
Metal Complexes of a 5-Nitro-8-Hydroxyquinoline-Proline Hybrid with Enhanced Water Solubility Targeting Multidrug Resistant Cancer Cells

 , ,
, ,  , , , ,
, , , ,  , and
, and
Abstract
1. Introduction
2. Results and Discussion
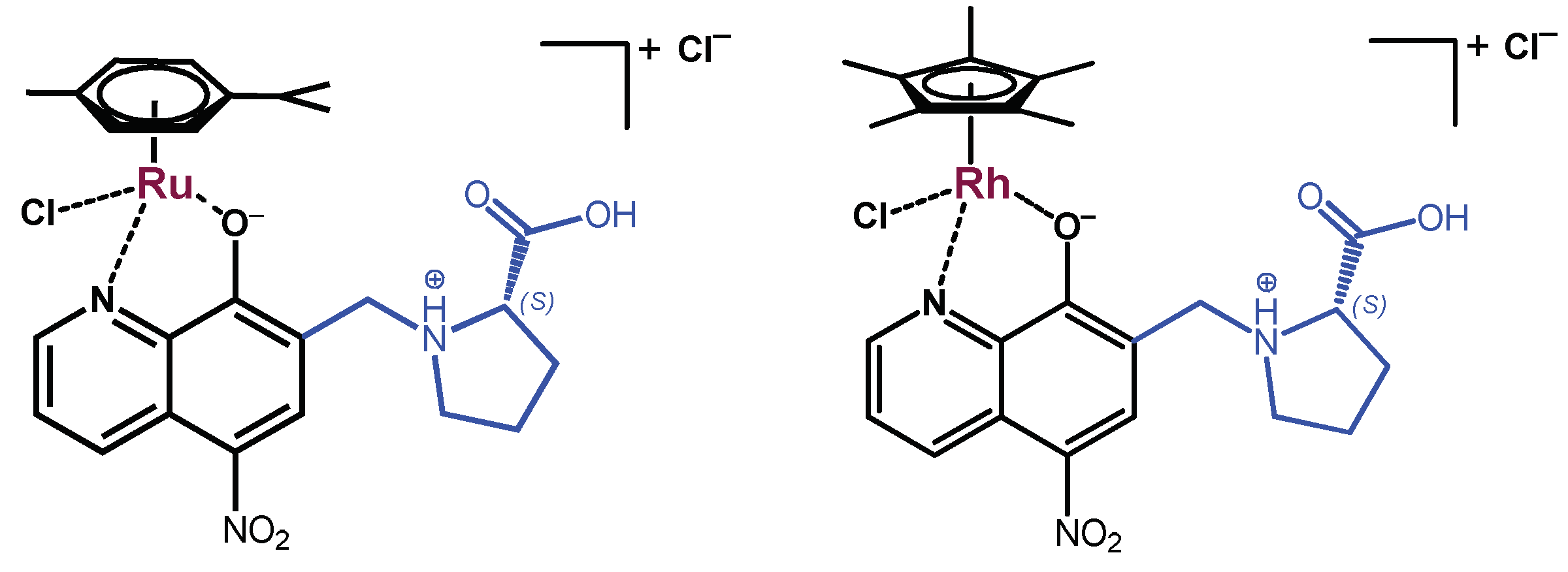
2.1. Synthesis and Characterization of the HQNO2-L-Pro ligand and Its Ru(η6-p-cymene) and Rh(η5-C5Me5) Complexes
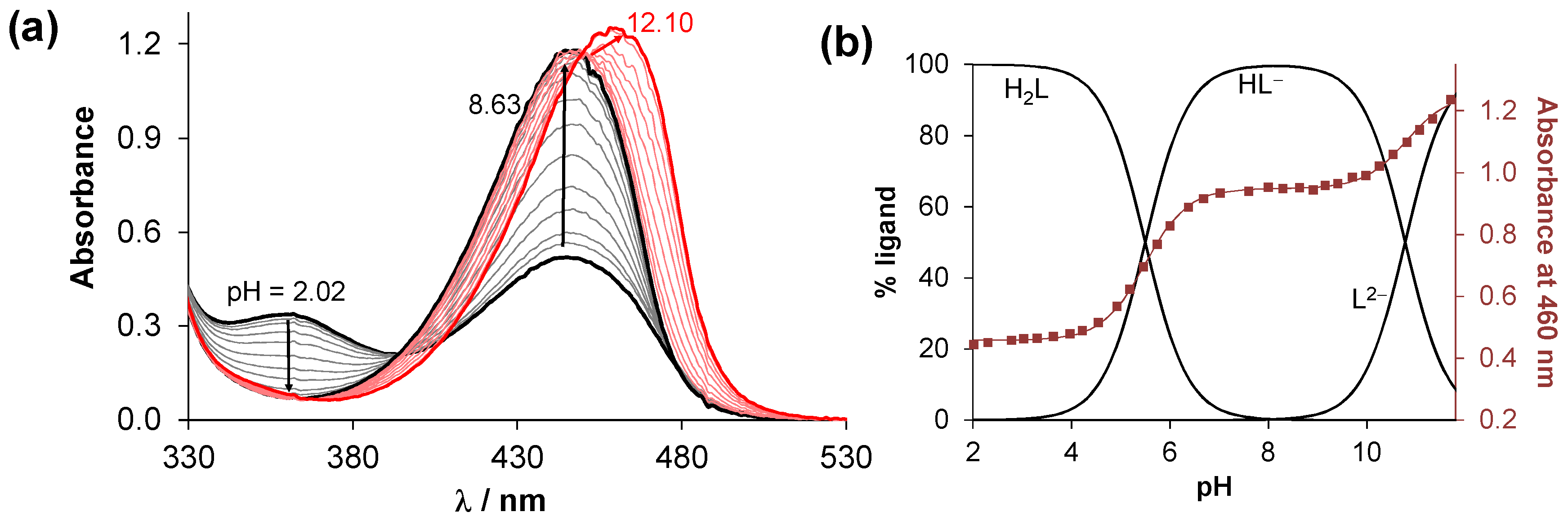
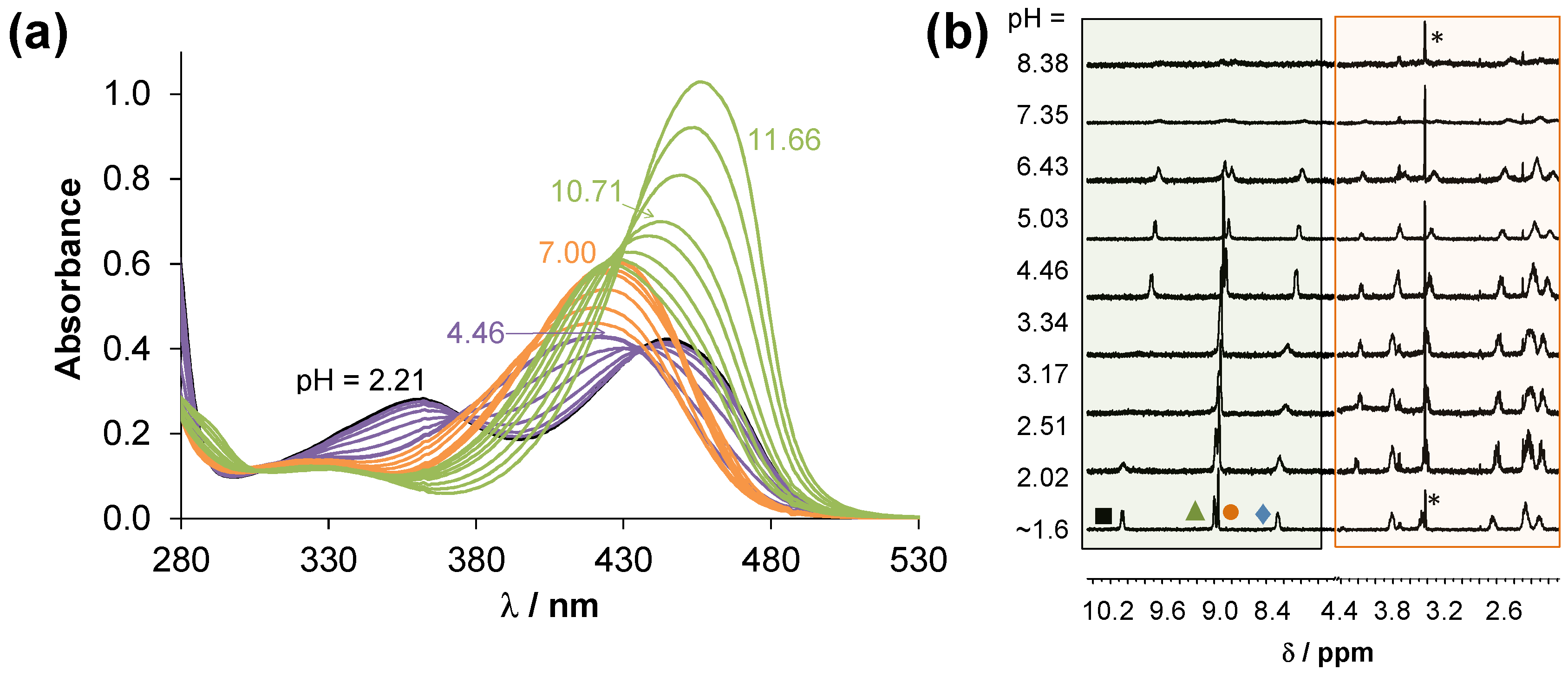
2.2. Solution Characterization of the HQNO2-L-Pro
2.3. Complex Formation of the HQNO2-L-Pro Ligand with Essential Metal Ions
2.3.1. Solution Equilibrium of the Fe(III) and Fe(II) Complexes
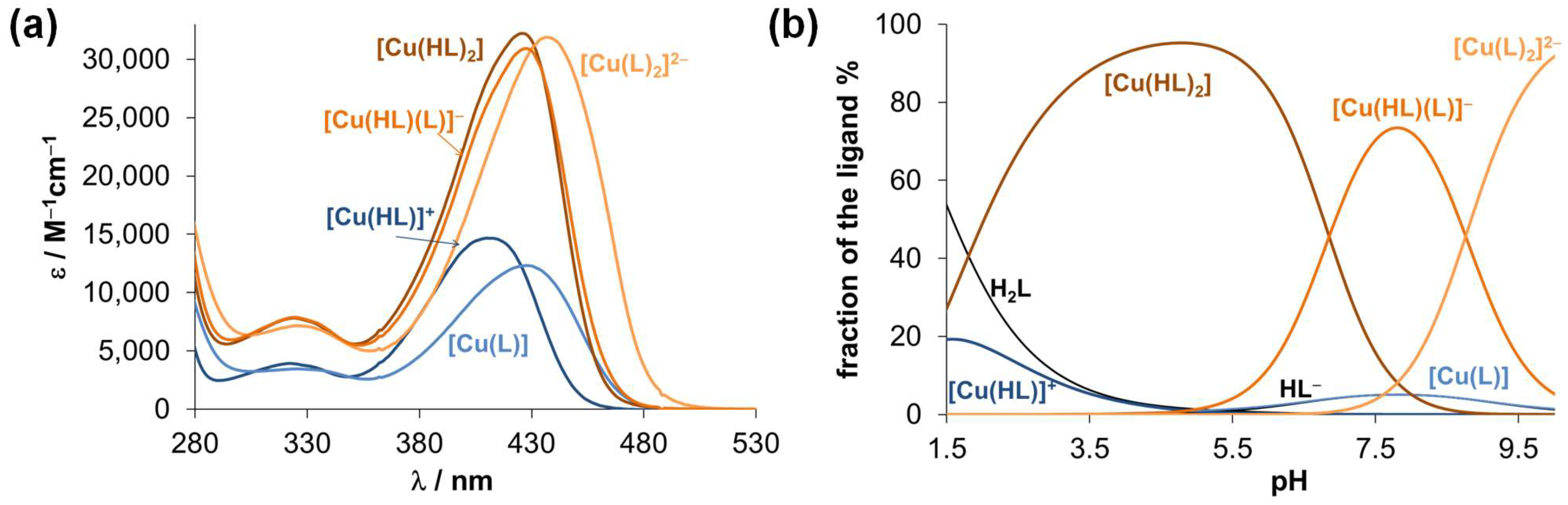
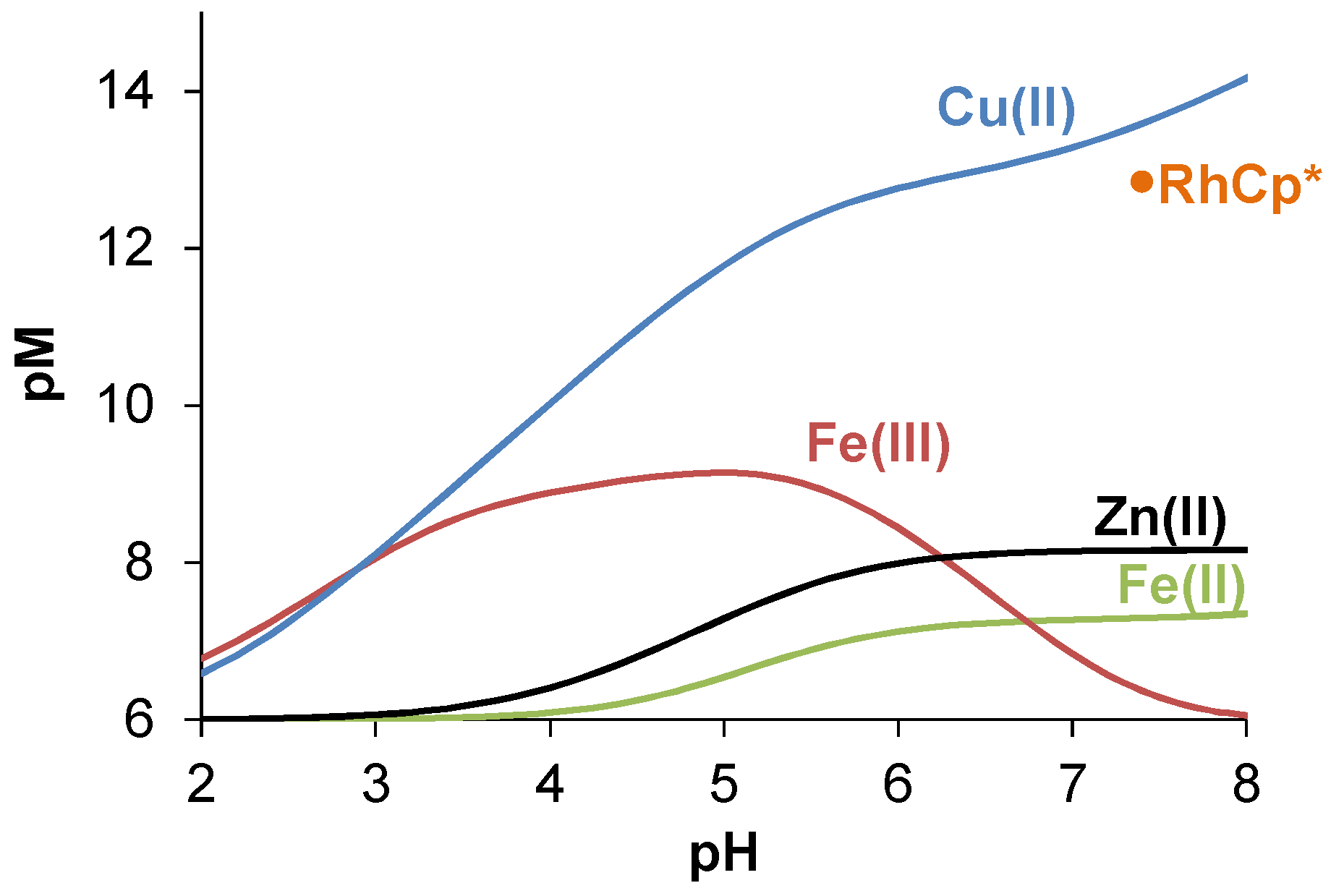
2.3.2. Solution Equilibrium of the Cu(II) and Zn(II) Complexes
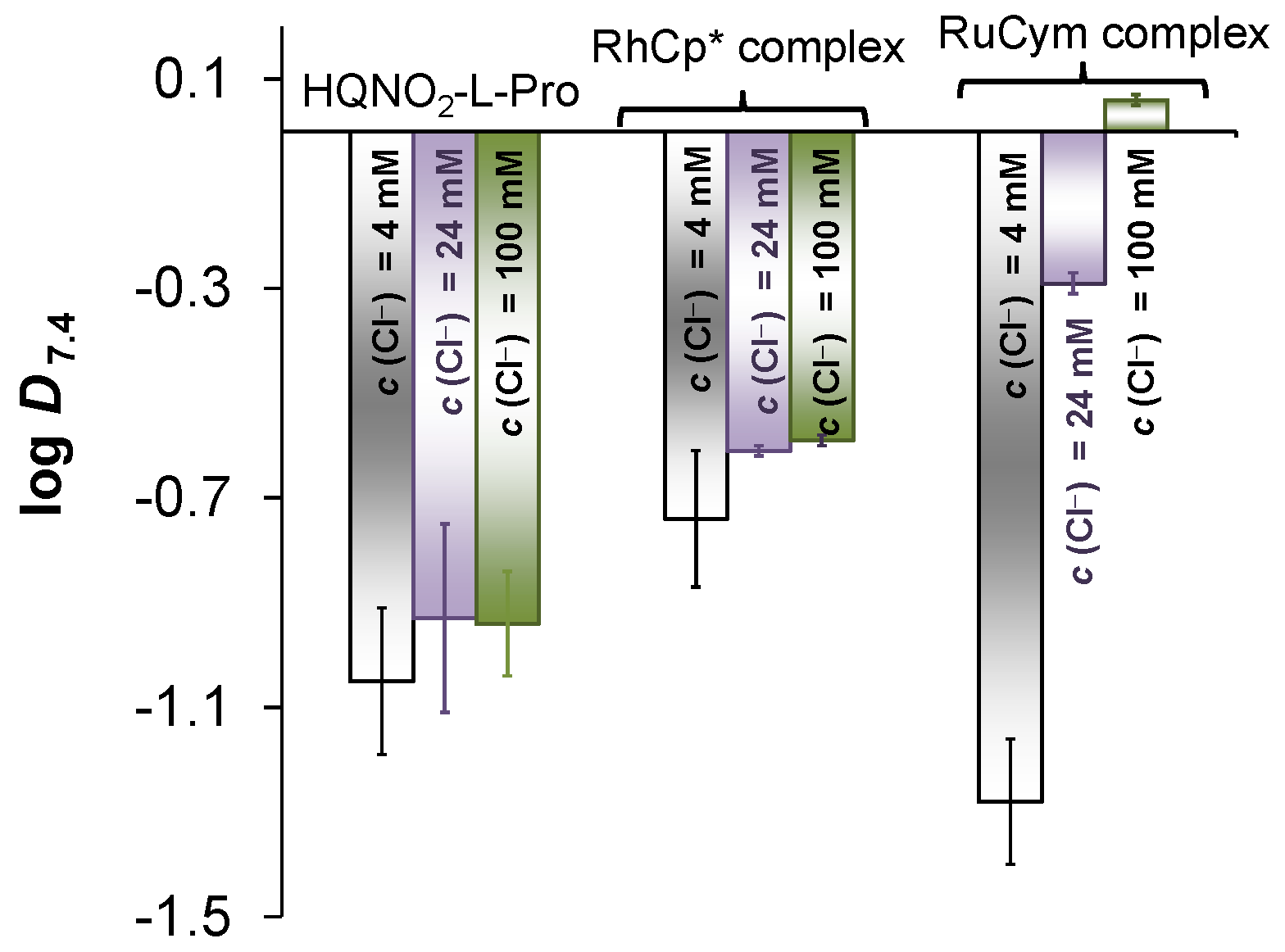
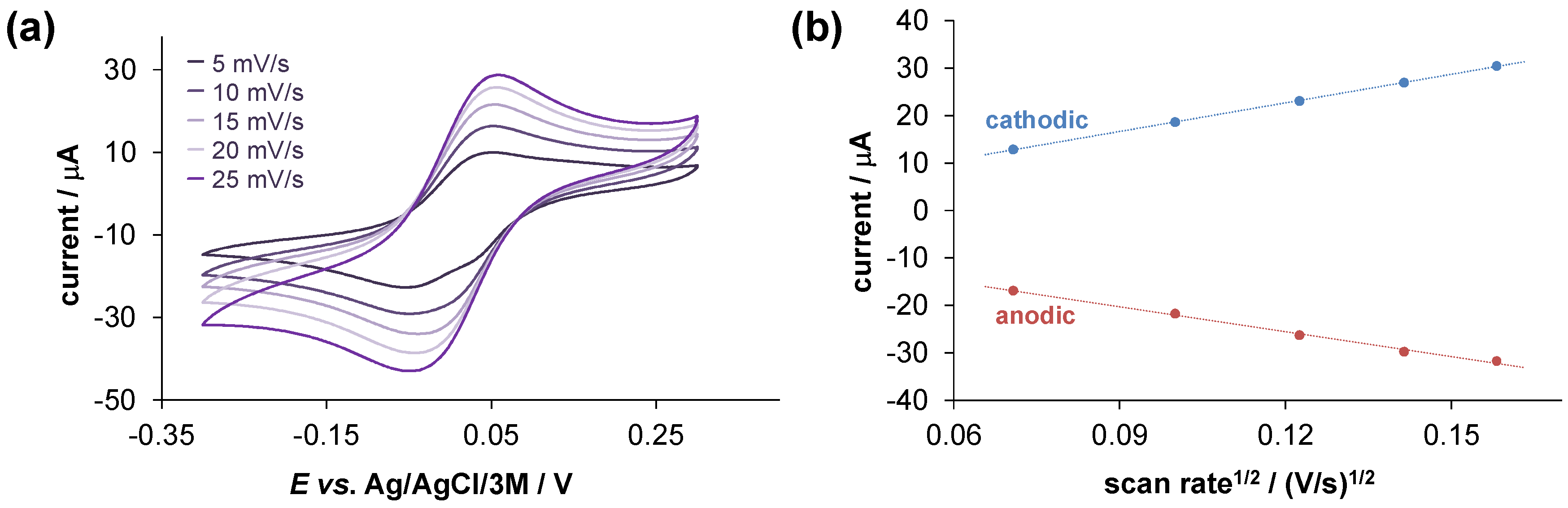
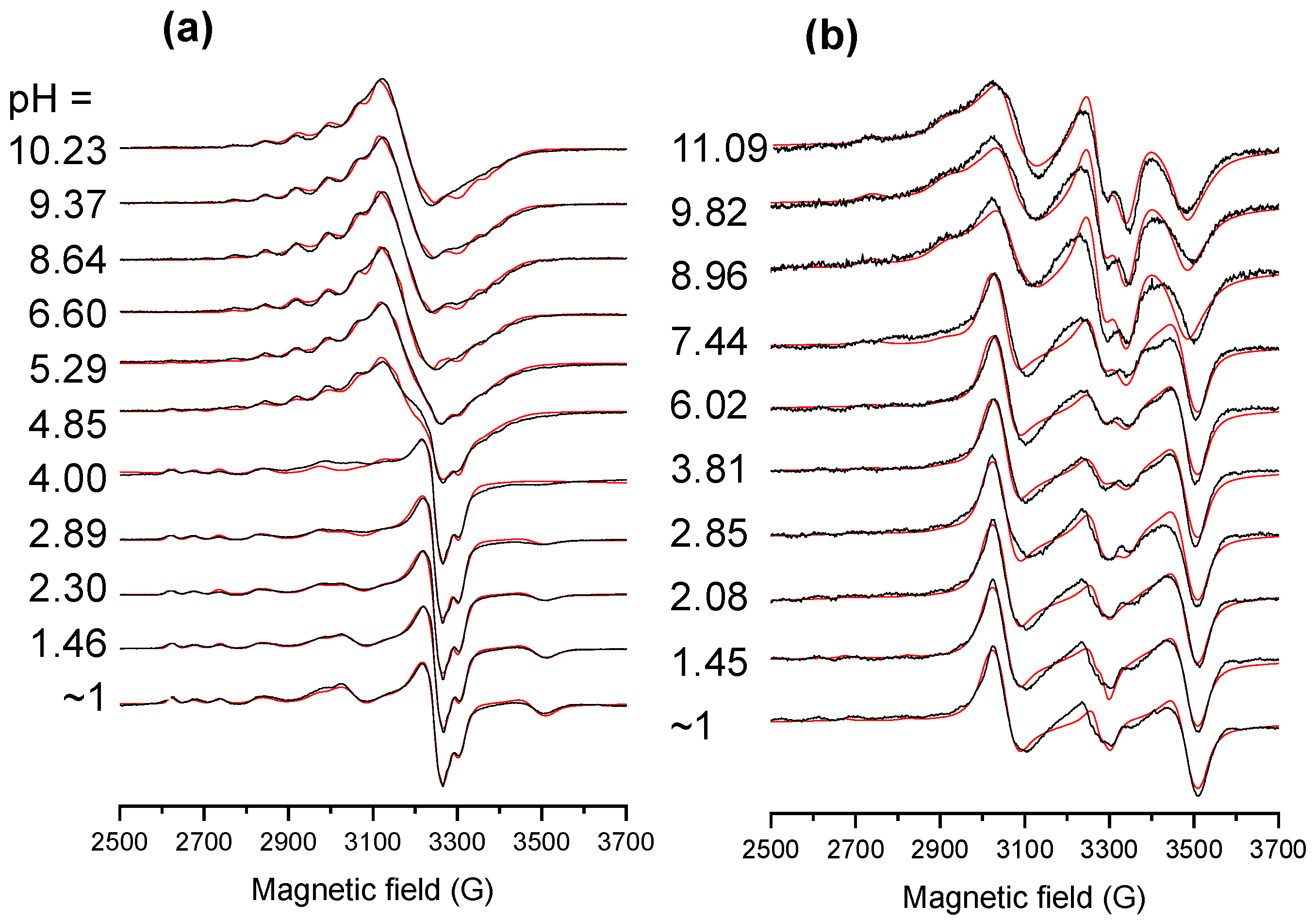
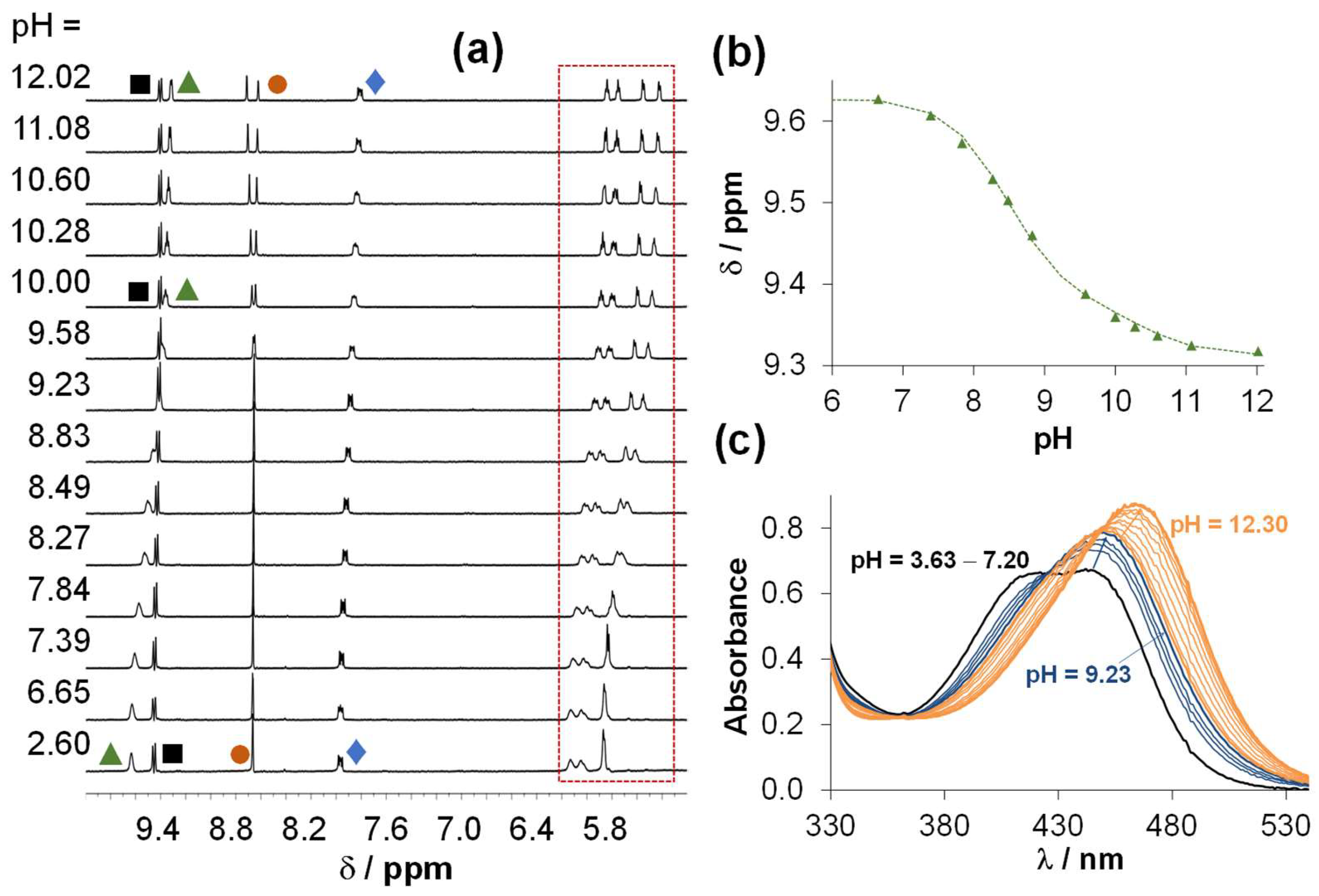
2.4. Solution Speciation of the RhCp* and RuCym Complexes of HQNO2-L-Pro
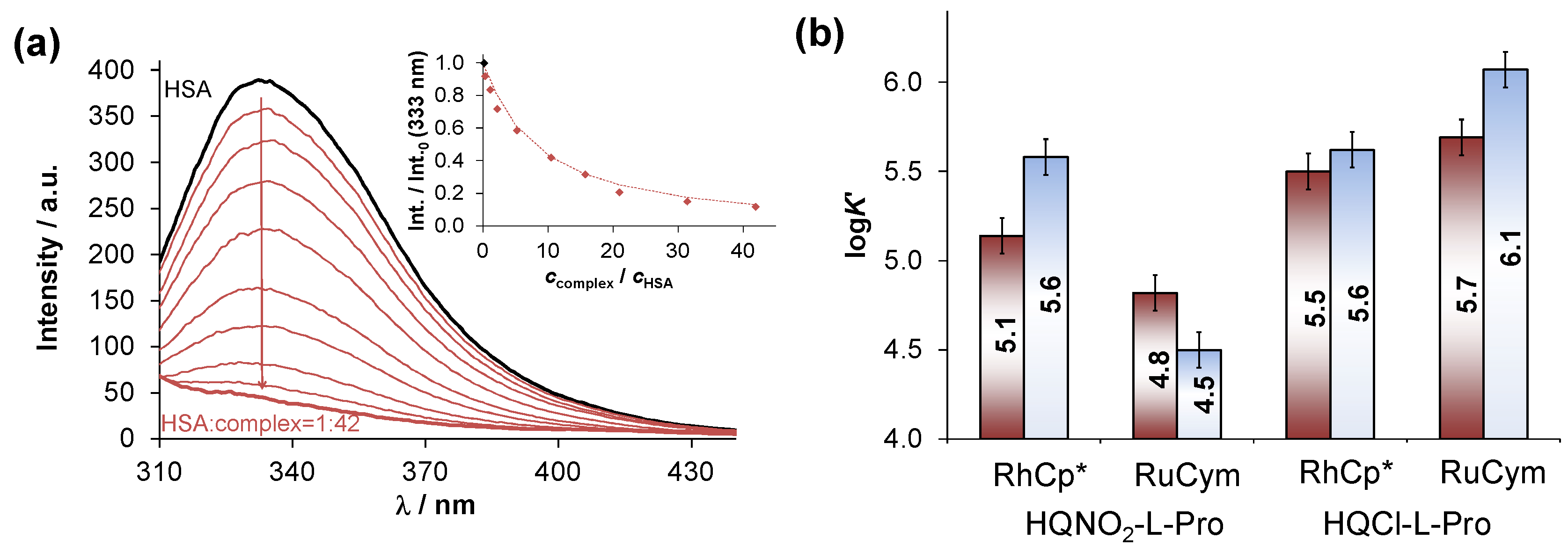
2.5. Interaction of the RuCym and RhCp* Complexes of HQNO2-L-Pro with HSA
2.6. Cellular Effects
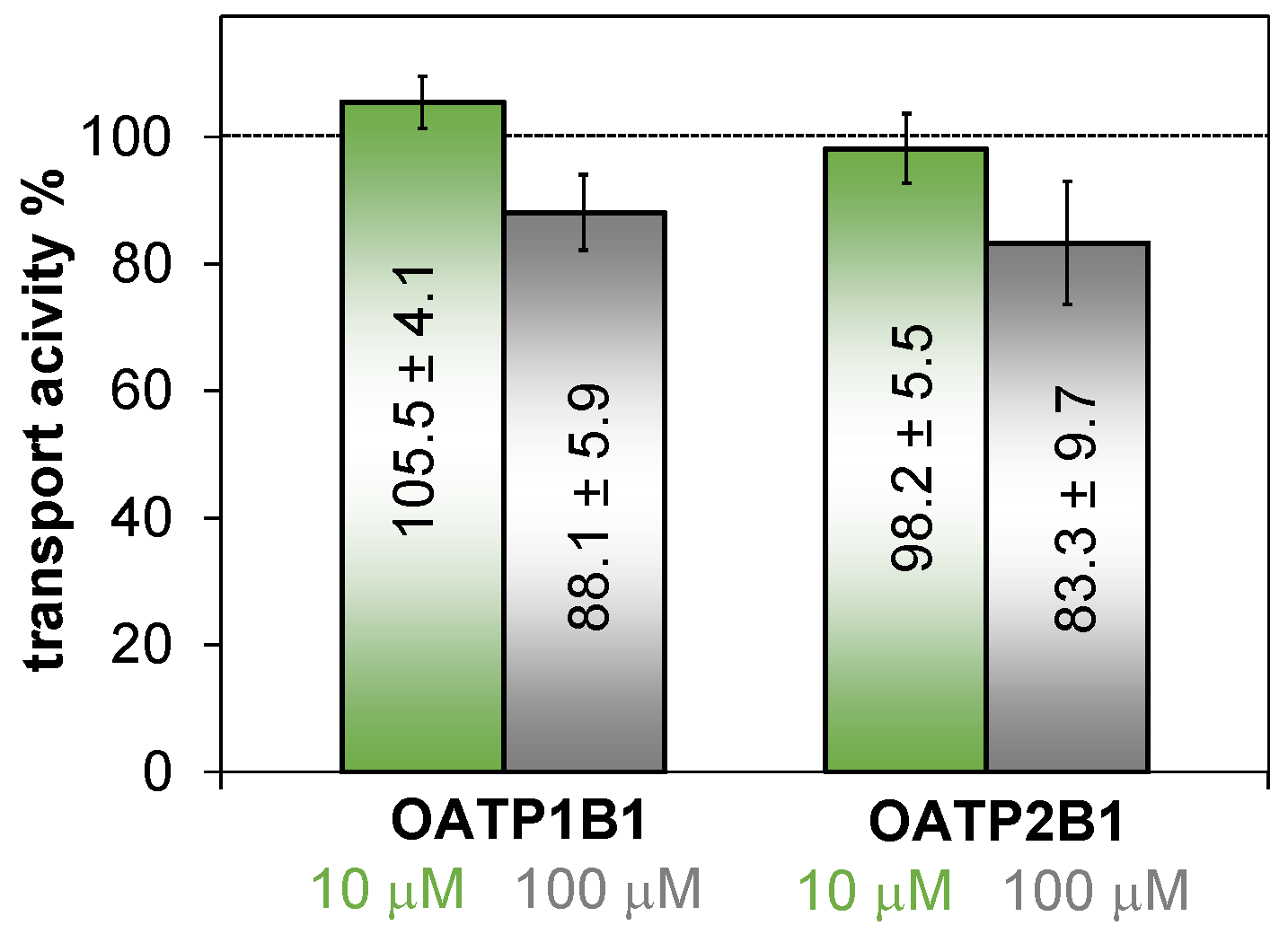
2.6.1. Inhibitory Effect of HQNO2-L-Pro on Organic Anion Transporting Polypeptides
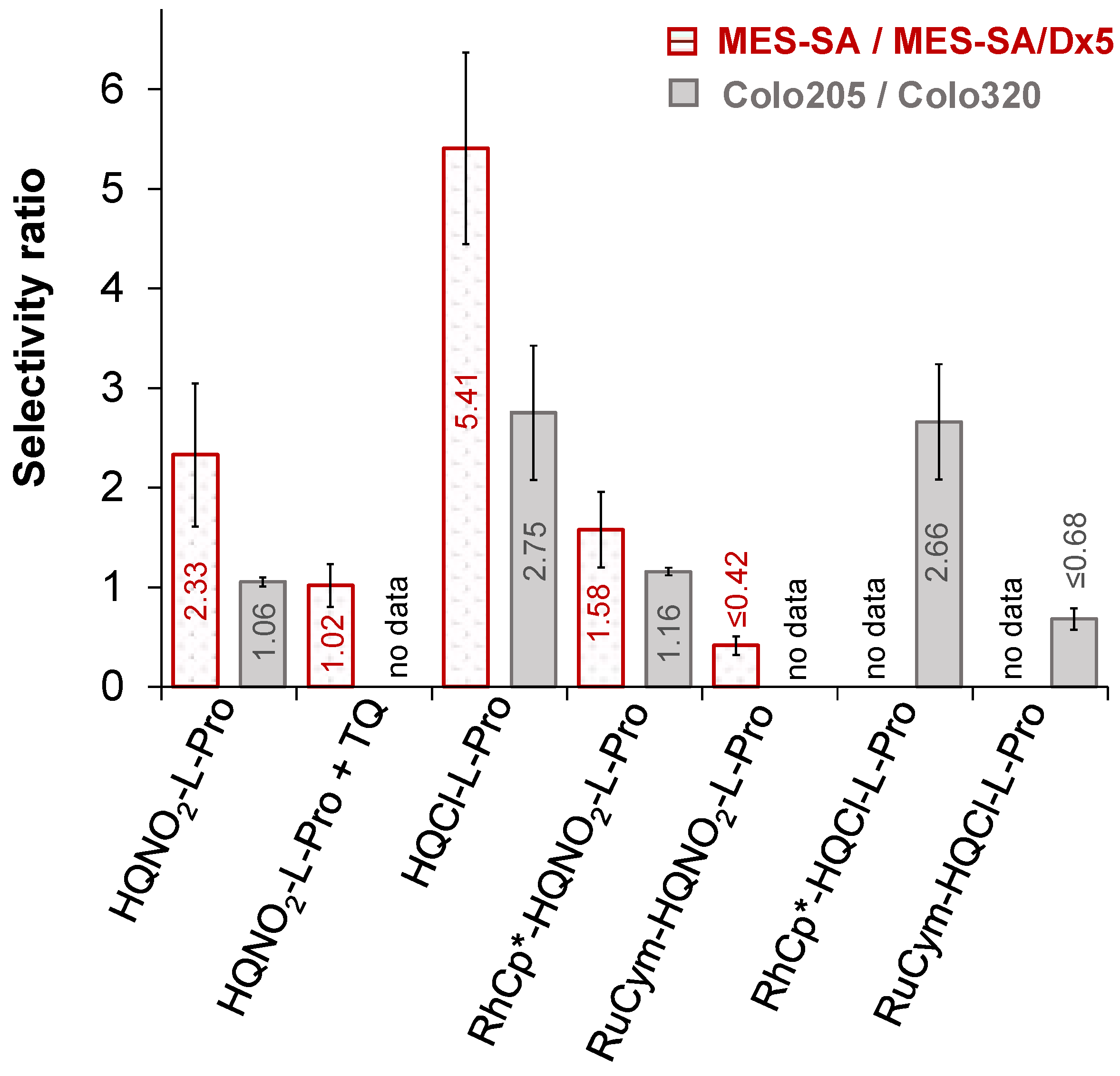
2.6.2. Anticancer Activity of the HQNO2-L-Pro and Its Half-Sandwich Organometallic Complexes
2.6.3. Antibacterial Activity of the HQNO2-L-Pro and Its Half-Sandwich Organometallic Complexes
3. Materials and Methods
3.1. Chemicals
3.2. Stock Solutions and Sample Preparation
3.3. Synthesis and Characterization of Ligands and Their Complexes
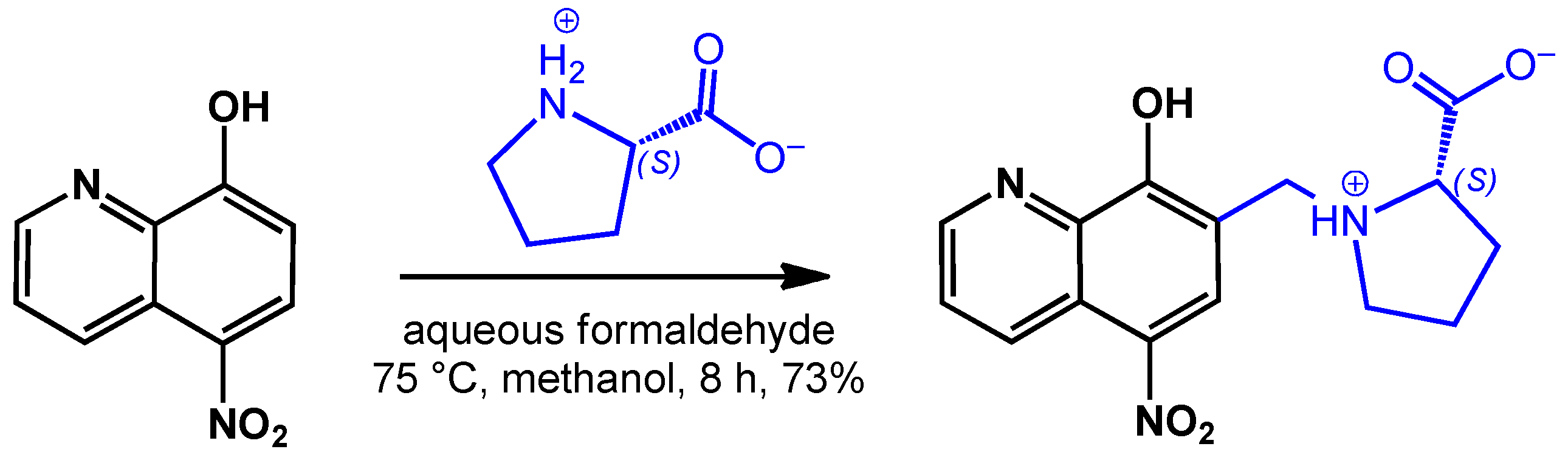
3.3.1. Synthesis of HQNO2-L-Pro
3.3.2. Synthesis of [RuCym(HQNO2-L-Pro)Cl]Cl
3.3.3. Synthesis of [RhCp*(HQNO2-L-Pro)Cl]Cl
3.4. Electrospray Mass Spectrometry
3.5. PH-Potentiometric Measurements
3.6. UV-Visible Spectrophotometry, Spectrofluorometry, and Circular Dichroism Spectroscopy
3.7. NMR and EPR Spectroscopy
3.8. Determination of Distribution Coefficients
3.9. Cyclic Voltammetry
3.10. Capillary Zone Electrophoresis
3.11. OATP Uptake Assay
3.12. In Vitro Cell Studies: Cell Lines and Culture Conditions and Cytotoxicity Assay
3.13. Antibacterial Effect: Bacterial Culture and Determination of MIC Values
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Szakács, G.; Paterson, J.K.; Ludwig, J.A.; Booth-Genthe, C.; Gottesman, M.M. Targeting multidrug resistance in cancer. Nat. Rev. Drug Discov. 2006, 5, 219–234. [Google Scholar] [CrossRef] [PubMed]
- Chiba, P.; Ecker, G.F. Inhibitors of ABC-type drug efflux pumps: An overview of the current patent situation. Expert Opin. Ther. Pat. 2004, 14, 499–508. [Google Scholar] [CrossRef]
- Gottesman, M.M.; Fojo, T.; Bates, S.E. Multidrug resistance in cancer: Role of ATP-dependent transporters. Nat. Rev. Cancer 2002, 2, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Tamaki, A.; Ierano, C.; Szakács, G.; Robey, R.W.; Bates, S.E. The controversial role of ABC transporters in clinical oncology. Essays Biochem. 2011, 50, 209–232. [Google Scholar] [PubMed]
- Pape, V.F.S.; Gaál, A.; Szatmári, I.; Kucsma, N.; Szoboszlai, N.; Streli, C.; Fülöp, F.; Enyedy, É.A.; Szakács, G. Relation of metal-binding property and selective toxicity of 8-hydroxyquinoline derived Mannich bases targeting multidrug resistant cancer cells. Cancers 2021, 13, 154. [Google Scholar] [CrossRef]
- Szakács, G.; Hall, M.D.; Gottesman, M.M.; Boumendjel, A.; Kachadourian, R.; Day, B.J.; Baubichon-Cortay, H.; Di Pietro, A. Targeting the Achilles heel of multidrug-resistant cancer by exploiting the fitness cost of resistance. Chem. Rev. 2014, 114, 5753–5774. [Google Scholar] [CrossRef]
- Pape, V.F.S.; Palkó, R.; Tóth, S.; Szabó, M.J.; Sessler, J.; Dormán, G.; Enyedy, É.A.; Soós, T.; Szatmári, I.; Szakács, G. Structure–activity relationships of 8-hydroxyquinoline-derived Mannich bases with tertiary amines targeting multidrug-resistant cancer. J. Med. Chem. 2022, 65, 7729–7745. [Google Scholar] [CrossRef]
- Türk, D.; Hall, M.D.; Chu, B.F.; Ludwig, J.A.; Fales, H.M.; Gottesman, M.M.; Szakács, G. Identification of compounds selectively killing multidrug-resistant cancer cells. Cancer Res. 2009, 69, 8293–8301. [Google Scholar] [CrossRef]
- Oliveri, V.; Vecchio, G. 8-Hydroxyquinolines in medicinal chemistry: A structural perspective. Eur. J. Med. Chem. 2016, 120, 252–274. [Google Scholar] [CrossRef]
- Joaquim, A.R.; Gionbelli, M.P.; Gosmann, G.; Fuentefria, A.M.; Lopes, M.S.; Fernandes de Andrade, S. Novel antimicrobial 8-hydroxyquinoline-based agents: Current development, structure–activity relationships, and perspectives. J. Med. Chem. 2021, 64, 16349–16379. [Google Scholar] [CrossRef]
- Kos, J.; Ku, C.F.; Kapustikova, I.; Oravec, M.; Zhang, H.J.; Jampilek, J. 8-Hydroxyquinoline-2-carboxanilides as antiviral agents against avian influenza virus. ChemistrySelect 2019, 4, 4582–4587. [Google Scholar] [CrossRef]
- Mészáros, J.P.; Poljarevic, J.M.; Szatmári, I.; Csuvik, O.; Fülöp, F.; Szoboszlai, N.; Spengler, G.; Enyedy, É.A. An 8-hydroxyquinoline-proline hybrid with multidrug resistance reversal activity and the solution chemistry of its half-sandwich organometallic Ru and Rh complexes. Dalton Trans. 2020, 49, 7977–7992. [Google Scholar] [CrossRef] [PubMed]
- Pivarcsik, T.; Dömötör, O.; Mészáros, J.P.; May, N.V.; Spengler, G.; Csuvik, O.; Szatmári, I.; Enyedy, É.A. 8-Hydroxyquinoline-amino acid hybrids and their half-sandwich Rh and Ru complexes: Synthesis, anticancer activities, solution chemistry and interaction with biomolecules. Int. J. Mol. Sci. 2021, 22, 11281. [Google Scholar] [CrossRef] [PubMed]
- Pape, V.F.S.; May, N.V.; Gál, G.T.; Szatmári, I.; Szeri, F.; Fülöp, F.; Szakács, G.; Enyedy, É.A. Impact of copper and iron binding properties on the anticancer activity of 8-hydroxyquinoline derived Mannich bases. Dalton Trans. 2018, 47, 17032–17045. [Google Scholar] [CrossRef]
- Cserepes, M.; Türk, D.; Tóth, S.; Pape, V.F.; Gaál, A.; Gera, M.; Szabó, J.E.; Kucsma, N.; Várady, G.; Vértessy, B.G.; et al. Unshielding Multidrug Resistant Cancer through Selective Iron Depletion of P-Glycoprotein-Expressing Cells Targeting MDR by the Pgp-Dependent Efflux of Iron Complexes. Cancer Res. 2020, 80, 663–674. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Zhou, Y.; Lind, S.E.; Ding, W.Q. Clioquinol targets zinc to lysosomes in human cancer cells. Biochem. J. 2009, 417, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Taggart, J.E.; Zhang, X.; Benbrook, D.M.; Lind, S.E.; Ding, W.Q. Nitroxoline (8-hydroxy-5-nitroquinoline) is more a potent anti-cancer agent than clioquinol (5-chloro-7-iodo-8-quinoline). Cancer Lett. 2011, 312, 11–17. [Google Scholar] [CrossRef]
- Barilli, A.; Atzeri, C.; Bassanetti, I.; Ingoglia, F.; Dall’Asta, V.; Bussolati, O.; Maffini, M.; Mucchino, C.; Marchiò, L. Oxidative stress induced by copper and iron complexes with 8-hydroxyquinoline derivatives causes paraptotic death of HeLa cancer cells. Mol. Pharm. 2014, 11, 1151–1163. [Google Scholar] [CrossRef]
- Scalese, G.; Machado, I.; Correia, I.; Pessoa, J.C.; Bilbao, L.; Pérez-Diaz, L.; Gambino, D. Exploring oxidovanadium (IV) homoleptic complexes with 8-hydroxyquinoline derivatives as prospective antitrypanosomal agents. New J. Chem. 2019, 43, 17756–17773. [Google Scholar] [CrossRef]
- Qin, Q.P.; Chen, Z.F.; Qin, J.L.; He, X.J.; Li, Y.L.; Liu, Y.C.; Huang, K.B.; Liang, H. Studies on antitumor mechanism of two planar platinum (II) complexes with 8-hydroxyquinoline: Synthesis, characterization, cytotoxicity, cell cycle and apoptosis. Eur. J. Med. Chem. 2015, 92, 302–313. [Google Scholar] [CrossRef]
- Wilfinger, N.; Austin, S.; Scheiber-Mojdehkar, B.; Berger, W.; Reipert, S.; Praschberger, M.; Paur, J.; Trondl, R.; Keppler, B.K.; Zielinski, C.C.; et al. Novel p53-dependent anticancer strategy by targeting iron signaling and BNIP3L-induced mitophagy. Oncotarget 2016, 7, 1242–1261. [Google Scholar] [CrossRef] [PubMed]
- Kubanik, M.; Holtkamp, H.; Söhnel, T.; Jamieson, S.M.F.; Hartinger, C.G. Impact of the halogen substitution pattern on the biological activity of organoruthenium 8-hydroxyquinoline anticancer agents. Organometallics 2015, 34, 5658–5668. [Google Scholar] [CrossRef]
- Dömötör, O.; Pape, V.F.S.; May, N.V.; Szakács, G.; Enyedy, É.A. Comparative solution equilibrium studies of antitumor ruthenium(η6-p-cymene) and rhodium (η5-C5Me5) complexes of 8-hydroxyquinolines. Dalton Trans. 2017, 46, 4382–4396. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, M.C.; Kljun, J.; Turel, I.; Di Virgilio, A.L.; León, I.E. Comparative antitumor studies of organoruthenium complexes with 8-hydroxyquinolines on 2D and 3D cell models of bone, lung and breast cancer. Metallomics 2019, 11, 666–675. [Google Scholar] [CrossRef] [PubMed]
- Kljun, J.; León, I.E.; Peršič, Š.; Cadavid-Vargas, J.F.; Etcheverry, S.B.; He, W.; Bai, Y.; Turel, I. Synthesis and biological characterization of organoruthenium complexes with 8-hydroxyquinolines. J. Inorg. Biochem. 2018, 186, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Meng, T.; Qin, Q.P.; Chen, Z.L.; Zou, H.H.; Wang, K.; Liang, F.P. Discovery of high in vitro and in vivo antitumor activities of organometallic ruthenium (II)–arene complexes with 5, 7-dihalogenated-2-methyl-8-quinolinol. Dalton Trans. 2019, 48, 5352–5360. [Google Scholar] [CrossRef] [PubMed]
- Yano, E.; Riisom, M.; Tong, K.K.H.; Hanif, M.; Leung, E.; Hartinger, C.G. Tracing the anticancer compound [RuII(η6-p-cymene)(8-oxyquinolinato)Cl] in a biological environment by mass spectrometric methods. Anal. Methods 2021, 13, 1463–1469. [Google Scholar] [CrossRef]
- Antić, D.R.; Parčina, M.; Gobin, I.; Didović, M.P. Chelation in antibacterial drugs: From nitroxoline to cefiderocol and beyond. Antibiotics 2022, 11, 1105. [Google Scholar] [CrossRef]
- Nepali, K.; Lee, H.Y.; Liou, J.P. Nitro-group-containing drugs. J. Med. Chem. 2019, 62, 2851–2893. [Google Scholar] [CrossRef]
- Ramenskaya, L.M.; Kraeva, O.V. Dissociation constants for the zinc (II)-8-hydroxyquinoline complexes in aqueous and sodium dodecyl sulfate micellar solutions. Russ. J. Phys. Chem. 2006, 80, 90–94. [Google Scholar] [CrossRef]
- Smith, R.M.; Martell, A.E.; Motekaitis, R.J. NIST Critically Selected Stability Constants of Metal Complexes Database; National Institute of Standards and Technology, U.S. Department of Commerce: Gaithersburg, MD, USA, 2004.
- Chen, H.; Parkinson, J.A.; Morris, R.E.; Sadler, P.J. Highly selective binding of organometallic ruthenium ethylenediamine complexes to nucleic acids: Novel recognition mechanisms. J. Am. Chem. Soc. 2003, 125, 173–186. [Google Scholar] [CrossRef] [PubMed]
- Fanali, G.; di Masi, A.; Trezza, V.; Marino, M.; Fasano, M.; Ascenzi, P. Human serum albumin: From bench to bedside. Mol. Aspects Med. 2012, 33, 209–290. [Google Scholar] [CrossRef] [PubMed]
- Elsadek, B.; Kratz, F. Impact of albumin on drug delivery—New applications on the horizon. J. Control Release 2012, 157, 4–28. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Luo, Q.; Ma, X.; Wu, K.; Liu, J.; Chen, Y.; Xiong, S.; Wang, J.; Sadler, P.J.; Wang, F. Arene control over thiolate to sulfinate oxidation in albumin by organometallic ruthenium anticancer complexes. Chem. Eur. J. 2009, 15, 6586–6594. [Google Scholar] [CrossRef]
- Dömötör, O.; Enyedy, É.A. Binding mechanisms of half-sandwich Rh(III) and Ru(II) arene complexes on human serum albumin: A comparative study. J. Biol. Inorg. Chem. 2019, 24, 703–719. [Google Scholar] [CrossRef]
- Dömötör, O.; Pivarcsik, T.; Mészáros, J.P.; Szatmári, I.; Fülöp, F.; Enyedy, É.A. Critical factors affecting the albumin binding of half-sandwich Ru(II) and Rh(III) complexes of 8-hydroxyquinolines and oligopyridines. Dalton Trans. 2021, 50, 11918–11930. [Google Scholar] [CrossRef]
- Hagenbuch, B.; Stieger, B. The SLCO (former SLC21) superfamily of transporters. Mol. Aspects Med. 2013, 34, 396–412. [Google Scholar] [CrossRef]
- König, J.; Cui, Y.; Nies, A.T.; Keppler, D. A novel human organic anion transporting polypeptide localized to the basolateral hepatocyte membrane. Am. J. Physiol. Gastrointest. Liver Physiol. 2000, 278, 156–164. [Google Scholar] [CrossRef]
- Tamai, I.; Nezu, J.I.; Uchino, H.; Sai, Y.; Oku, A.; Shimane, M.; Tsuji, A. Molecular identification and characterization of novel members of the human organic anion transporter (OATP) family. Biochem. Biophys. Res. Comm. 2000, 273, 251–260. [Google Scholar] [CrossRef]
- Kovacsics, D.; Patik, I.; Özvegy-Laczka, C. The role of organic anion transporting polypeptides in drug absorption, distribution, excretion and drug-drug interactions. Expert Opin. Drug Metab. Toxicol. 2017, 13, 409–424. [Google Scholar] [CrossRef]
- Guideline on the Investigation of Drug Interactions. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-investigation-drug-interactions-revision-1_en.pdf (accessed on 27 November 2022).
- In Vitro Metabolism and Transporter Mediated Drug-Drug Interaction Studies, Guidance for Industry. Available online: https://www.fda.gov/files/drugs/published/In-Vitro-Metabolism--and-Transporter--Mediated-Drug-Drug-Interaction-Studies-Guidance-for-Industry.pdf (accessed on 27 November 2022).
- Shitara, Y.; Maeda, K.; Ikejiri, K.; Yoshida, K.; Horie, T.; Sugiyama, Y. Clinical significance of organic anion transporting polypeptides (OATPs) in drug disposition: Their roles in hepatic clearance and intestinal absorption. Biopharm. Drug Dispos. 2013, 34, 45–78. [Google Scholar] [CrossRef] [PubMed]
- Droździk, M.; Lapczuk-Romanska, J.; Wenzel, C.; Skalski, Ł.; Szeląg-Pieniek, S.; Post, M.; Syczewska, M.; Kurzawski, M.; Oswald, S. Protein abundance of drug transporters in human Hepatitis C livers. Int. J. Mol. Sci. 2022, 23, 7947. [Google Scholar] [CrossRef] [PubMed]
- Nozawa, T.; Imai, K.; Nezu, J.I.; Tsuji, A.; Tamai, I. Functional characterization of pH-sensitive organic anion transporting polypeptide OATP-B in human. J. Pharmacol. Exp. Ther. 2004, 308, 438–445. [Google Scholar] [CrossRef] [PubMed]
- Ungvári, O.; Király, L.; Bakos, É.; Özvegy-Laczka, C. 8-acetoxy-trisulfopyrene as the first activatable fluorogenic probe for add-and-read assessment of organic anion-transporting polypeptides, OATP1B1, OATP1B3, and OATP2B1. FASEB J. 2021, 35, e21863. [Google Scholar] [CrossRef]
- Balázs, B.; Tóth, Z.; Kacsir, I.; Sipos, A.; Buglyó, P.; Somsák, L.; Bokor, É.; Kardos, B.; Bai, P. Targeting multiresistant Gram-positive bacteria by ruthenium, osmium, iridium and rhodium half-sandwich type complexes with bidentate monosaccharide ligands. Front. Chem. 2022, 10, 868234. [Google Scholar] [CrossRef]
- Bal, A.M.; David, M.Z.; Garau, J.; Gottlieb, T.; Mazzei, T.; Scaglione, F.; Tattevin, P.; Gould, I.M. Future trends in the treatment of methicillin-resistant Staphylococcus aureus (MRSA) infection: An in-depth review of newer antibiotics active against an enduring pathogen. J. Glob. Antimicrob. Resist. 2017, 10, 295–303. [Google Scholar] [CrossRef]
- Gans, P.; Sabatini, A.; Vacca, A. Investigation of equilibria in solution. Determination of equilibrium constants with the HYPERQUAD suite of programs. Talanta 1996, 43, 1739–1753. [Google Scholar] [CrossRef]
- Beaven, G.H.; Chen, S.-H.; D’albis, A.; Gratzer, W.B. A Spectroscopic Study of the Haemin–Human-Serum-Albumin System. Eur. J. Biochem. 1974, 41, 539–546. [Google Scholar] [CrossRef]
- Irving, H.M.; Miles, M.G.; Pettit, L.D. A study of some problems in determining the stoicheiometric proton dissociation constants of complexes by potentiometric titrations using a glass electrode. Anal. Chim. Acta 1967, 38, 475–488. [Google Scholar] [CrossRef]
- Pettit, L.D.; Powell, H.K.J. SCQuery. The IUPAC Stability Constants Database; Academic Software, Version 5.5; Royal Society of Chemistry: London, UK, 1993. [Google Scholar]
- Enyedy, É.A.; May, N.V.; Pape, V.F.S.; Heffeter, P.; Szakács, G.; Keppler, B.K.; Kowol, C.R. Complex formation and cytotoxicity of Triapine derivatives: A comparative solution study on the effect of the chalcogen atom and NH-methylation. Dalton Trans. 2020, 49, 16887–16902. [Google Scholar] [CrossRef]
- Enyedy, É.A.; Primik, M.F.; Kowol, C.R.; Arion, V.B.; Kiss, T.; Keppler, B.K. Interaction of Triapine and related thiosemicarbazones with iron(III)/(II) and gallium(III): A comparative solution equilibrium study. Dalton Trans. 2011, 40, 5895–5905. [Google Scholar] [CrossRef] [PubMed]
- Baes, C.F.; Mesmer, R.E. The Hydrolysis of Cations; Wiley: New York, NY, USA, 1976. [Google Scholar]
- Brown, P.L.; Ekberg, C. Hydrolysis of Metal Ions; Wiley: New York, NY, USA, 2016; pp. 573–585. [Google Scholar]
- Bíró, L.; Godó, A.J.; Bihari, Z.; Garribba, E.; Buglyó, P. Tuning the hydrolytic properties of half-sandwich-type organometallic cations in aqueous solution. Eur. J. Inorg. Chem. 2013, 2013, 3090–3100. [Google Scholar] [CrossRef]
- Dömötör, O.; Aicher, S.; Schmidlehner, M.; Novak, M.S.; Roller, A.; Jakupec, M.A.; Kandioller, W.; Hartinger, C.G.; Keppler, B.K.; Enyedy, É.A. Antitumor pentamethylcyclopentadienyl rhodium complexes of maltol and allomaltol: Synthesis, solution speciation and bioactivity. J. Inorg. Biochem. 2014, 134, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Lakowicz, J.R. Principles of Fluorescence Spectroscopy; Springer: New York, NY, USA, 2006. [Google Scholar]
- Rockenbauer, A.; Korecz, L. Automatic computer simulations of ESR spectra. Appl. Magn. Reson. 1996, 10, 29–43. [Google Scholar] [CrossRef]
- Zhai, W.; Feng, Y.; Liu, H.; Rockenbauer, A.; Mance, D.; Li, S.; Song, Y.; Baldus, M.; Liu, Y. Diastereoisomers of L-proline-linked trityl-nitroxide biradicals: Synthesis and effect of chiral configurations on exchange interactions. Chem. Sci. 2018, 9, 4381–4391. [Google Scholar] [CrossRef]
- Patik, I.; Székely, V.; Német, O.; Szepesi, Á.; Kucsma, N.; Várady, G.; Szakács, G.; Bakos, É.; Özvegy-Laczka, C. Identification of novel cell-impermeant fluorescent substrates for testing the function and drug interaction of organic anion-transporting polypeptides, OATP1B1/1B3 and 2B1. Sci. Rep. 2018, 8, 2630. [Google Scholar] [CrossRef]
- Bottke, D.; Koychev, D.; Busse, A.; Heufelder, K.; Wiegel, T.; Thiel, E.; Hinkelbein, W.; Keilholz, U. Fractionated irradiation can induce functionally relevant multidrug resistance gene and protein expression in human tumor cell lines. Radiat. Res. 2008, 170, 41–48. [Google Scholar] [CrossRef]
- Windt, T.; Tóth, S.; Patik, I.; Sessler, J.; Kucsma, N.; Szepesi, Á.; Zdrazil, B.; Özvegy-Laczka, C.; Szakács, G. Identification of anticancer OATP2B1 substrates by an in vitro triple-fluorescence-based cytotoxicity screen. Arch. Toxicol. 2019, 93, 953–964. [Google Scholar] [CrossRef]
- CLSI. Susceptibility testing process. In Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically, 10th ed.; Christopher, P.J., Polgar, E.P., Eds.; Clinical and Laboratory Standards Institute: Wayne, MI, USA, 2015; Volume 32, pp. 15–19. [Google Scholar]
- Rózga, M.; Sokołowska, M.; Protas, A.M.; Bal, W. Human serum albumin coordinates Cu(II) at its N-terminal binding site with 1 pM affinity. Biol. Inorg. Chem. 2007, 12, 913–918. [Google Scholar] [CrossRef]
- Linder, M.C. Ceruloplasmin and other copper binding components of blood plasma and their functions: An update. Metallomics 2016, 8, 887–905. [Google Scholar] [CrossRef]
- Boyett, J.D.; Sullivan, J.F. Distribution of protein-bound zinc in normal and cirrhotic serum. Metabolism 1970, 19, 148–157. [Google Scholar] [CrossRef]
- Harris, W.R. Thermodynamic binding constants of the zinc-human serum transferrin complex. Biochemistry 1983, 22, 3920–3926. [Google Scholar] [CrossRef] [PubMed]
- Masuoka, J.; Saltman, P.J. Zinc(II) and copper(II) binding to serum albumin. A comparative study of dog, bovine, and human albumin. J. Biol. Chem. 1994, 269, 25557–25561. [Google Scholar] [CrossRef] [PubMed]
- Ohyoshi, E.; Hamada, Y.; Nakata, K.; Kohata, S. The interaction between human and bovine serum albumin and zinc studied by a competitive spectrophotometry. J. Inorg. Chem. 1999, 75, 213–218. [Google Scholar] [CrossRef] [PubMed]
- Aisen, P.; Leibman, A.; Zweier, J. Stoichiometric and site characteristics of the binding of iron to human transferrin. J. Biol. Chem. 1978, 253, 1930–1937. [Google Scholar] [CrossRef] [PubMed]
- Enyedy, É.A.; Mészáros, J.P.; Dömötör, O.; Hackl, C.M.; Roller, A.; Keppler, B.K.; Kandioller, W. Comparative solution equilibrium studies on pentamethylcyclopentadienyl rhodium complexes of 2,2′-bipyridine and ethylenediamine and their interaction with human serum albumin. J. Inorg. Biochem. 2015, 152, 93–103. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | Ionic Strength (I) | pKa (OH) | pKa (NProH+) | cligand/μM |
|---|---|---|---|---|
| pH-potentiometry | 0.20 M KNO3 | 5.57 ± 0.09 | 10.68 ± 0.07 | 1700 |
| 1H NMR | 0.20 M KNO3 | 5.56 ± 0.03 | >10.5 | 1000 |
| UV-vis | 0.20 M KNO3 | 5.50 ± 0.03 | 10.78 ± 0.03 | 40 |
| UV-vis | 0.10 M KCl | 5.43 ± 0.03 | 10.75 ± 0.03 | 40 |
| logβ | Fe(III) | Fe(II) | Cu(II) | Zn(II) |
|---|---|---|---|---|
| [M(HL)] | 19.75 ± 0.03 | 16.52 ± 0.03 | 19.40 ± 0.03 | 17.36 ± 0.03 |
| [M(L)] | − | 8.39 ± 0.03 | 14.45 ± 0.03 | n.d. a |
| [M(HL)2] | 38.76 ± 0.03 | 32.74 ± 0.03 | 38.60 ± 0.03 | 33.71 ± 0.06 |
| [M(HL)(L)] | − | − | 31.75 ± 0.03 | n.d. a |
| [M(L)2] | − | − | 22.99 ± 0.03 | n.d. a |
| [M(HL)3] | 55.66 ± 0.03 | 47.60 ± 0.06 | − | − |
| RhCp* Complex | RuCym Complex | |
|---|---|---|
| pKa (1) | 9.33 ± 0.03 | 8.55 ± 0.03 |
| pKa (2) | 10.34 ± 0.03 | 10.21 ± 0.03 |
| logK’ (H2O/Cl−) a | 2.07 ± 0.03 | 1.47 ± 0.01 |
| IC50 (μM) | ||||
|---|---|---|---|---|
| MES-SA | MES-SA/Dx5 | Colo205 | Colo320 | |
| ligand alone | ||||
| HQNO2-L-Pro | 226 ± 17 | 97 ± 29 | 34 ± 1 | 32.4 ± 0.4 |
| HQNO2-L-Pro + TQ | 223 ± 6 | 219 ± 46 | - | - |
| HQCl-L-Pro | 23 ± 4 | 4.3 ± 0.1 | 23 ± 3 a | 9 ± 2 a |
| complexes | ||||
| RuCym-HQNO2-L-Pro | 166 ± 37 | >400 | >100 | >100 |
| RhCp*-HQNO2-L-Pro | 167 ± 38 | 106 ± 8 | 35 ± 1 | 29.8 ± 0.2 |
| RuCym-HQCl-L-Pro | - | - | 68 ± 11 a | >100 a |
| RhCp*-HQCl-L-Pro | - | - | 26 ± 5 a | 10 ± 1 a |
| incubation of HQNO2-L-Pro with metal salts | ||||
| +1 equiv. Cu(II) | 40 ± 7 | 25 ± 6 | - | - |
| +0.5 equiv. Cu(II) | 49 ± 15 | 35 ± 11 | - | - |
| +1 equiv. Zn(II) | 164 ± 24 | >400 | - | - |
| +0.5 equiv. Zn(II) | >400 | >400 | - | - |
| +1 equiv. Fe(III) | 89 ± 9 | 47 ± 9 | - | - |
| +0.33 equiv. Fe(III) | 121 ± 27 | 53 ± 9 | - | - |
| doxorubicin | 0.03 ± 0.01 | 4 ± 1 | 0.71 ± 0.02 | 5.4 ± 0.9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pivarcsik, T.; Pósa, V.; Kovács, H.; May, N.V.; Spengler, G.; Pósa, S.P.; Tóth, S.; Nezafat Yazdi, Z.; Özvegy-Laczka, C.; Ugrai, I.; et al. Metal Complexes of a 5-Nitro-8-Hydroxyquinoline-Proline Hybrid with Enhanced Water Solubility Targeting Multidrug Resistant Cancer Cells. Int. J. Mol. Sci. 2023, 24, 593. https://doi.org/10.3390/ijms24010593
Pivarcsik T, Pósa V, Kovács H, May NV, Spengler G, Pósa SP, Tóth S, Nezafat Yazdi Z, Özvegy-Laczka C, Ugrai I, et al. Metal Complexes of a 5-Nitro-8-Hydroxyquinoline-Proline Hybrid with Enhanced Water Solubility Targeting Multidrug Resistant Cancer Cells. International Journal of Molecular Sciences. 2023; 24(1):593. https://doi.org/10.3390/ijms24010593
Chicago/Turabian StylePivarcsik, Tamás, Vivien Pósa, Hilda Kovács, Nóra V. May, Gabriella Spengler, Szonja P. Pósa, Szilárd Tóth, Zeinab Nezafat Yazdi, Csilla Özvegy-Laczka, Imre Ugrai, and et al. 2023. "Metal Complexes of a 5-Nitro-8-Hydroxyquinoline-Proline Hybrid with Enhanced Water Solubility Targeting Multidrug Resistant Cancer Cells" International Journal of Molecular Sciences 24, no. 1: 593. https://doi.org/10.3390/ijms24010593
APA StylePivarcsik, T., Pósa, V., Kovács, H., May, N. V., Spengler, G., Pósa, S. P., Tóth, S., Nezafat Yazdi, Z., Özvegy-Laczka, C., Ugrai, I., Szatmári, I., Szakács, G., & Enyedy, É. A. (2023). Metal Complexes of a 5-Nitro-8-Hydroxyquinoline-Proline Hybrid with Enhanced Water Solubility Targeting Multidrug Resistant Cancer Cells. International Journal of Molecular Sciences, 24(1), 593. https://doi.org/10.3390/ijms24010593