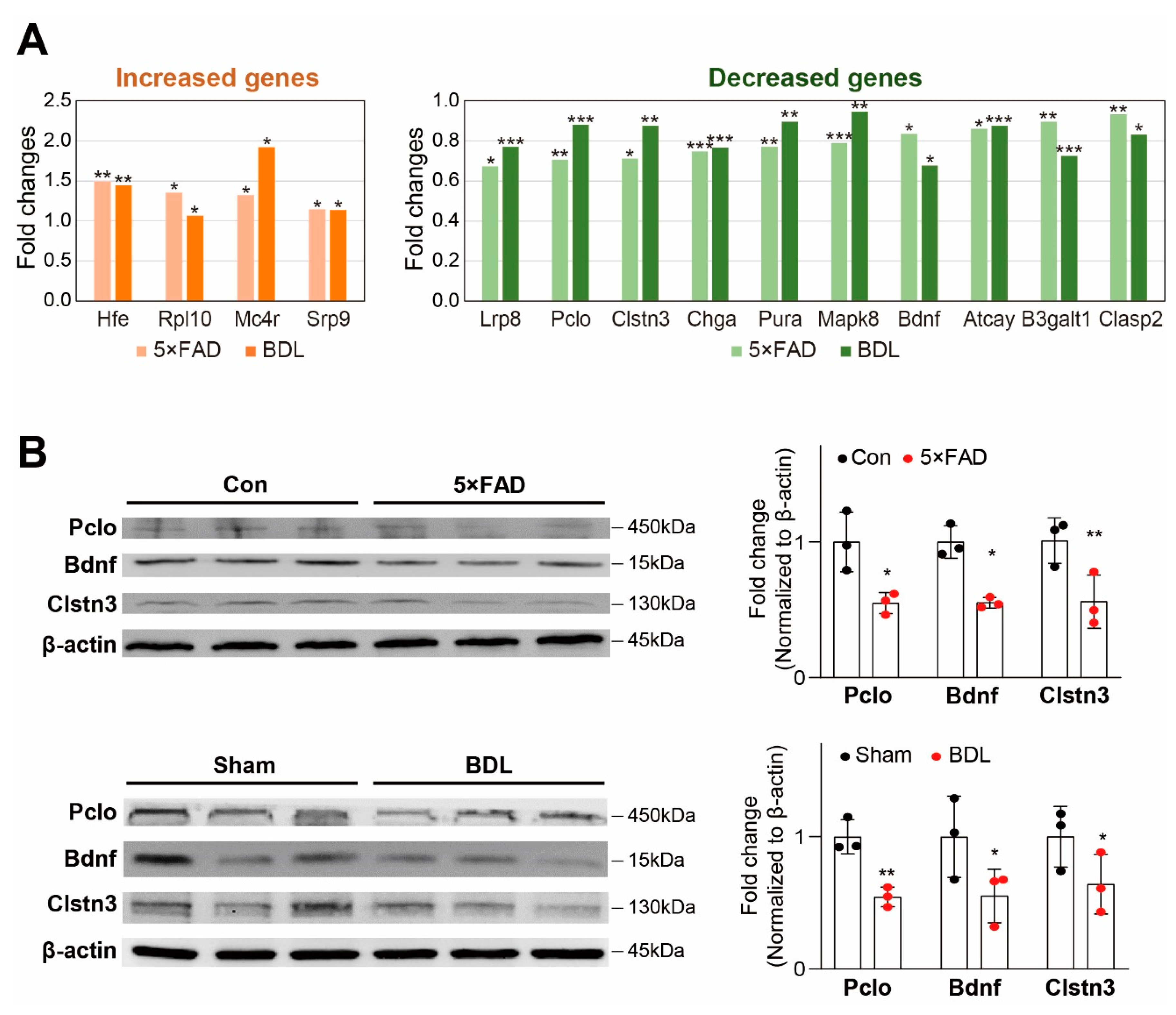
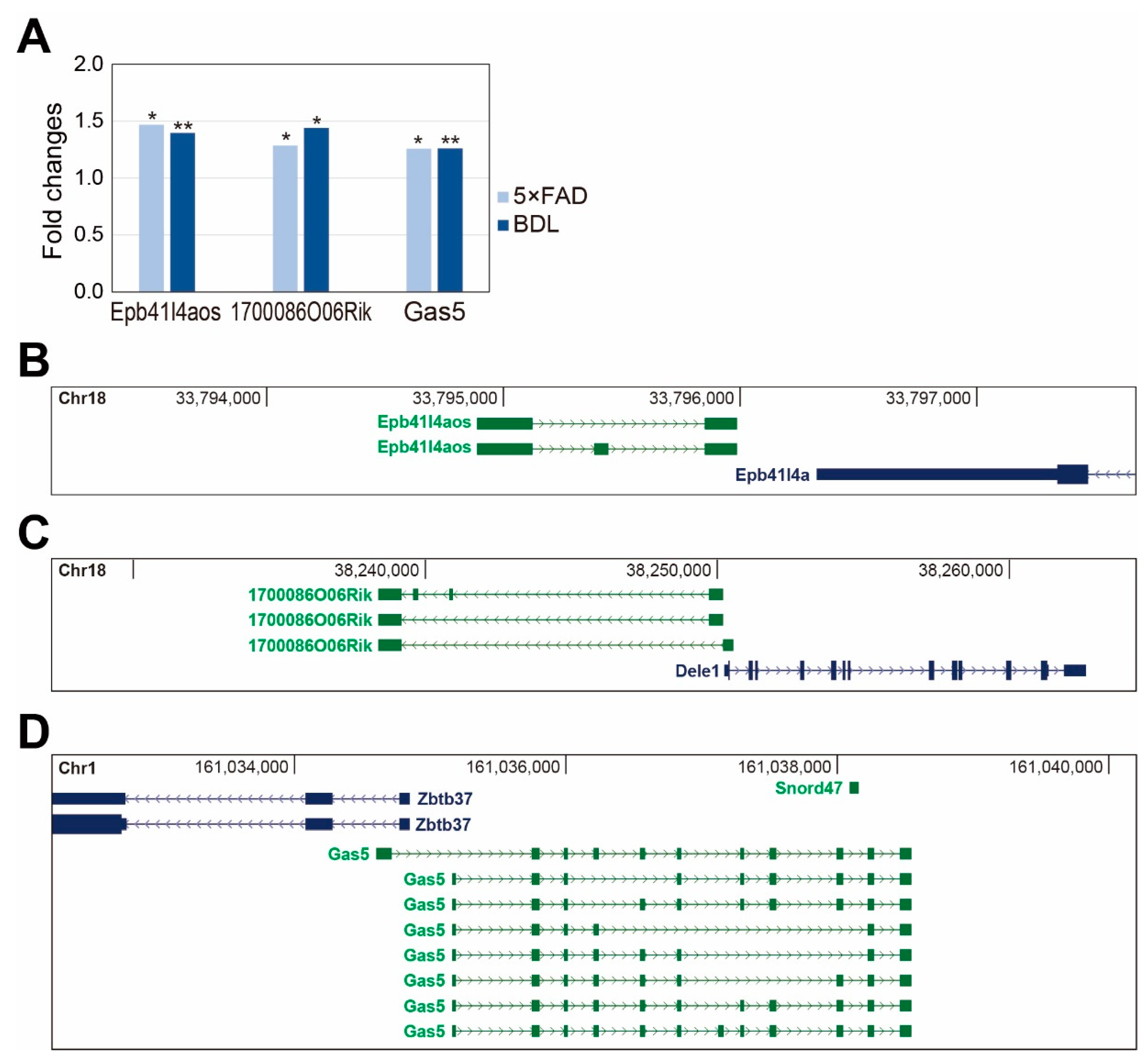
2. Results
3. Discussion
4. Materials and Methods
4.1. Preparation of Animals for BDL Surgery
4.2. Preparation of 5×FAD Brain Cortex
4.3. RNA Sequencing
4.4. The Data Used to Analyze the Transcriptome of the 5×FAD Mouse
4.5. Analysis of RNA Sequencing Data
4.6. Functional Analysis of Differentially Expressed Genes
4.7. Western Blot Analysis
4.8. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Vilstrup, H.; Amodio, P.; Bajaj, J.; Cordoba, J.; Ferenci, P.; Mullen, K.D.; Weissenborn, K.; Wong, P. Hepatic encephalopathy in chronic liver disease: 2014 practice guideline by the american association for the study of liver diseases and the european association for the study of the liver. Hepatology 2014, 60, 715–735. [Google Scholar] [CrossRef] [PubMed]
- Yanny, B.; Winters, A.; Boutros, S.; Saab, S. Hepatic encephalopathy challenges, burden, and diagnostic and therapeutic approach. Clin. Liver Dis. 2019, 23, 607–623. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Franco, O.; Morin, J.P.; Cortes-Sol, A.; Molina-Jimenez, T.; Del Moral, D.I.; Flores-Munoz, M.; Roldan-Roldan, G.; Juarez-Portilla, C.; Zepeda, R.C. Cognitive impairment after resolution of hepatic encephalopathy: A systematic review and meta-analysis. Front. Neurosci. 2021, 15, 579263. [Google Scholar] [CrossRef] [PubMed]
- Basu, P.P.; Shah, N.J. Clinical and neurologic manifestation of minimal hepatic encephalopathy and overt hepatic encephalopathy. Clin. Liver Dis. 2015, 19, 461–472. [Google Scholar] [CrossRef] [PubMed]
- Torres, D.S.; Abrantes, J.; Brandao-Mello, C.E. Cognitive and neurophysiological assessment of patients with minimal hepatic encephalopathy in brazil. Sci. Rep. 2020, 10, 8610. [Google Scholar] [CrossRef]
- Ferenci, P. Hepatic encephalopathy. Gastroenterol. Rep. 2017, 5, 138–147. [Google Scholar] [CrossRef]
- Garcia-Garcia, R.; Cruz-Gomez, A.J.; Urios, A.; Mangas-Losada, A.; Forn, C.; Escudero-Garcia, D.; Kosenko, E.; Torregrosa, I.; Tosca, J.; Giner-Duran, R.; et al. Learning and memory impairments in patients with minimal hepatic encephalopathy are associated with structural and functional connectivity alterations in hippocampus. Sci. Rep. 2018, 8, 9664. [Google Scholar] [CrossRef]
- Claeys, W.; Van Hoecke, L.; Geerts, A.; Van Vlierberghe, H.; Lefere, S.; Van Imschoot, G.; Van Wonterghem, E.; Ghesquiere, B.; Vandenbroucke, R.E.; Van Steenkiste, C. A mouse model of hepatic encephalopathy: Bile duct ligation induces brain ammonia overload, glial cell activation and neuroinflammation. Sci. Rep. 2022, 12, 17558. [Google Scholar] [CrossRef]
- Drews, L.; Zimmermann, M.; Westhoff, P.; Brilhaus, D.; Poss, R.E.; Bergmann, L.; Wiek, C.; Brenneisen, P.; Piekorz, R.P.; Mettler-Altmann, T.; et al. Ammonia inhibits energy metabolism in astrocytes in a rapid and glutamate dehydrogenase 2-dependent manner. Dis. Model. Mech. 2020, 13, dmm047134. [Google Scholar] [CrossRef]
- Magen, I.; Avraham, Y.; Ackerman, Z.; Vorobiev, L.; Mechoulam, R.; Berry, E.M. Cannabidiol ameliorates cognitive and motor impairments in mice with bile duct ligation. J. Hepatol. 2009, 51, 528–534. [Google Scholar] [CrossRef]
- Butterworth, R.F.; Norenberg, M.D.; Felipo, V.; Ferenci, P.; Albrecht, J.; Blei, A.T.; ISHEN Commission on Experimental Models of HE. Experimental models of hepatic encephalopathy: Ishen guidelines. Liver Int. 2009, 29, 783–788. [Google Scholar] [CrossRef] [PubMed]
- Nasehi, M.; Mafi, F.; Ebrahimi-Ghiri, M.; Zarrindast, M.R. Function of opioidergic and dopaminergic antagonists on both spatial and object novelty detection deficits induced in rodent model of hepatic encephalopathy. Behav. Brain Res. 2016, 313, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Cho, I.; Koo, B.N.; Kam, E.H.; Lee, S.K.; Oh, H.; Kim, S.Y. Bile duct ligation of c57bl/6 mice as a model of hepatic encephalopathy. Anesth. Pain Med. 2020, 15, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Starkel, P.; Leclercq, I.A. Animal models for the study of hepatic fibrosis. Best Pract. Res. Clin. Gastroenterol. 2011, 25, 319–333. [Google Scholar] [CrossRef]
- Tag, C.G.; Weiskirchen, S.; Hittatiya, K.; Tacke, F.; Tolba, R.H.; Weiskirchen, R. Induction of experimental obstructive cholestasis in mice. Lab. Anim. 2015, 49, 70–80. [Google Scholar] [CrossRef]
- Tag, C.G.; Sauer-Lehnen, S.; Weiskirchen, S.; Borkham-Kamphorst, E.; Tolba, R.H.; Tacke, F.; Weiskirchen, R. Bile duct ligation in mice: Induction of inflammatory liver injury and fibrosis by obstructive cholestasis. J. Vis. Exp. 2015, 96, e52438. [Google Scholar] [CrossRef]
- Heron, M. Deaths: Leading causes for 2014. Natl. Vital Stat. Rep. Cent. Dis. Control. Prev. Natl. Cent. Health Stat. Natl. Vital Stat. Syst. 2016, 65, 1–96. [Google Scholar]
- Kapogiannis, D.; Mattson, M.P. Disrupted energy metabolism and neuronal circuit dysfunction in cognitive impairment and alzheimer’s disease. Lancet Neurol. 2011, 10, 187–198. [Google Scholar] [CrossRef]
- Craft, S. The role of metabolic disorders in alzheimer disease and vascular dementia: Two roads converged. Arch. Neurol. 2009, 66, 300–305. [Google Scholar] [CrossRef]
- Zhang, J.; Chen, C.; Hua, S.; Liao, H.; Wang, M.; Xiong, Y.; Cao, F. An updated meta-analysis of cohort studies: Diabetes and risk of alzheimer’s disease. Diabetes Res. Clin. Pract. 2017, 124, 41–47. [Google Scholar] [CrossRef]
- Sookoian, S.; Castano, G.O.; Scian, R.; Fernandez Gianotti, T.; Dopazo, H.; Rohr, C.; Gaj, G.; San Martino, J.; Sevic, I.; Flichman, D.; et al. Serum aminotransferases in nonalcoholic fatty liver disease are a signature of liver metabolic perturbations at the amino acid and krebs cycle level. Am. J. Clin. Nutr. 2016, 103, 422–434. [Google Scholar] [CrossRef] [PubMed]
- Fillit, H.; Nash, D.T.; Rundek, T.; Zuckerman, A. Cardiovascular risk factors and dementia. Am. J. Geriatr. Pharmacother. 2008, 6, 100–118. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wen, D.X.; Zhao, Y.H.; Hang, Y.N.; Mandell, M.S. Increase of beta-amyloid and c-reactive protein in liver transplant recipients with postoperative cognitive dysfunction. Hepatobiliary Pancreat. Dis. Int. 2013, 12, 370–376. [Google Scholar] [CrossRef] [PubMed]
- Nho, K.; Kueider-Paisley, A.; Ahmad, S.; MahmoudianDehkordi, S.; Arnold, M.; Risacher, S.L.; Louie, G.; Blach, C.; Baillie, R.; Han, X.; et al. Association of altered liver enzymes with alzheimer disease diagnosis, cognition, neuroimaging measures, and cerebrospinal fluid biomarkers. JAMA Netw. Open 2019, 2, e197978. [Google Scholar] [CrossRef]
- Musiek, E.S.; Xiong, D.D.; Holtzman, D.M. Sleep, circadian rhythms, and the pathogenesis of alzheimer disease. Exp. Mol. Med. 2015, 47, e148. [Google Scholar] [CrossRef] [PubMed]
- Chiu, W.C.; Tsan, Y.T.; Tsai, S.L.; Chang, C.J.; Wang, J.D.; Chen, P.C.; Health Data Analysis in Taiwan (hDATa) Research Group. Hepatitis c viral infection and the risk of dementia. Eur. J. Neurol. 2014, 21, 1068-e59. [Google Scholar] [CrossRef]
- Huang da, W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using david bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. String v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef]
- Fang, Y.; Fullwood, M.J. Roles, functions, and mechanisms of long non-coding rnas in cancer. Genom. Proteom. Bioinform. 2016, 14, 42–54. [Google Scholar] [CrossRef]
- Gui, S.; Chen, P.; Liu, Y.; Chen, Q.; Cheng, T.; Lv, S.; Zhou, T.; Song, Z.; Xiao, J.; He, W.; et al. Tuba1c expression promotes proliferation by regulating the cell cycle and indicates poor prognosis in glioma. Biochem. Biophys. Res. Commun. 2021, 577, 130–138. [Google Scholar] [CrossRef]
- Zhu, H.; Hu, X.; Gu, L.; Jian, Z.; Li, L.; Hu, S.; Qiu, S.; Xiong, X. Tuba1c is a prognostic marker in low-grade glioma and correlates with immune cell infiltration in the tumor microenvironment. Front. Genet. 2021, 12, 759953. [Google Scholar] [CrossRef] [PubMed]
- Albahde, M.A.H.; Zhang, P.; Zhang, Q.; Li, G.; Wang, W. Upregulated expression of tuba1c predicts poor prognosis and promotes oncogenesis in pancreatic ductal adenocarcinoma via regulating the cell cycle. Front. Oncol. 2020, 10, 49. [Google Scholar] [CrossRef] [PubMed]
- Shinohara, H.; Inoue, A.; Toyama-Sorimachi, N.; Nagai, Y.; Yasuda, T.; Suzuki, H.; Horai, R.; Iwakura, Y.; Yamamoto, T.; Karasuyama, H.; et al. Dok-1 and dok-2 are negative regulators of lipopolysaccharide-induced signaling. J. Exp. Med. 2005, 201, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Yamanashi, Y.; Tamura, T.; Kanamori, T.; Yamane, H.; Nariuchi, H.; Yamamoto, T.; Baltimore, D. Role of the rasgap-associated docking protein p62(dok) in negative regulation of b cell receptor-mediated signaling. Genes Dev. 2000, 14, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Phillips, J.H. Identification of tyrosine residues crucial for cd200r-mediated inhibition of mast cell activation. J. Leukoc. Biol. 2006, 79, 363–368. [Google Scholar] [CrossRef]
- Downer, E.J.; Johnston, D.G.; Lynch, M.A. Differential role of dok1 and dok2 in tlr2-induced inflammatory signaling in glia. Mol. Cell. Neurosci. 2013, 56, 148–158. [Google Scholar] [CrossRef]
- Gaetani, L.; Bellomo, G.; Parnetti, L.; Blennow, K.; Zetterberg, H.; Di Filippo, M. Neuroinflammation and alzheimer’s disease: A machine learning approach to csf proteomics. Cells 2021, 10, 1930. [Google Scholar] [CrossRef]
- Landgren, S.; von Otter, M.; Palmer, M.S.; Zetterstrom, C.; Nilsson, S.; Skoog, I.; Gustafson, D.R.; Minthon, L.; Wallin, A.; Andreasen, N.; et al. A novel arc gene polymorphism is associated with reduced risk of alzheimer’s disease. J. Neural Transm. 2012, 119, 833–842. [Google Scholar] [CrossRef]
- Leung, H.W.; Foo, G.; VanDongen, A. Arc regulates transcription of genes for plasticity, excitability and alzheimer’s disease. Biomedicines 2022, 10, 1946. [Google Scholar] [CrossRef]
- Correa, B.R.; de Araujo, P.R.; Qiao, M.; Burns, S.C.; Chen, C.; Schlegel, R.; Agarwal, S.; Galante, P.A.; Penalva, L.O. Functional genomics analyses of rna-binding proteins reveal the splicing regulator snrpb as an oncogenic candidate in glioblastoma. Genome Biol. 2016, 17, 125. [Google Scholar] [CrossRef]
- Zhu, X.; Yan, J.; Bregere, C.; Zelmer, A.; Goerne, T.; Kapfhammer, J.P.; Guzman, R.; Wellmann, S. Rbm3 promotes neurogenesis in a niche-dependent manner via imp2-igf2 signaling pathway after hypoxic-ischemic brain injury. Nat. Commun. 2019, 10, 3983. [Google Scholar] [CrossRef] [PubMed]
- Sertel, S.M.; von Elling-Tammen, M.S.; Rizzoli, S.O. The mrna-binding protein rbm3 regulates activity patterns and local synaptic translation in cultured hippocampal neurons. J. Neurosci. Off. J. Soc. Neurosci. 2021, 41, 1157–1173. [Google Scholar] [CrossRef]
- Lin, C.H.; Hansen, S.; Wang, Z.; Storm, D.R.; Tapscott, S.J.; Olson, J.M. The dosage of the neurod2 transcription factor regulates amygdala development and emotional learning. Proc. Natl. Acad. Sci. USA 2005, 102, 14877–14882. [Google Scholar] [CrossRef] [PubMed]
- Wilke, S.A.; Hall, B.J.; Antonios, J.K.; Denardo, L.A.; Otto, S.; Yuan, B.; Chen, F.; Robbins, E.M.; Tiglio, K.; Williams, M.E.; et al. Neurod2 regulates the development of hippocampal mossy fiber synapses. Neural Dev. 2012, 7, 9. [Google Scholar] [CrossRef] [PubMed]
- Noubade, R.; del Rio, R.; McElvany, B.; Zachary, J.F.; Millward, J.M.; Wagner, D.D.; Offner, H.; Blankenhorn, E.P.; Teuscher, C. Von-willebrand factor influences blood brain barrier permeability and brain inflammation in experimental allergic encephalomyelitis. Am. J. Pathol. 2008, 173, 892–900. [Google Scholar] [CrossRef] [PubMed]
- Hollestelle, M.J.; Donkor, C.; Mantey, E.A.; Chakravorty, S.J.; Craig, A.; Akoto, A.O.; O’Donnell, J.; van Mourik, J.A.; Bunn, J. Von willebrand factor propeptide in malaria: Evidence of acute endothelial cell activation. Br. J. Haematol. 2006, 133, 562–569. [Google Scholar] [CrossRef]
- Sweeney, M.D.; Sagare, A.P.; Zlokovic, B.V. Blood-brain barrier breakdown in alzheimer disease and other neurodegenerative disorders. Nat. Rev. Neurol. 2018, 14, 133–150. [Google Scholar] [CrossRef]
- Papassotiropoulos, A.; Lambert, J.C.; Wavrant-De Vrieze, F.; Wollmer, M.A.; von der Kammer, H.; Streffer, J.R.; Maddalena, A.; Huynh, K.D.; Wolleb, S.; Lutjohann, D.; et al. Cholesterol 25-hydroxylase on chromosome 10q is a susceptibility gene for sporadic alzheimer’s disease. Neurodegener. Dis. 2005, 2, 233–241. [Google Scholar] [CrossRef]
- Jang, J.; Park, S.; Hur, H.J.; Cho, H.J.; Hwang, I.; Kang, Y.P.; Im, I.; Lee, H.; Lee, E.; Yang, W.; et al. 25-hydroxycholesterol contributes to cerebral inflammation of x-linked adrenoleukodystrophy through activation of the nlrp3 inflammasome. Nat. Commun. 2016, 7, 13129. [Google Scholar] [CrossRef]
- Yamada, K.; Ono, M.; Bensaddek, D.; Lamond, A.I.; Rocha, S. Fmn2 is a novel regulator of the cyclin-dependent kinase inhibitor p21. Cell Cycle 2013, 12, 2348–2354. [Google Scholar] [CrossRef]
- Agis-Balboa, R.C.; Pinheiro, P.S.; Rebola, N.; Kerimoglu, C.; Benito, E.; Gertig, M.; Bahari-Javan, S.; Jain, G.; Burkhardt, S.; Delalle, I.; et al. Formin 2 links neuropsychiatric phenotypes at young age to an increased risk for dementia. EMBO J. 2017, 36, 2815–2828. [Google Scholar] [CrossRef] [PubMed]
- Wen, S.; Schroeter, A.; Klocker, N. Synaptic plasticity in hepatic encephalopathy—A molecular perspective. Arch. Biochem. Biophys. 2013, 536, 183–188. [Google Scholar] [CrossRef]
- Sawai, M.; Uchida, Y.; Ohno, Y.; Miyamoto, M.; Nishioka, C.; Itohara, S.; Sassa, T.; Kihara, A. The 3-hydroxyacyl-coa dehydratases hacd1 and hacd2 exhibit functional redundancy and are active in a wide range of fatty acid elongation pathways. J. Biol. Chem. 2017, 292, 15538–15551. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Zhang, J.; He, S.; Xiao, B.; Peng, X. Slc39a1 contribute to malignant progression and have clinical prognostic impact in gliomas. Cancer Cell Int. 2020, 20, 573. [Google Scholar] [CrossRef] [PubMed]
- Scarr, E.; Udawela, M.; Greenough, M.A.; Neo, J.; Seo, M.S.; Money, T.T.; Upadhyay, A.; Bush, A.I.; Everall, I.P.; Thomas, E.A.; et al. Increased cortical expression of the zinc transporter slc39a12 suggests a breakdown in zinc cellular homeostasis as part of the pathophysiology of schizophrenia. NPJ Schizophr. 2016, 2, 16002. [Google Scholar] [CrossRef]
- Ni, W.; Li, H.F.; Zheng, Y.C.; Wu, Z.Y. Ftl mutation in a chinese pedigree with neuroferritinopathy. Neurol. Genet. 2016, 2, e74. [Google Scholar] [CrossRef]
- Yoon, S.H.; Kim, N.Y.; Kim, Y.J.; Lyoo, C.H. Novel ferritin light chain gene mutation in a korean patient with neuroferritinopathy. J. Mov. Disord. 2019, 12, 63–65. [Google Scholar] [CrossRef]
- David, S.; Jhelum, P.; Ryan, F.; Jeong, S.Y.; Kroner, A. Dysregulation of iron homeostasis in the central nervous system and the role of ferroptosis in neurodegenerative disorders. Antioxid. Redox Signal. 2022, 37, 150–170. [Google Scholar] [CrossRef]
- Tetz, V.; Tetz, G. Effect of deoxyribonuclease i treatment for dementia in end-stage alzheimer’s disease: A case report. J. Med. Case Rep. 2016, 10, 131. [Google Scholar] [CrossRef]
- Gibson, R.L.; Burns, J.L.; Ramsey, B.W. Pathophysiology and management of pulmonary infections in cystic fibrosis. Am. J. Respir. Crit. Care Med. 2003, 168, 918–951. [Google Scholar] [CrossRef]
- Wen, F.; Shen, A.; Choi, A.; Gerner, E.W.; Shi, J. Extracellular DNA in pancreatic cancer promotes cell invasion and metastasis. Cancer Res. 2013, 73, 4256–4266. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, F.S.; Kobori, J.A.; Orellana, C.; Calvo, I.; Rosello, M.; Martinez, F.; Lopez, B.; Xu, M.; Pignolo, R.J.; Shore, E.M.; et al. Multi-system involvement in a severe variant of fibrodysplasia ossificans progressiva (acvr1 c.772g>a; r258g): A report of two patients. Am. J. Med. Genet. A 2015, 167A, 2265–2271. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, F.S.; Xu, M.; Seemann, P.; Connor, J.M.; Glaser, D.L.; Carroll, L.; Delai, P.; Fastnacht-Urban, E.; Forman, S.J.; Gillessen-Kaesbach, G.; et al. Classic and atypical fibrodysplasia ossificans progressiva (fop) phenotypes are caused by mutations in the bone morphogenetic protein (bmp) type i receptor acvr1. Hum. Mutat. 2009, 30, 379–390. [Google Scholar] [CrossRef]
- Kitterman, J.A.; Strober, J.B.; Kan, L.; Rocke, D.M.; Cali, A.; Peeper, J.; Snow, J.; Delai, P.L.; Morhart, R.; Pignolo, R.J.; et al. Neurological symptoms in individuals with fibrodysplasia ossificans progressiva. J. Neurol. 2012, 259, 2636–2643. [Google Scholar] [CrossRef] [PubMed]
- Horgusluoglu-Moloch, E.; Risacher, S.L.; Crane, P.K.; Hibar, D.; Thompson, P.M.; Saykin, A.J.; Nho, K.; Alzheimer’s Disease Neuroimaging Initiative (ADNI). Genome-wide association analysis of hippocampal volume identifies enrichment of neurogenesis-related pathways. Sci. Rep. 2019, 9, 14498. [Google Scholar] [CrossRef] [PubMed]
- Hewitt, V.L.; Miller-Fleming, L.; Twyning, M.J.; Andreazza, S.; Mattedi, F.; Prudent, J.; Polleux, F.; Vagnoni, A.; Whitworth, A.J. Decreasing pdzd8-mediated mito-er contacts improves organismal fitness and mitigates abeta42 toxicity. Life Sci. Alliance 2022, 5, e202201531. [Google Scholar] [CrossRef]
- Artemov, A.V.; Boulygina, E.S.; Tsygankova, S.V.; Nedoluzhko, A.V.; Chekanov, N.N.; Gruzdeva, N.M.; Selezneva, N.D.; Roshchina, I.F.; Gavrilova, S.I.; Velichkovsky, B.B.; et al. Study of alzheimer family case reveals hemochromotosis-associated hfe mutation. Hum. Genome Var. 2014, 1, 14004. [Google Scholar] [CrossRef]
- Abo El Gheit, R.E.; Atef, M.M.; Badawi, G.A.; Elwan, W.M.; Alshenawy, H.A.; Emam, M.N. Role of serine protease inhibitor, ulinastatin, in rat model of hepatic encephalopathy: Aquaporin 4 molecular targeting and therapeutic implication. J. Physiol. Biochem. 2020, 76, 573–586. [Google Scholar] [CrossRef]
- Hessel, E.V.; de Wit, M.; Wolterink-Donselaar, I.G.; Karst, H.; de Graaff, E.; van Lith, H.A.; de Bruijn, E.; de Sonnaville, S.; Verbeek, N.E.; Lindhout, D.; et al. Identification of srp9 as a febrile seizure susceptibility gene. Ann. Clin. Transl. Neurol. 2014, 1, 239–250. [Google Scholar] [CrossRef]
- Helbecque, N.; Cottel, D.; Amouyel, P. Low-density lipoprotein receptor-related protein 8 gene polymorphisms and dementia. Neurobiol. Aging 2009, 30, 266–271. [Google Scholar] [CrossRef]
- Molitor, L.; Bacher, S.; Burczyk, S.; Niessing, D. The molecular function of pura and its implications in neurological diseases. Front. Genet. 2021, 12, 638217. [Google Scholar] [CrossRef] [PubMed]
- Igata, R.; Katsuki, A.; Kakeda, S.; Watanabe, K.; Igata, N.; Hori, H.; Konishi, Y.; Atake, K.; Kawasaki, Y.; Korogi, Y.; et al. Pclo rs2522833-mediated gray matter volume reduction in patients with drug-naive, first-episode major depressive disorder. Transl. Psychiatry 2017, 7, e1140. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.H.; Bylykbashi, E.; Chatila, Z.K.; Lee, S.W.; Pulli, B.; Clemenson, G.D.; Kim, E.; Rompala, A.; Oram, M.K.; Asselin, C.; et al. Combined adult neurogenesis and bdnf mimic exercise effects on cognition in an alzheimer’s mouse model. Science 2018, 361, eaan8821. [Google Scholar] [CrossRef]
- Kim, M.; Lee, Y.S.; Yoo, Y.M.; Choi, J.J.; Kim, H.N.; Kang, C.; Yu, J.M.; Moon, S.H.; Kim, A.; Kim, C.W. Exogenous clasp2 protein treatment enhances wound healing in vitro and in vivo. Wound Repair Regen. 2019, 27, 345–359. [Google Scholar] [CrossRef] [PubMed]
- Kleinman, J.E. Genetic variation in pclo is associated with prefrontal cortex expression and bipolar disorder. Biol. Psychiatry 2011, 69, 298. [Google Scholar] [CrossRef] [PubMed]
- Mori, Y.; Tsuji, M.; Oguchi, T.; Kasuga, K.; Kimura, A.; Futamura, A.; Sugimoto, A.; Kasai, H.; Kuroda, T.; Yano, S.; et al. Serum bdnf as a potential biomarker of alzheimer’s disease: Verification through assessment of serum, cerebrospinal fluid, and medial temporal lobe atrophy. Front. Neurol. 2021, 12, 653267. [Google Scholar] [CrossRef]
- Uchida, Y.; Nakano, S.; Gomi, F.; Takahashi, H. Up-regulation of calsyntenin-3 by beta-amyloid increases vulnerability of cortical neurons. FEBS Lett. 2011, 585, 651–656. [Google Scholar] [CrossRef]
- Uchida, Y.; Gomi, F.; Murayama, S.; Takahashi, H. Calsyntenin-3 c-terminal fragment accumulates in dystrophic neurites surrounding abeta plaques in tg2576 mouse and alzheimer disease brains: Its neurotoxic role in mediating dystrophic neurite formation. Am. J. Pathol. 2013, 182, 1718–1726. [Google Scholar] [CrossRef]
- van Niel, G.; D’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef]
- Wang, Y.; Balaji, V.; Kaniyappan, S.; Kruger, L.; Irsen, S.; Tepper, K.; Chandupatla, R.; Maetzler, W.; Schneider, A.; Mandelkow, E.; et al. The release and trans-synaptic transmission of tau via exosomes. Mol. Neurodegener. 2017, 12, 5. [Google Scholar] [CrossRef]
- Eitan, E.; Hutchison, E.R.; Marosi, K.; Comotto, J.; Mustapic, M.; Nigam, S.M.; Suire, C.; Maharana, C.; Jicha, G.A.; Liu, D.; et al. Extracellular vesicle-associated abeta mediates trans-neuronal bioenergetic and Ca2+-handling deficits in alzheimer’s disease models. NPJ Aging Mech. Dis. 2016, 2, 16019. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, L.; Honsho, M.; Zahn, T.R.; Keller, P.; Geiger, K.D.; Verkade, P.; Simons, K. Alzheimer’s disease beta-amyloid peptides are released in association with exosomes. Proc. Natl. Acad. Sci. USA 2006, 103, 11172–11177. [Google Scholar] [CrossRef] [PubMed]
- Vandendriessche, C.; Balusu, S.; Van Cauwenberghe, C.; Brkic, M.; Pauwels, M.; Plehiers, N.; Bruggeman, A.; Dujardin, P.; Van Imschoot, G.; Van Wonterghem, E.; et al. Importance of extracellular vesicle secretion at the blood-cerebrospinal fluid interface in the pathogenesis of alzheimer’s disease. Acta Neuropathol. Commun. 2021, 9, 143. [Google Scholar] [CrossRef]
- Balusu, S.; Van Wonterghem, E.; De Rycke, R.; Raemdonck, K.; Stremersch, S.; Gevaert, K.; Brkic, M.; Demeestere, D.; Vanhooren, V.; Hendrix, A.; et al. Identification of a novel mechanism of blood-brain communication during peripheral inflammation via choroid plexus-derived extracellular vesicles. EMBO Mol. Med. 2016, 8, 1162–1183. [Google Scholar] [CrossRef]
- Brkic, M.; Balusu, S.; Van Wonterghem, E.; Gorle, N.; Benilova, I.; Kremer, A.; Van Hove, I.; Moons, L.; De Strooper, B.; Kanazir, S.; et al. Amyloid beta oligomers disrupt blood-csf barrier integrity by activating matrix metalloproteinases. J. Neurosci. Off. J. Soc. Neurosci. 2015, 35, 12766–12778. [Google Scholar] [CrossRef]
- Salminen, A.; Ojala, J.; Kauppinen, A.; Kaarniranta, K.; Suuronen, T. Inflammation in alzheimer’s disease: Amyloid-beta oligomers trigger innate immunity defence via pattern recognition receptors. Prog. Neurobiol. 2009, 87, 181–194. [Google Scholar] [CrossRef]
- Ding, Y.; Zhao, J.; Zhang, X.; Wang, S.; Viola, K.L.; Chow, F.E.; Zhang, Y.; Lippa, C.; Klein, W.L.; Gong, Y. Amyloid beta oligomers target to extracellular and intracellular neuronal synaptic proteins in alzheimer’s disease. Front. Neurol. 2019, 10, 1140. [Google Scholar] [CrossRef]
- Marsh, J.; Alifragis, P. Synaptic dysfunction in alzheimer’s disease: The effects of amyloid beta on synaptic vesicle dynamics as a novel target for therapeutic intervention. Neural Regen. Res. 2018, 13, 616–623. [Google Scholar] [PubMed]
- Chepkova, A.N.; Sergeeva, O.A.; Gorg, B.; Haas, H.L.; Klocker, N.; Haussinger, D. Impaired novelty acquisition and synaptic plasticity in congenital hyperammonemia caused by hepatic glutamine synthetase deficiency. Sci. Rep. 2017, 7, 40190. [Google Scholar] [CrossRef]
- Subramanian, J.; Savage, J.C.; Tremblay, M.E. Synaptic loss in alzheimer’s disease: Mechanistic insights provided by two-photon in vivo imaging of transgenic mouse models. Front. Cell. Neurosci. 2020, 14, 592607. [Google Scholar] [CrossRef]
- Jack, C.R., Jr.; Knopman, D.S.; Jagust, W.J.; Shaw, L.M.; Aisen, P.S.; Weiner, M.W.; Petersen, R.C.; Trojanowski, J.Q. Hypothetical model of dynamic biomarkers of the alzheimer’s pathological cascade. Lancet Neurol. 2010, 9, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Kashyap, G.; Bapat, D.; Das, D.; Gowaikar, R.; Amritkar, R.E.; Rangarajan, G.; Ravindranath, V.; Ambika, G. Synapse loss and progress of alzheimer’s disease—A network model. Sci. Rep. 2019, 9, 6555. [Google Scholar] [CrossRef]
- Palop, J.J.; Chin, J.; Mucke, L. A network dysfunction perspective on neurodegenerative diseases. Nature 2006, 443, 768–773. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J. Alzheimer’s disease is a synaptic failure. Science 2002, 298, 789–791. [Google Scholar] [CrossRef]
- Bastrikova, N.; Gardner, G.A.; Reece, J.M.; Jeromin, A.; Dudek, S.M. Synapse elimination accompanies functional plasticity in hippocampal neurons. Proc. Natl. Acad. Sci. USA 2008, 105, 3123–3127. [Google Scholar] [CrossRef] [PubMed]
- Lamprecht, R.; LeDoux, J. Structural plasticity and memory. Nat. Rev. Neurosci. 2004, 5, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Morfini, G.A.; Burns, M.; Binder, L.I.; Kanaan, N.M.; LaPointe, N.; Bosco, D.A.; Brown, R.H., Jr.; Brown, H.; Tiwari, A.; Hayward, L.; et al. Axonal transport defects in neurodegenerative diseases. J. Neurosci. Off. J. Soc. Neurosci. 2009, 29, 12776–12786. [Google Scholar] [CrossRef]
- Coleman, M. Molecular signaling how do axons die? Adv. Genet. 2011, 73, 185–217. [Google Scholar]
- Witter, M.P. The perforant path: Projections from the entorhinal cortex to the dentate gyrus. Prog. Brain Res. 2007, 163, 43–61. [Google Scholar]
- Stebbins, G.T.; Murphy, C.M. Diffusion tensor imaging in alzheimer’s disease and mild cognitive impairment. Behav. Neurol. 2009, 21, 39–49. [Google Scholar] [CrossRef]
- Tran, M.; Reddy, P.H. Defective autophagy and mitophagy in aging and alzheimer’s disease. Front. Neurosci. 2020, 14, 612757. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.J.; Pu, J.L.; Krafft, P.R.; Zhang, J.M.; Chen, S. The molecular mechanisms between autophagy and apoptosis: Potential role in central nervous system disorders. Cell. Mol. Neurobiol. 2015, 35, 85–99. [Google Scholar] [CrossRef] [PubMed]
- Knott, G.W.; Holtmaat, A.; Wilbrecht, L.; Welker, E.; Svoboda, K. Spine growth precedes synapse formation in the adult neocortex in vivo. Nat. Neurosci. 2006, 9, 1117–1124. [Google Scholar] [CrossRef] [PubMed]
- Shim, K.S.; Lubec, G. Drebrin, a dendritic spine protein, is manifold decreased in brains of patients with alzheimer’s disease and down syndrome. Neurosci. Lett. 2002, 324, 209–212. [Google Scholar] [CrossRef]
- Yu, K.; Lin, C.J.; Hatcher, A.; Lozzi, B.; Kong, K.; Huang-Hobbs, E.; Cheng, Y.T.; Beechar, V.B.; Zhu, W.; Zhang, Y.; et al. Pik3ca variants selectively initiate brain hyperactivity during gliomagenesis. Nature 2020, 578, 166–171. [Google Scholar] [CrossRef]
- Colucci-D’Amato, L.; Speranza, L.; Volpicelli, F. Neurotrophic factor bdnf, physiological functions and therapeutic potential in depression, neurodegeneration and brain cancer. Int. J. Mol. Sci. 2020, 21, 7777. [Google Scholar] [CrossRef]
- Poursaei, E.; Daneshmandpour, Y.; Aghaei Moghadam, E.; Abolghasemi, M.; Jamshidi, J.; Baradaran, B.; Asadi, M.; Kazeminasab, S.; Emamalizadeh, B. Lrp8 (rs5177) and cep85l (rs11756438) are contributed to schizophrenia susceptibility in iranian population. Psychiatr. Genet. 2020, 30, 162–165. [Google Scholar] [CrossRef]
- Xu, P.; Zhang, G.; Hou, S.; Sha, L.G. Mapk8 mediates resistance to temozolomide and apoptosis of glioblastoma cells through mapk signaling pathway. Biomed. Pharmacother. 2018, 106, 1419–1427. [Google Scholar] [CrossRef]
- Yin, R.H.; Yu, J.T.; Tan, L. The role of sorl1 in alzheimer’s disease. Mol. Neurobiol. 2015, 51, 909–918. [Google Scholar] [CrossRef]
- Ding, S.; Mehrabi, R.; Koten, C.; Kang, Z.; Wei, Y.; Seong, K.; Kistler, H.C.; Xu, J.R. Transducin beta-like gene ftl1 is essential for pathogenesis in fusarium graminearum. Eukaryot. Cell 2009, 8, 867–876. [Google Scholar] [CrossRef]
- Klauck, S.M.; Felder, B.; Kolb-Kokocinski, A.; Schuster, C.; Chiocchetti, A.; Schupp, I.; Wellenreuther, R.; Schmotzer, G.; Poustka, F.; Breitenbach-Koller, L.; et al. Mutations in the ribosomal protein gene rpl10 suggest a novel modulating disease mechanism for autism. Mol. Psychiatry 2006, 11, 1073–1084. [Google Scholar] [CrossRef] [PubMed]
- Slattery, C.F.; Zhang, J.; Paterson, R.W.; Foulkes, A.J.M.; Carton, A.; Macpherson, K.; Mancini, L.; Thomas, D.L.; Modat, M.; Toussaint, N.; et al. Apoe influences regional white-matter axonal density loss in alzheimer’s disease. Neurobiol. Aging 2017, 57, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Nutma, E.; Ceyzeriat, K.; Amor, S.; Tsartsalis, S.; Millet, P.; Owen, D.R.; Papadopoulos, V.; Tournier, B.B. Cellular sources of tspo expression in healthy and diseased brain. Eur. J. Nucl. Med. Mol. Imaging 2021, 49, 146–163. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Zhuang, H.; Wang, Q.; Yang, L.; Xie, Z.; Zhang, Z.; Tan, W.; Tang, C.; Chen, Y.; Shang, C. Slc39a1 overexpression is associated with immune infiltration in hepatocellular carcinoma and promotes its malignant progression. J. Hepatocell. Carcinoma 2022, 9, 83–98. [Google Scholar] [CrossRef]
- Strunz, M.; Jarrell, J.T.; Cohen, D.S.; Rosin, E.R.; Vanderburg, C.R.; Huang, X. Modulation of sparc/hevin proteins in alzheimer’s disease brain injury. J. Alzheimer’s Dis. 2019, 68, 695–710. [Google Scholar] [CrossRef] [PubMed]
- Liang, F.; Wang, B.; Bao, L.; Zhao, Y.S.; Zhang, S.M.; Zhang, S.Q. Overexpression of ilk promotes temozolomide resistance in glioma cells. Mol. Med. Rep. 2017, 15, 1297–1304. [Google Scholar] [CrossRef] [PubMed]
- Villegas, V.E.; Zaphiropoulos, P.G. Neighboring gene regulation by antisense long non-coding rnas. Int. J. Mol. Sci. 2015, 16, 3251–3266. [Google Scholar] [CrossRef]
- Guo, X.; Aviles, G.; Liu, Y.; Tian, R.; Unger, B.A.; Lin, Y.T.; Wiita, A.P.; Xu, K.; Correia, M.A.; Kampmann, M. Mitochondrial stress is relayed to the cytosol by an oma1-dele1-hri pathway. Nature 2020, 579, 427–432. [Google Scholar] [CrossRef]
- Sun, D.; Yu, Z.; Fang, X.; Liu, M.; Pu, Y.; Shao, Q.; Wang, D.; Zhao, X.; Huang, A.; Xiang, Z.; et al. Lncrna gas5 inhibits microglial m2 polarization and exacerbates demyelination. EMBO Rep. 2017, 18, 1801–1816. [Google Scholar] [CrossRef]
- Li, J.; Lv, H.; Che, Y.Q. Long non-coding rna gas5 potentiates the effects of microrna-21 downregulation in response to ischaemic brain injury. Neuroscience 2020, 437, 87–97. [Google Scholar] [CrossRef]
- Forner, S.; Kawauchi, S.; Balderrama-Gutierrez, G.; Kramar, E.A.; Matheos, D.P.; Phan, J.; Javonillo, D.I.; Tran, K.M.; Hingco, E.; da Cunha, C.; et al. Systematic phenotyping and characterization of the 5×FAD mouse model of alzheimer’s disease. Sci. Data 2021, 8, 270. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. Star: Ultrafast universal rna-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of rna-seq experiments with tophat and cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef]
- Liberzon, A.; Subramanian, A.; Pinchback, R.; Thorvaldsdottir, H.; Tamayo, P.; Mesirov, J.P. Molecular signatures database (msigdb) 3.0. Bioinformatics 2011, 27, 1739–1740. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, Y.-K.; Jung, Y.S.; Song, J. Transcriptome Profile in the Mouse Brain of Hepatic Encephalopathy and Alzheimer’s Disease. Int. J. Mol. Sci. 2023, 24, 675. https://doi.org/10.3390/ijms24010675
Kim Y-K, Jung YS, Song J. Transcriptome Profile in the Mouse Brain of Hepatic Encephalopathy and Alzheimer’s Disease. International Journal of Molecular Sciences. 2023; 24(1):675. https://doi.org/10.3390/ijms24010675
Chicago/Turabian StyleKim, Young-Kook, Yoon Seok Jung, and Juhyun Song. 2023. "Transcriptome Profile in the Mouse Brain of Hepatic Encephalopathy and Alzheimer’s Disease" International Journal of Molecular Sciences 24, no. 1: 675. https://doi.org/10.3390/ijms24010675
APA StyleKim, Y.-K., Jung, Y. S., & Song, J. (2023). Transcriptome Profile in the Mouse Brain of Hepatic Encephalopathy and Alzheimer’s Disease. International Journal of Molecular Sciences, 24(1), 675. https://doi.org/10.3390/ijms24010675